Back to Journals » OncoTargets and Therapy » Volume 11
RIP1 regulates TNF-α-mediated lymphangiogenesis and lymphatic metastasis in gallbladder cancer by modulating the NF-κB-VEGF-C pathway
Authors Li C ![]() , Jiang X, Lin B, Hong H, Zhu S, Jiang L, Wang X, Tang N, She F, Chen Y
, Jiang X, Lin B, Hong H, Zhu S, Jiang L, Wang X, Tang N, She F, Chen Y
Received 6 December 2017
Accepted for publication 9 March 2018
Published 16 May 2018 Volume 2018:11 Pages 2875—2890
DOI https://doi.org/10.2147/OTT.S159026
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Ingrid Espinoza
Cheng-Zong Li,1–3,* Xiao-Jie Jiang,1,2,* Bin Lin,1,2 Hai-Jie Hong,1,2 Si-Yuan Zhu,1,2 Lei Jiang,1,2 Xiao-Qian Wang,1 Nan-Hong Tang,1 Fei-Fei She,2 Yan-Ling Chen1,2
1Department of Hepatobiliary Surgery and Fujian Institute of Hepatobiliary Surgery, Fujian Medical University Union Hospital, Fuzhou, People’s Republic of China; 2Key Laboratory of the Ministry of Education for Gastrointestinal Cancer and Key Laboratory of Tumour Microbiology, School of Basic Medical Sciences, Fujian Medical University, Fuzhou, People’s Republic of China; 3Department of General Surgery, The Second Affiliated Hospital Of Fujian Medical University, Quanzhou, People’s Republic of China
*These authors contributed equally to this work
Background: Tumor necrosis factor alpha (TNF-α) enhances lymphangiogenesis in gallbladder carcinoma (GBC) via activation of nuclear factor (NF-κB)-dependent vascular endothelial growth factor-C (VEGF-C). Receptor-interacting protein 1 (RIP1) is a multifunctional protein in the TNF-α signaling pathway and is highly expressed in GBC. However, whether RIP1 participates in the signaling pathway of TNF-α-mediated VEGF-C expression that enhances lymphangiogenesis in GBC remains unclear.
Methods: The RIP1 protein levels in the GBC-SD and NOZ cells upon stimulation with increasing concentrations of TNF-α as indicated was examined using Western blot. Lentiviral RIP1 shRNA and siIκBα were constructed and transduced respectively them into NOZ and GBC-SD cells, and then PcDNA3.1-RIP1 vectors was transduced into siRIP1 cell lines to reverse RIP1 expression. The protein expression of RIP1, inhibitor of NF-κB alpha (IκBα), p-IκBα, TAK1, NF-κB essential modulator were examined through immunoblotting or immunoprecipitation. Moreover, VEGF-C mRNA levels were measured by quantitative real-time polymerase chain reaction, VEGF-C protein levels were measured by immunoblotting and enzyme-linked immunosorbent assay, and VEGF-C promoter and NF-κB activities were quantified using a dual luciferase reporter assay. The association of NF-κB with the VEGF-C promoter was analysed by chromatin immunoprecipitation assay. A three-dimensional coculture method and orthotopic transplantation nude mice model were used to evaluate lymphatic tube-forming and metastasis ability in GBC cells. The expression of RIP1 protein, TNF-α protein and lymphatic vessels in human GBC tissues was examined by immunohistochemistry, and the dependence between RIP1 protein with TNF-α protein and lymphatic vessel density was analysed.
Results: TNF-α dose- and time-dependently increased RIP1 protein expression in the GBC-SD and NOZ cells of GBC, and the strongest effect was observed with a concentration of 50 ng/ml.
RIP1 is fundamental for TNF-α-mediated NF-κB activation in GBC cells and can regulate TNF-α-mediated VEGF-C expression at the protein and transcriptional levels through the NF-κB pathway. RIP1 can regulate TNF-α-mediated lymphatic tube formation and metastasis in GBC cells both in vitro and vivo. The average optical density of RIP1 was linearly related to that of TNF-α protein and the lymphatic vessel density in GBC tissues.
Conclusion: We conclude that RIP1 regulates TNF-α-mediated lymphangiogenesis and lymph node metastasis in GBC by modulating the NF-κB-VEGF-C pathway.
Keywords: gallbladder cancer, RIP1, TNF-α, NF-κB, VEGF-C, lymphangiogenesis
Introduction
Gallbladder cancer (GBC) is a relatively infrequent but fatal disease, and <10% of cases have the opportunity to undergo surgical treatment.1 Moreover, the overall 5-year survival rate is <5%.2 This malignancy exhibits a propensity for lymphatic metastasis; the incidence of regional lymphatic metastasis at stages II and III/IV is in the range of 19%–62% and 75%–85%, respectively.1,3,4 Thus, lymph node metastasis (LNM) serves as a significant factor in the prognosis of GBC.5 However, the mechanism of lymphatic metastasis in GBC is poorly understood.
Studies have confirmed that cancer development and progression are closely associated with chronic inflammation.6 We previously reported that tumor necrosis factor alpha (TNF-α), a well-characterized proinflammatory factor, can activate nuclear factor (NF)-κB-dependent vascular endothelial growth factor-C (VEGF-C) to enhance lymphangiogenesis in GBC.7,8 However, the signaling pathways that participate in the TNF-α-mediated activation of NF-κB-dependent VEGF-C are complicated.9–11 Therefore, elucidating the precise molecular mechanism underlying TNF-α-mediated lymphatic signaling is critical for the clinical treatment of lymphatic metastasis in GBC.
TNF-α can activate death receptors such as TNF-α receptor 1 (TNFR1) and TNF-α receptor 2 (TNFR2).12 Upon stimulation of TNFR1, receptor-interacting protein 1 (RIP1) forms a complex with other proteins and factors referred to as Complex I.13,14 Furthermore, RIP1 acts as a scaffold for combination with NF-κB essential modulator (NEMO), transforming growth factor beta (TGFβ)-activated kinase 1 (TAK1), and the TAK1-binding proteins TAB1 and TAB2, which leads to phosphorylation of inhibitor of NF-κB alpha (IκBα), as well as degradation and induction of NF-κB-dependent genetic transcription.15 However, whether RIP1 participates in the signaling pathways associated with TNF-α-mediated VEGF-C expression leading to the enhancement of lymphangiogenesis in GBC remains unclear.
In the present study, TNF-α dose and time dependently increased RIP1 mRNA and protein expression in the human GBC cell lines GBC-SD and NOZ. RIP1 was essential for TNF-α-mediated NF-κB activation and could regulate TNF-α-mediated VEGF-C expression at the protein and transcriptional levels through the NF-κB pathway in GBC cells. Subsequently, we showed that RIP1 could regulate TNF-α-mediated lymphangiogenesis and lymphatic metastasis in vivo and ex vivo. Finally, our findings confirmed that expression of the RIP1 protein was linearly associated with that of the TNF-α protein and the lymphatic vessels in human GBC specimens.
Materials and methods
Cell lines and human specimens
Human dermal lymphatic endothelial cell line (HDLEC, purchased from ScienCell, San Diego, CA, USA), Jurkat cell line (purchased from ScienCell), and human GBC cell lines, including NOZ (purchased from Health Science Research Resources Bank, Osaka, Japan) and GBC-SD (purchased from Shanghai Institute of Cell Bank, Chinese Academy of Sciences) were purchased and cultured as previously described.16 Thirty GBC samples were obtained from patients admitted to the Hepatopancreatobiliary Surgery Department of Fujian Union Hospital. Written informed consent was obtained from all patients prior to tumor resection. All experiments using human specimens were authorized by the Ethics Review Committee of Fujian Union Hospital.
Quantitative real-time polymerase chain reaction (qPCR)
Extraction of total RNA was performed using the TRIzol reagent from Thermo Fisher Scientific (Waltham, MA, USA). To synthesize complementary DNA, 1 μg of total RNA extract and a reverse transcription reagent (Thermo Fisher Scientific) was used. Then, 0.5 μL of the complementary DNA (cDNA) sample and qPCR Master Mix (Roche, Indianapolis, IN, USA) were used for quantitative real-time (RT) PCR with the ABI 7500 RT fluorescent qPCR system (Applied Biosystems; Thermo Fisher Scientific), according to the instructions provided with the SYBR@ Green reagent. Each condition was analyzed in triplicate.
Immunoblotting
Immunoblotting analysis was conducted as previously described.17 The antibodies used in these analyses included anti-RIP1 (ab72139, 1:1,000; Abcam, Cambridge, UK), anti-IκBα (ab32518, 1:1,000; Abcam), anti-phospho-IκBα (ab133462, 1:10,000; Abcam), anti-VEGF-C (ab9546, 1:1,000; Abcam), anti-TAK1 (sc-7967, 1:1,000; Santa Cruz Biotechnology Inc., Dallas, TX, USA), anti-NEMO (ab97642, Abcam, 1:1,000), and anti-GAPDH (ab8245, 1:1,000; Abcam). The detected proteins were semiquantified relative to GAPDH expression in each gel.
Immunoprecipitation
Immunoprecipitation lysis buffer containing a proteasome inhibitor mix was used to lyse the collected cells for 30 min on ice. The cell lysates were then centrifuged at 12,000 rpm at 4°C for 30 min, and the lysate supernatant, which was immunoprecipitated with 2 μg of the corresponding antibody and protein A/G-beads, was slowly shaken and subsequently incubated overnight in a 4°C refrigerator. After immunoprecipitation, the protein A/G beads were centrifuged at 3,000 rpm at 4°C for 5 min to collect them at the bottom of the tube. The collected protein A/G beads were rinsed four times with 1 mL of immunoprecipitation buffer, and the 2× sample-loading buffer was then added, followed by boiling for 10 min. These samples were then prepared for further immunoblotting.
siRNA and shRNA
The RIP1 siRNA sequence (5′-GCACAAATACGAACTTCAA-3′) used in this work has been described previously.18 The IκBα siRNA sequence (GenBank accession no NM_020529) was synthesized by Suzhou GenePharma Co, Ltd (Suzhou, People’s Republic of China). Plasmids containing the shRNA targeting RIP1 or a negative control were purchased from Suzhou GenePharma Co, Ltd.
Plasmid vectors and transfection
We constructed lentiviral vectors (LV3-H1/GFP&Puro; GenePharma, Suzhou, People’s Republic of China) expressing either RIP1-targeting shRNA or a negative control-targeting shRNA and transduced them into GBC cells. PcDNA3.1-RIP1 plasmid vectors were previously constructed in our laboratory.19 The pGL3-Basic and pRL-TK vectors were purchased from Promega Corporation (Fitchburg, WI, USA), and the pGL3B-332 vector with the VEGF-C promoter sequence (−332/+1) was constructed as previously described.8 NF-κB-luc reporter plasmids were synthesized by the Beyotime Institute of Biotechnology (Shanghai, People’s Republic of China). Transfection of the siIκBα, PcDNA3.1-RIP1, pRL-TK, pGL3-Basic, pGL3B-332, and NF-κB-luc reporter plasmids was conducted in Opti-MEM medium (Thermo Fisher Scientific) using Lipofectamine reagent (Thermo Fisher Scientific).
Dual-luciferase reporter assay
After cotransfection with the pRL-TK plasmid vector (containing the Renilla and firefly reporter gene) and the NF-κB, pGL3Basic, or pGL3B-332 (containing the VEGF-C promoter sequence) luciferase reporter plasmid vectors, the cells were either stimulated with recombinant human TNF-α (50 ng/mL) or left unstimulated for 24 h. Thereafter, the cells were harvested and lysed with lysis buffer, and the activity of Renilla and firefly luciferase was examined with the Dual-Luciferase Reporter (DLR™) Assay System (Promega Corporation) using a Glo-Max Microplate Luminometer (Promega Corporation) according to the manufacturer’s instructions.
Enzyme-linked immunosorbent assay (ELISA)
The VEGF-C concentration in culture supernatants was analyzed via an ELISA method using an ELISA kit from R&D Systems, Inc. (Minneapolis, MN, USA) as previously described.8 A multimode reader (Bio-Rad Laboratories Inc., Hercules, CA, USA) was used to read the absorbance at 450 nm. Each measurement was performed in triplicate.
Chromatin immunoprecipitation (ChIP)
ChIP was performed using a ChIP kit from Merck Millipore (Billerica, MA, USA) as previously described.8 First, cells were cross-linked and fixed with 1% formaldehyde for 10 min at 37°C in an incubator. Then, 1 M glycine was added for 5 min at room temperature to terminate the reaction, and the sample was washed with ice-cold PBS 2 times. Thereafter, the cells were harvested in sodium dodecyl sulfate (SDS) lysis buffer containing a protease inhibitor cocktail containing phenylmethylsulfonyl fluoride. Then, the chromatin in the cell lysates was sheared into fragments of 400–600 bp via ultrasonic fragmentation. NF-κB immunoprecipitation reactions were performed with 5 μg of NF-κB antibody, and rabbit serum was used as a negative control. The VEGF-C promoter fragment (−389/−278) containing the NF-κB binding sites has been previously identified, and it was amplified with the following primers: F: 5′-gac agg ggc ggg gag gga ga-3′ and R: 5′-ctc act ctc cct cgg aag ccg tct c-3′.
Lymphangiogenesis assay
The HDLEC tube formation assay was performed as previously described.20 The μ-slides (ibidi, Martinsried, Germany) were previously coated with 10 μL of Matrigel (BD Biosciences, San Jose, CA, USA) and incubated in a 37°C incubator for 30 min. HDLECs were also previously labeled with a red fluorescent cell membrane probe (Thermo Fisher Scientific). Thereafter, GBC cells (7.5 × 103 cells per well) were cocultured with HDLECs (7.5 × 103 cells per well) on the Matrigel-based μ-slides and polymerized in serum-free medium with or without 50 ng/mL of TNF-α in a 37°C incubator. After 1, 3, 6, 8, and 24 h, tube formation was photographed under a fluorescence microscope. The data and images of tube formation were analyzed using Carl Zeiss Axiovision imaging system Release 4.8 (Carl Zeiss Meditec AG, Jena, Germany).
Animal experiments
The orthotopic murine transplantation model of GBC was constructed based on the method described previously.21 Briefly, about 20 μL cell suspension (containing 4 × 105 cells) mixed with Matrigel was injected into the gallbladder with a 26G insulin syringe (BD, Franklin Lakes, NJ, USA). Then, 2 weeks after establishing the xenograft mouse model, intraperitoneal injection of recombinant human TNF-α (2 μg/kg) was performed 2 days a week for 3 weeks. Then, the mice were sacrificed via decapitation, and the primary tumors were removed and soaked in formalin liquid for further hematoxylin and eosin staining and immunohistochemical assays. All experiments involving GBC xenografts were authorized by the Ethics Review Committee of Fujian Union Hospital. Moreover, the animal care procedures and health program were performed according to the criteria of the Animal Welfare Act and the Public Health Services “Guide for the Care and Use of Laboratory Animals”.
Immunohistochemistry
The primary antibodies used for immunohistochemistry targeted the following proteins: TNF-α (sc-52746, 1:100; Santa Cruz Biotechnology Inc.), RIP1 (ab72139, 1:500; Abcam), D2-40 (1:150; Maixin-Bio, Fuzhou, People’s Republic of China), and LYVE-1 (AF2125, 1:250; R&D Systems, Inc). Immunohistochemistry was performed as previously described.18 Expression of TNF-α and RIP1 proteins was semiquantitatively analyzed in 15 visual fields by calculating the average optical density. The expression of D2-40-positive and LYVE-1-positive vessels was analyzed as previously described.8
Statistical analyses
The data are expressed as the mean ± SEM. Differences between groups were determined with Student’s t-test. A p-value of <0.05 was considered significant.
Results
TNF-α enhances RIP1 mRNA and protein expression in vitro
From previous studies, we found that the Jurkat cell line was often used in the study of the TNF-α-RIP1 pathway.22,23 Therefore, we took the cell line as a positive control group and first detected the level of RIP1 mRNA by qPCR in the Jurkat cell line and the GBC lines upon stimulation by TNF-α. The results of qPCR showed that TNF-α could dose (the strongest effect was observed with a concentration of 50 ng/mL) and time dependently (treated with 50 ng/mL TNF-α) promote the expression of RIP1 mRNA in GBC cells, just as in the Jurkat cell line (Figure 1A and B). We further examined RIP1 protein levels in the GBC-SD and NOZ cells upon stimulation with increasing concentrations of TNF-α as indicated. The results showed that TNF-α dose dependently enhanced the expression of RIP1 protein in GBC-SD and NOZ cells, and the strongest effect was also observed with a concentration of 50 ng/mL (Figure 1C and E). We finally measured the expression of the RIP1 protein in these two cell lines treated with 50 ng/mL of recombinant human TNF-α for 6, 12, and 24 h, and found that TNF-α time dependently enhanced RIP1 protein expression in GBC-SD and NOZ cells (Figure 1D and F).
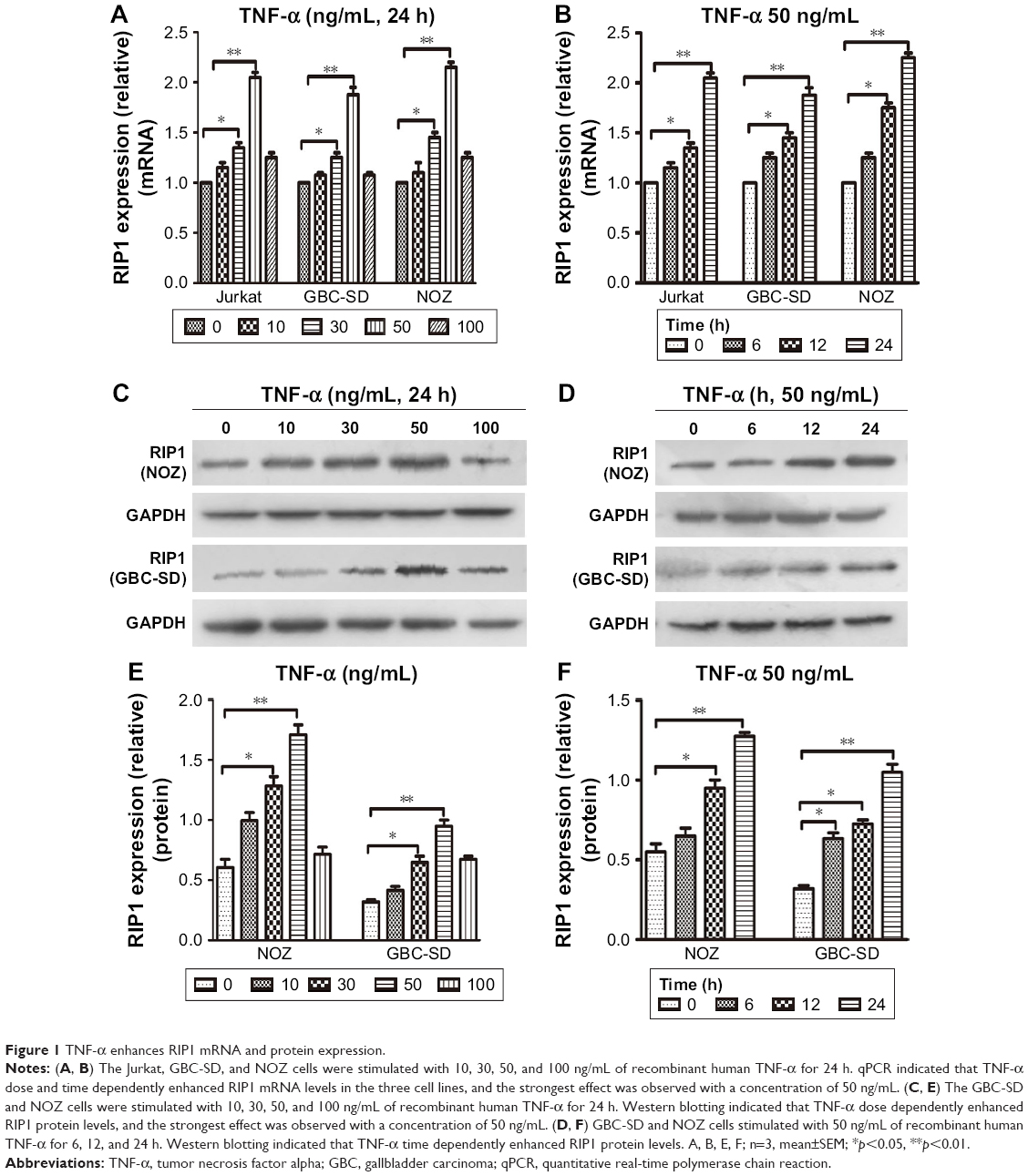 | Figure 1 TNF-α enhances RIP1 mRNA and protein expression. |
RIP1 is indispensable for TNF-α-mediated activation of NF-κB in vitro
To determine whether RIP1 contributes to NF-κB activation in TNF-α-stimulated GBC-SD and NOZ cells, we stably transduced these two cell lines with a lentiviral vector (LV3-H1/GFP&Puro) expressing RIP1 shRNA (LV-siRIP1) (Figure 2A) and negative control shRNA (LV-siNC) and used qPCR and Western blotting to examine RIP1 mRNA and protein levels in the control, LV-siNC, and LV-siRIP1 cell groups. We found that the levels of RIP1 mRNA and protein were markedly lower in the LV-siRIP1 group than in the LV-siNC and control groups (Figure 2B–D).
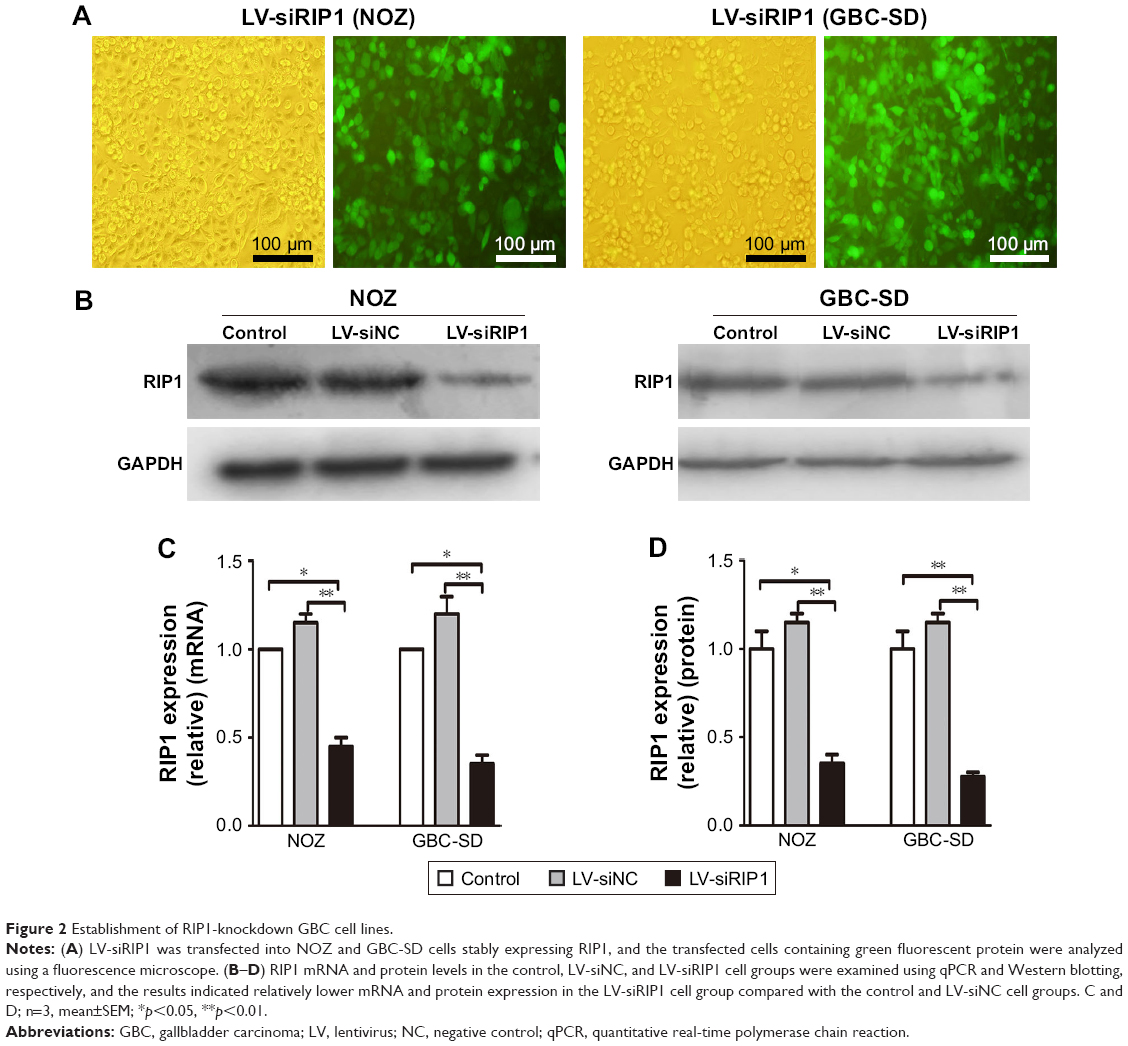 | Figure 2 Establishment of RIP1-knockdown GBC cell lines. |
Furthermore, we explored the function of RIP1 in the TNF-α-mediated activation of the NF-κB signaling pathway. We found that NF-κB activation was significantly enhanced in GBC-SD and NOZ cells upon TNF-α stimulation, and this enhancement by TNF-α was markedly abolished in RIP1-knockdown GBC-SD and NOZ cells (Figure 3A). Transfection with PcDNA3.1-RIP1 vector in the siRIP1 group reversed the abolished effect of RIP1 knockdown on the TNF-α-mediated activation of NF-κB (Figure 3A). Accordingly, the phosphorylation of IκBα was also significantly enhanced in GBC-SD and NOZ cells upon TNF-α stimulation, and this enhancement by TNF-α was also markedly abolished in RIP1-knockdown GBC-SD and NOZ cells (Figure 3B). Importantly, when IκBα was co-knocked down with RIP1 (Figure 3C–E), the inhibitory effect of RIP1 knockdown on the TNF-α-mediated activation of NF-κB was again reversed (Figure 3F). Moreover, as shown in Figure 3G, TAK1 and NEMO were coprecipitated with RIP1 in GBC-SD and NOZ cells. These findings further confirmed the function of RIP1 in the TNF-α-mediated activation of the NF-κB signaling pathway in GBC.
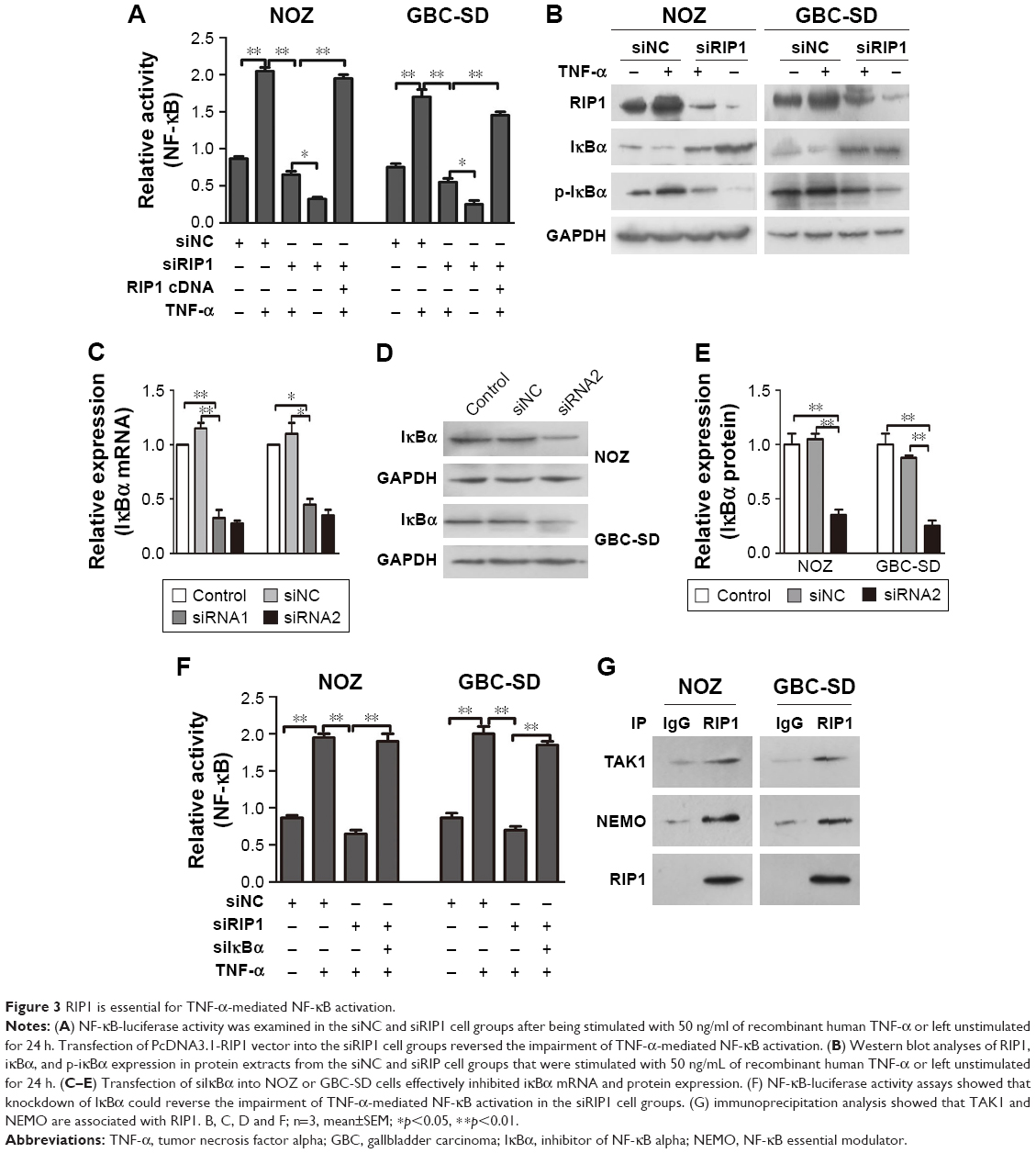 | Figure 3 RIP1 is essential for TNF-α-mediated NF-κB activation. |
RIP1 regulates TNF-α-mediated VEGF-C expression in GBC cells through the NF-κB pathway
TNF-α can promote the expression of VEGF-C mRNA and protein in some nontumor cells such as fibroblasts and rheumatoid synoviocytes.24,25 In our previous study, we found that TNF-α had a dramatic impact on the expression profile of VEGF-C in GBC cells, just as in human lung fibroblasts (Figure S1).8 Meanwhile, we also found that TNF-α can activate VEGF-D and CCR7 in GBC cells.16,26 Therefore, we first measured the expression of VEGF-A, VEGF-C, VEGF-D, and CCR7 mRNA in GBC cells and RIP1-knockdown GBC cells upon TNF-α stimulation. The results showed that the VEGF-C mRNA level was markedly impaired in RIP1-knockdown GBC-SD and NOZ cells, but VEGF-A, VEGF-D, and CCR7 mRNA levels were not markedly impaired (Figure S2).
In the present study, we further explored whether RIP1/NF-κB signaling participates in the TNF-α-mediated expression of VEGF-C mRNA and protein and confirmed that its expression was markedly enhanced in GBC-SD and NOZ cells upon TNF-α stimulation. This enhancement by TNF-α was markedly abolished in RIP1-knockdown GBC-SD and NOZ cells (Figure 4A, C, and D). We further demonstrated that NF-κB plays a role in TNF-α-mediated VEGF-C expression based on a significant decrease in VEGF-C mRNA and protein levels following treatment with Bay-11-7082, an NF-κB (p65) inhibitor (Figure 4B, E, and F).
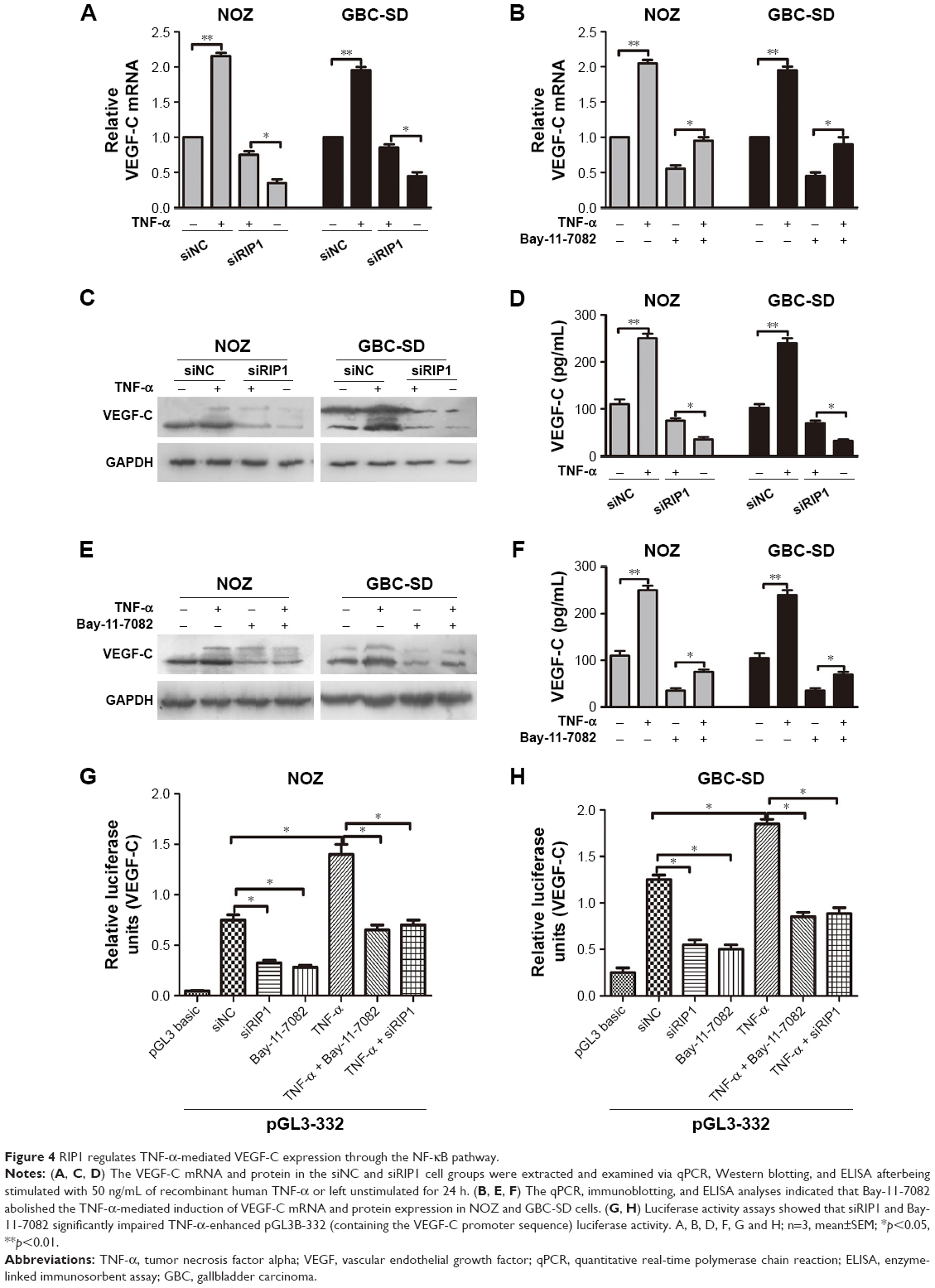 | Figure 4 RIP1 regulates TNF-α-mediated VEGF-C expression through the NF-κB pathway. |
Moreover, we examined the promoter activity of VEGF-C to analyze whether siRIP1 and Bay-11-7082 markedly impaired TNF-α-mediated VEGF-C expression at the transcriptional level. Indeed, we found that siRIP1 and Bay-11-7082 markedly impaired the TNF-α-mediated enhancement of VEGF-C promoter-driven luciferase activity in GBC-SD and NOZ cells (Figure 4G and H). To determine whether siRIP1 impaired the TNF-α-enhanced association of NF-κB with the endogenous VEGF-C promoter region, ChIP analysis was performed. As shown in Figure 5, TNF-α enhanced the association of NF-κB with the VEGF-C promoter region in GBC-SD and NOZ cells, and this enhancement by TNF-α was markedly impaired in RIP1-knockdown GBC-SD and NOZ cells. These observations suggest that TNF-α mediates VEGF-C expression through the RIP1/NF-κB pathway in GBC-SD and NOZ cells.
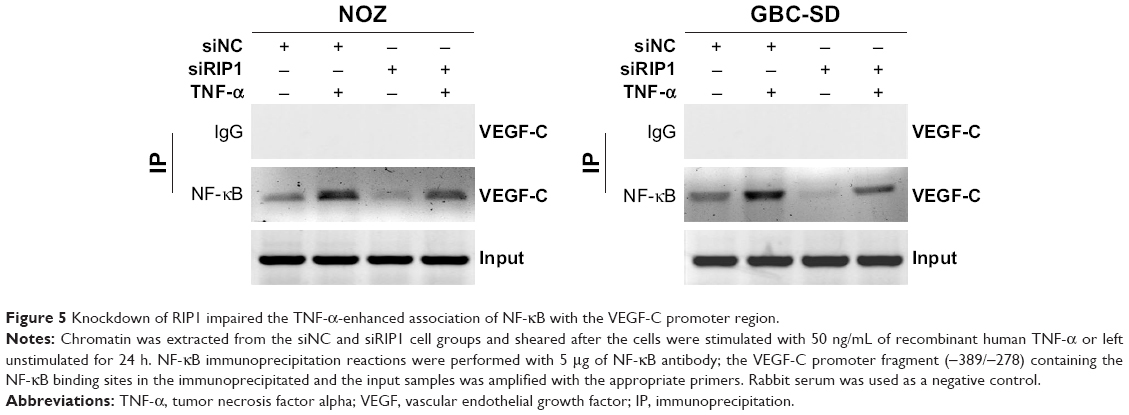 | Figure 5 Knockdown of RIP1 impaired the TNF-α-enhanced association of NF-κB with the VEGF-C promoter region. |
RIP1 regulates TNF-α-mediated lymphangiogenesis in HDLECs in vitro
We further tested and verified the role of RIP1 in the tube-forming ability of HDLECs. In our previous study, we have demonstrated using the MTT assay that 50 ng/mL of TNF-α (compared with untreated group) did not affect the growth of HDLECs.8 Therefore, red fluorescent probe-labeled HDLECs were cocultured with the established GBC-SD or NOZ cell groups (control, LV-siNC, and LV-siRIP1) on Matrigel-based μ-slides and then either not stimulated or stimulated with 50 ng/mL of TNF-α from the beginning. Tube formation was detected 1, 3, 6, 8, and 24 h after coculture initiation. As shown in Figure 6A and C, the best tube formation effect occurred 6 h after coculture. In addition, TNF-α markedly enhanced the tube-forming ability of HDLECs in the control and LV-siNC groups, and this enhancement by TNF-α was markedly impaired in RIP1-knockdown GBC-SD and NOZ cells. Importantly, transfection of PcDNA3.1-RIP1 vector into RIP1-knockdown GBC-SD and NOZ cells could balance the impaired effect of RIP1 knockdown on TNF-α-mediated lymphangiogenesis in HDLECs.
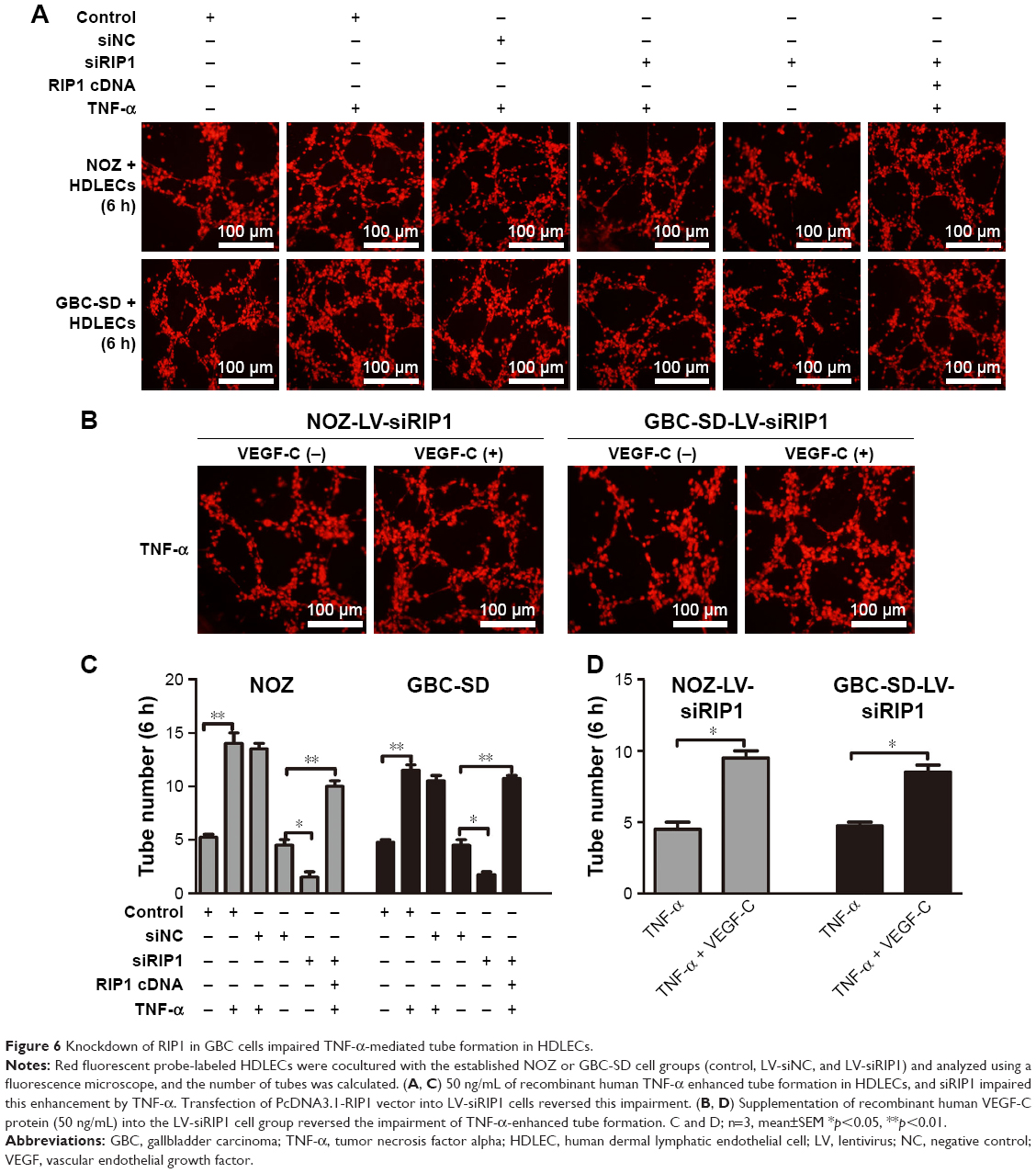 | Figure 6 Knockdown of RIP1 in GBC cells impaired TNF-α-mediated tube formation in HDLECs. |
In addition, to further explore whether the impairment of the TNF-α-mediated tube-forming ability of HDLECs in RIP1-knockdown GBC-SD and NOZ cells was related to decreased VEGF-C protein levels, 50 ng/mL of recombinant human VEGF-C protein (ab83573, Abcam) was supplemented into the culture media of RIP1-knockdown GBC-SD and NOZ cells. As shown in Figure 6B and D, supplementation with VEGF-C protein reversed the impairment of TNF-α-mediated tube-forming ability in the RIP1-knockdown GBC-SD and NOZ cells.
RIP1 regulates TNF-α-mediated lymphangiogenesis promoting lymphatic metastasis in GBC in vivo
In addition to our in vitro experiments, we examined the function of RIP1 in TNF-α-mediated lymphangiogenesis promoting lymphatic metastasis in vivo. First, we generated a murine orthotopic transplantation model of GBC using three established cell groups (NOZ-control, NOZ-LV-siNC, and NOZ-LV-siRIP1) (Figure 7A). After 2 weeks, the mice received intra-abdominal injections of 2 μg/kg of recombinant human TNF-α 2 days a week for 3 weeks. Orthotopic tumor sizes were measured, and the results showed that tumor sizes were markedly increased in the control and LV-siNC groups upon TNF-α stimulation; this increase induced by TNF-α was markedly abolished in RIP1-knockdown NOZ cells (Figure S3). And lymph node metastases were subsequently detected on a gross scale (Figure 7B) and further confirmed via hematoxylin and eosin staining (Figure 7C). Additionally, the size of the lymph nodes was measured, and the result showed that there was no significant difference in size among the different groups (Figure 7F). Meanwhile, the lymphatic vessels of the tumors of the nude mice were examined through immunohistochemical analysis using an LYVE-1 antibody (Figure 7D). As shown in Figure 7E, the lymphatic vessel number (LVN) was markedly increased in the control and LV-siNC groups upon TNF-α stimulation, and this increase induced by TNF-α was markedly abolished in RIP1-knockdown NOZ cells. The data on lymphatic vessel density (LVD) and LNM in the mouse tumors are presented in Table 1.
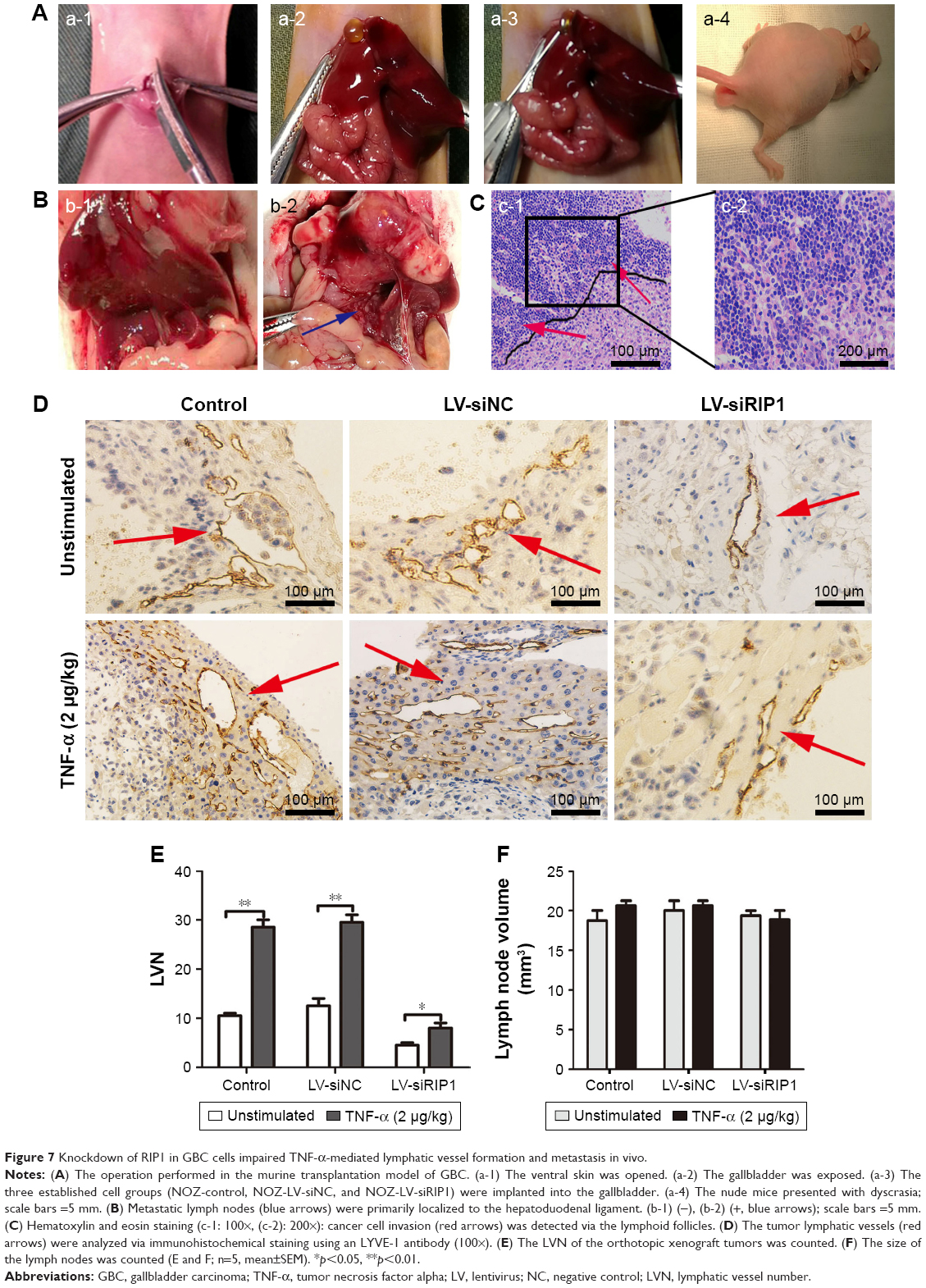 | Figure 7 Knockdown of RIP1 in GBC cells impaired TNF-α-mediated lymphatic vessel formation and metastasis in vivo. |
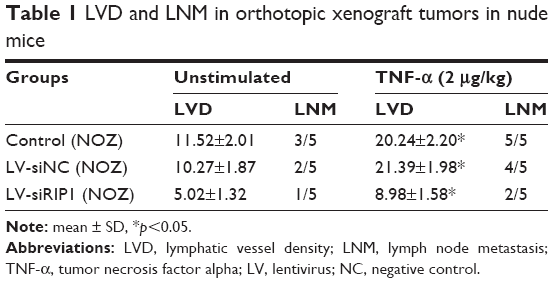 | Table 1 LVD and LNM in orthotopic xenograft tumors in nude mice |
Relationships of RIP1 with TNF-α and LVD in clinical GBC specimens
Finally, we explored whether the expression of the RIP1 protein was correlated with that of the TNF-α protein and with LVD in human GBC tissues. We first examined the expression of the RIP1 and TNF-α proteins in GBC tissues using immunohistochemistry (Figure 8A). As shown in Figure 8B, the average optical density of RIP1 was linearly related to that of TNF-α in GBC tissues. In addition, lymphatic vessels, based on D2-40 immunostaining, were flattened and invaded by GBC cells (Figure 8C), and linear dependence was also detected between the average optical density of RIP1 and LVD in GBC tissues (Figure 8D). Taken together, these results provide evidence of a potential association of RIP1 protein expression with TNF-α protein expression and LVD in human GBC tissues.
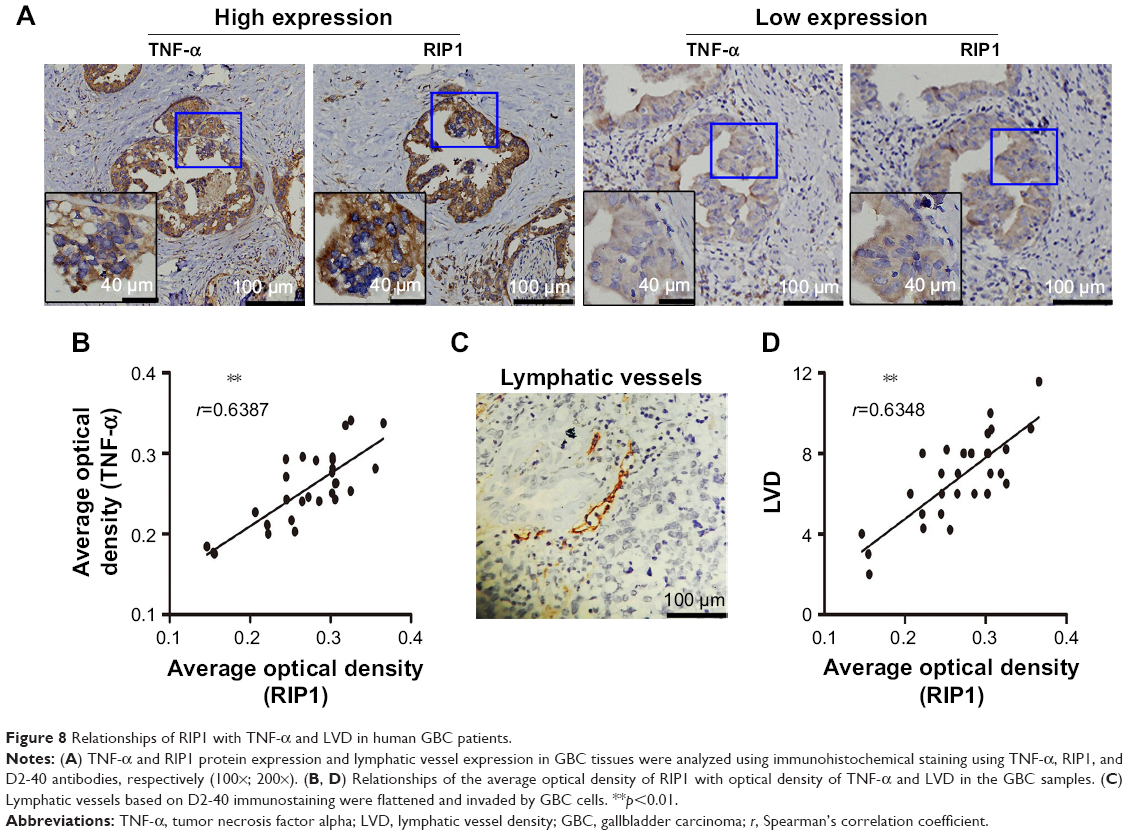 | Figure 8 Relationships of RIP1 with TNF-α and LVD in human GBC patients. |
Discussion
Previous studies have shown that TNF-α-mediated VEGF-C expression can promote lymphangiogenesis in ovarian cancer and GBC, and that VEGF-C plays a major role in TNF-α-mediated lymphangiogenesis in these tissues.8,27,28 Therefore, anti-VEGF and TNF-α-targeted therapies have been designed to limit this activity.29 However, the overall survival of patients has not significantly improved,30 and multitargeted therapy has become increasingly popular in the development of new agents.31 Nevertheless, the entire TNF-α signaling pathway that regulates lymphangiogenesis and lymphatic metastasis has not yet been elucidated. RIP1 kinase is a vital regulator that controls multiple cell processes, including cell survival, by activating NF-κB, apoptosis, and necroptosis via the TNF-α signaling pathway.13,15,32 In the present study, we found that TNF-α dose and time dependently increased RIP1 mRNA and protein expression in the GBC-SD and NOZ cells (Figure 1). According to our previous work,8 the observed TNF-α-mediated RIP1 mRNA and protein expression occurred in a similar manner to the upregulation of NF-κB-dependent VEGF-C by TNF-α in GBC cells. These findings encouraged us to further explore whether RIP1 acts as a switch to control the fate of TNF-α–NF-κB–VEGFC axis-mediated lymphangiogenesis and lymphatic metastasis in GBC.
Whether RIP1 is indispensable for the TNF-α-mediated activation of NF-κB remains contentious. Filliol et al33 and Wong et al34 reported that RIP1 is dispensable for TNF-α-mediated NF-κB activation in liver parenchymal cells and mouse embryonic fibroblasts. However, the present study demonstrated that RIP1 is crucial for TNF-α-mediated NF-κB activation in GBC cells. This conclusion is supported by the following findings: 1) NF-κB-luciferase activity assays showed that siRIP1 markedly abolished TNF-α-mediated NF-κB activation, whereas additional RIP1 cDNA reversed this effect (Figure 3A); 2) knockdown of IκBα can also reverse the impairment of TNF-α-mediated NF-κB activation in RIP1-knockdown GBC cells (Figure 3F); and 3) TAK1 and NEMO are physically related to RIP1 (Figure 3G). Our study was in line with previous findings in melanoma cells3 and hepatic cancer cells.35 However, the mechanisms of the cell type-dependent activation of NF-κB remain unclear. Regardless, our findings showed that RIP1 is responsible for the TNF-α-mediated activation of NF-κB in GBC cells.
Then, we demonstrated that RIP1/NF-κB signaling contributes to VEGF-C activation in TNF-α-stimulated GBC cells. In our study, the results of qPCR, Western blotting, ELISA, and VEGF-C promoter-luciferase activity assays showed that knockdown of RIP1 or inhibition of NF-κB could impair the TNF-α-mediated upregulation of VEGF-C expression at the translational or transcriptional level. The relationship between RIP1 and NF-κB in TNF-α signaling was demonstrated in our study. Therefore, we deduced that RIP1/NF-κB signaling is required for TNF-α-mediated VEGF-C expression. Moreover, the results of ChIP assays showed that siRIP1 markedly abolished TNF-α-enhanced NF-κB binding to the VEGF-C promoter. This finding further supported our hypothesis.
Some studies have shown clear defects in lymphoid system development and maintenance in RIP1-deficient mice,36,37 indicating that RIP1 significantly influences the development and maintenance of the lymphoid system.38 However, information regarding the role of RIP1 in cancer development is lacking, as most reports have focused on the roles of RIP1 in proliferation,3,18,39 migration,40 and invasion.18 The relationship between RIP1 and lymphangiogenesis in cancer cells has not previously been reported. To evaluate the function of RIP1 in the tube-forming ability of HDLECs in GBC cells, we utilized a three-dimensional coculture method in the present study, and the results showed that knockdown of RIP1 can impair the TNF-α-mediated increase in tube formation in HDLECs in GBC cells. In addition, supplementation of the VEGF-C protein into the culture media of RIP1-knockdown GBC cells reversed the induction of tube formation. These results indicated that RIP1 has the ability to promote lymphangiogenesis, and this ability is related to VEGF-C levels. Therefore, this manuscript is the first report showing that RIP1 can regulate the TNF-α–VEGFC axis-induced lymphangiogenesis in human cancer cells. Moreover, knockdown of RIP1 inhibited TNF-α-mediated lymphangiogenesis and lymphatic metastasis in vivo, which further supported our conclusion based on vitro experiments.
Finally, we explored the correlations of the expression of the RIP1 protein with that of the TNF-α protein and with LVD in human GBC patients. TNF-α plays a significant role in the development of chronic inflammation associated with tumors.41,42 Our previous work showed that TNF-α concentration in the bile of GBC patients is significantly correlated with LVD in GBC tissue.8 Moreover, a prior study found that RIP1 is highly expressed in melanoma, glioblastoma, mammary cancer, and GBC tissues.3,18,43 However, the correlation between the expression of the RIP1 and TNF-α proteins and LVD in human cancer has not been studied previously. The research reported herein demonstrated that the average optical density of RIP1 was linearly associated with that of TNF-α and LVD in GBC specimens, based on immunohistochemistry. These results further supported our hypothesis regarding the role of RIP1 in TNF-α-mediated lymphangiogenesis in GBC.
Conclusion
Our study revealed that RIP1 exhibits a specific prosurvival function that might regulate TNF-α-mediated lymphangiogenesis and lymphatic metastasis by modulating the NF-κB–VEGF-C pathway in human GBC. Further studies aimed at identifying potential drug targets for disrupting the function of RIP1 could provide a novel therapeutic strategy to improve GBC treatment.
Acknowledgment
Our research was supported by the National Natural Science Foundation of China (number 81672468).
Disclosure
The authors report no conflicts of interest in this work.
References
Kanthan RS, Ahmed J-L, Kanthan S, Chandra S. Gallbladder cancer in the 21st century. J Oncol. 2015;2015:967472. | ||
Zhu AX, Hong TS, Hezel AF, Kooby DA. Current management of gallbladder carcinoma. Oncologist. 2010;15(2):168–181. | ||
Liu XY, Lai F, Yan XG, et al. RIP1 kinase is an oncogenic driver in melanoma. Cancer Res. 2015;75(8):1736–1748. | ||
Lai CH, Lau WY. Gallbladder cancer – a comprehensive review. Surgeon. 2008;6(2):101–110. | ||
Fuks D, Regimbeau JM, Pessaux P, et al. Is port-site resection necessary in the surgical management of gallbladder cancer? J Visc Surg. 2013;150(4):277–284. | ||
Cusson N, Oikemus S, Kilpatrick ED, Cunningham L, Kelliher M. The death domain kinase RIP protects thymocytes from tumor necrosis factor receptor type 2–induced cell death. J Exp Med. 2002;196(1):15–26. | ||
Lei Jiang FS, Chen Y. Expression of VEGF-C and VEGF-D and their correlation with lymphangiogenesis and angiogenesis in gallbladder carcinoma. Chin J Oncol. 2010;32(3):190–195. | ||
Du Q, Jiang L, Wang X, Wang M, She F, Chen Y. Tumor necrosis factor-alpha promotes the lymphangiogenesis of gallbladder carcinoma through nuclear factor-kappaB-mediated upregulation of vascular endothelial growth factor-C. Cancer Sci. 2014;105(10):1261–1271. | ||
Brenner D, Blaser H, Mak TW. Regulation of tumour necrosis factor signalling: live or let die. Nat Rev Immunol. 2015;15(6):362–374. | ||
Balkwill F. Tumour necrosis factor and cancer. Nat Rev Cancer. 2009;9(5):361–371. | ||
Silke J. The regulation of TNF signalling: what a tangled web we weave. Curr Opin Immunol. 2011;23(5):620–626. | ||
Croft M, Benedict CA, Ware CF. Clinical targeting of the TNF and TNFR superfamilies. Nat Rev Drug Discov. 2013;12(2):147–168. | ||
Christofferson DE, Li Y, Yuan J. Control of life-or-death decisions by RIP1 kinase. Annu Rev Physiol. 2014;76:129–150. | ||
Haas TL, Emmerich CH, Gerlach B, et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol Cell. 2009;36(5):831–844. | ||
Ofengeim D, Yuan J. Regulation of RIP1 kinase signalling at the crossroads of inflammation and cell death. Nat Rev Mol Cell Biol. 2013;14(11):727–736. | ||
Hong H, Jiang L, Lin Y, et al. TNF-alpha promotes lymphangiogenesis and lymphatic metastasis of gallbladder cancer through the ERK1/2/AP-1/VEGF-D pathway. BMC Cancer. 2016;16:240. | ||
Chen Y, Jiang L, She F, et al. Vascular endothelial growth factor-C promotes the growth and invasion of gallbladder cancer via an autocrine mechanism. Mol Cell Biochem. 2010;345(1–2):77–89. | ||
Zhu G, Chen X, Wang X, et al. Expression of the RIP-1 gene and its role in growth and invasion of human gallbladder carcinoma. Cell Physiol Biochem. 2014;34(4):1152–1165. | ||
Zhu G. The Roles and Mechanisms of RIP1 Promote the Proliferation, Invasion and Matastasis of Gallbladder Carcinoma [doctor dissertation]. Fujian Medical University; 2015: Fuzhou, People’s republic of China. | ||
Zeng Y, Opeskin K, Goad J, Williams ED. Tumor-induced activation of lymphatic endothelial cells via vascular endothelial growth factor receptor-2 is critical for prostate cancer lymphatic metastasis. Cancer Res. 2006;66(19):9566–9575. | ||
Du Q, Jiang L, Wang X-Q, Pan W, She F-F, Chen Y-L. Establishment of and comparison between orthotopic xenograft and subcutaneous xenograft models of gallbladder carcinoma. Asian Pac J Cancer Prev. 2014;15(8):3747–3752. | ||
Cho YS, Challa S, Moquin D, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;6(12):137, 1112–1123. | ||
Adrian T. Ting FXP-MaBS. RIP mediates tumor necrosis factor receptor 1 activation of NF-κB but not Fas/APO-1-initiated apoptosis. EMBO J. 1996;15(22):6189–6196. | ||
Ristimäki A, Narko K, Enholm B, Joukov V, Alitalo K. Proinflammatory cytokines regulate expression of the lymphatic endothelial mitogen vascular endothelial growth factor-C. J Biol Chem. 1998;273(14):8413. | ||
Cha HS, Bae EK, Koh JH, et al. Tumor necrosis factor-alpha induces vascular endothelial growth factor-C expression in rheumatoid synoviocytes. J Rheumatol. 2007;34(1):16–19. | ||
HaiJie H, CaiLong H, SiYuan Z, et al. CCR7 mediates the TNF-α-induced lymphatic metastasis of gallbladder cancer through the “ERK1/2– AP-1” and “JNK–AP-1” pathways. J Exp Clin Cancer Res. 2016;35(1):51. | ||
Ji H, Cao R, Yang Y, et al. TNFR1 mediates TNF-alpha-induced tumour lymphangiogenesis and metastasis by modulating VEGF-C-VEGFR3 signalling. Nat Commun. 2014;5:4944. | ||
Kimura H, Mikami D, Kamiyama K, et al. Telmisartan, a possible PPAR-delta agonist, reduces TNF-alpha-stimulated VEGF-C production by inhibiting the p38MAPK/HSP27 pathway in human proximal renal tubular cells. Biochem Biophys Res Commun. 2014;454(2):320–327. | ||
Shojaei F. Anti-angiogenesis therapy in cancer: current challenges and future perspectives. Cancer Lett. 2012;320(2):130–137. | ||
Ebos JM, Kerbel RS. Antiangiogenic therapy: impact on invasion, disease progression, and metastasis. Nat Rev Clin Oncol. 2011;8(4):210–221. | ||
Katsi VK, Psarros CT, Krokidis MG, et al. Anti-angiogenic therapy and cardiovascular diseases: current strategies and future perspectives. Anti Angiog Drug Discov Dev. 2014;2:1–43. | ||
Humphries F, Yang S, Wang B, Moynagh PN. RIP kinases: key decision makers in cell death and innate immunity. Cell Death Differ. 2015;22(2):225–236. | ||
Filliol A, Piquet-Pellorce C, Le Seyec J, et al. RIPK1 protects from TNF-alpha-mediated liver damage during hepatitis. Cell Death Dis. 2016;7(11):e2462. | ||
Wong WW, Gentle IE, Nachbur U, Anderton H, Vaux DL, Silke J. RIPK1 is not essential for TNFR1-induced activation of NF-kappaB. Cell Death Differ. 2010;17(3):482–487. | ||
Sun J, Yu X, Wang C, et al. RIP-1/c-FLIPL induce hepatic cancer cell apoptosis through regulating tumor necrosis factor-related apoptosis-inducing ligand (TRAIL). Med Sci Monit. 2017;23:1190–1199. | ||
Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420(6917):860–867. | ||
Zhang H, Zhou X, McQuade T, Li J, Chan FK-M, Zhang J. Corrigendum: functional complementation between FADD and RIP1 in embryos and lymphocytes. Nature. 2012;483(7390):498. | ||
Zhang J, Zhang H, Li J, et al. RIP1-mediated regulation of lymphocyte survival and death responses. Immunol Res. 2011;51(2–3):227–236. | ||
Luan Q, Jin L, Jiang CC, et al. RIPK1 regulates survival of human melanoma cells upon endoplasmic reticulum stress through autophagy. Autophagy. 2015;11(7):975–994. | ||
Bist P, Leow SC, Phua QH, et al. Annexin-1 interacts with NEMO and RIP1 to constitutively activate IKK complex and NF-kappaB: implication in breast cancer metastasis. Oncogene. 2011;30(28):3174–3185. | ||
Macarthur M, Hold GL, El-Omar EM. Inflammation and Cancer II. Role of chronic inflammation and cytokine gene polymorphisms in the pathogenesis of gastrointestinal malignancy. Am J Physiol Gastrointest Liver Physiol. 2004;286(4):G515–G520. | ||
Zhou LS, Shi JS, Wang ZR, Wang L. Expression of tumor necrosis factor in gallstone and gallbladder carcinoma tissue. Carcinog Teratog Mutagen. 2000;12(3):139–142. | ||
Park S, Hatanpaa KJ, Xie Y, et al. The receptor interacting protein 1 inhibits p53 induction through NF-κB activation and confers a worse prognosis in glioblastoma. Cancer Res. 2009;69(7):2809–2816. |
Supplementary materials
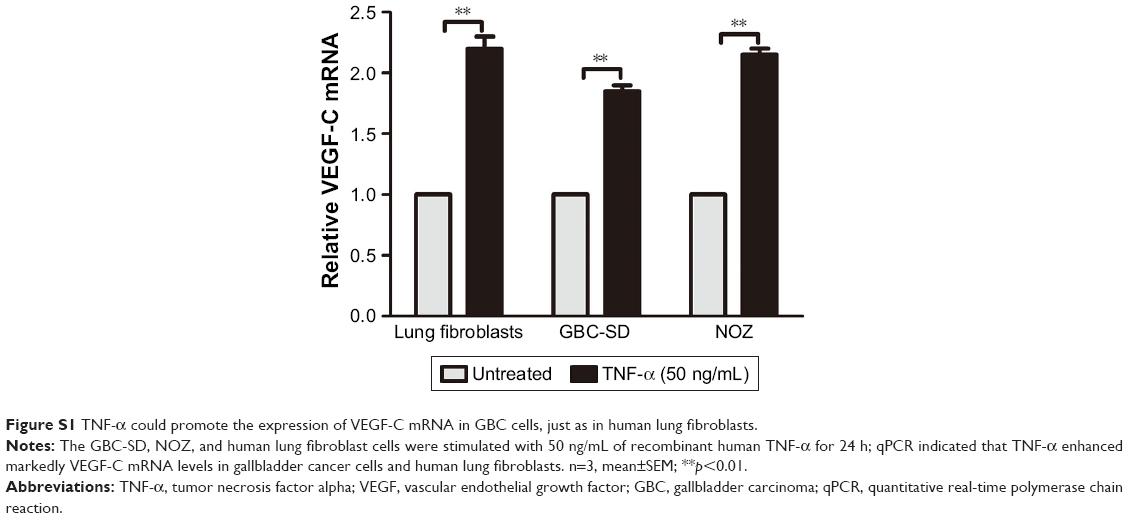 | Figure S1 TNF-α could promote the expression of VEGF-C mRNA in GBC cells, just as in human lung fibroblasts. |
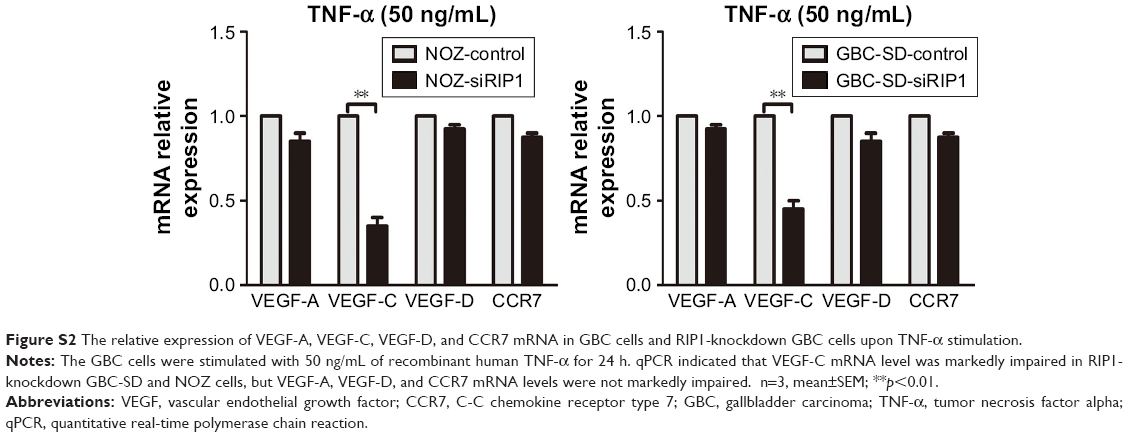 | Figure S2 The relative expression of VEGF-A, VEGF-C, VEGF-D, and CCR7 mRNA in GBC cells and RIP1-knockdown GBC cells upon TNF-α stimulation. |
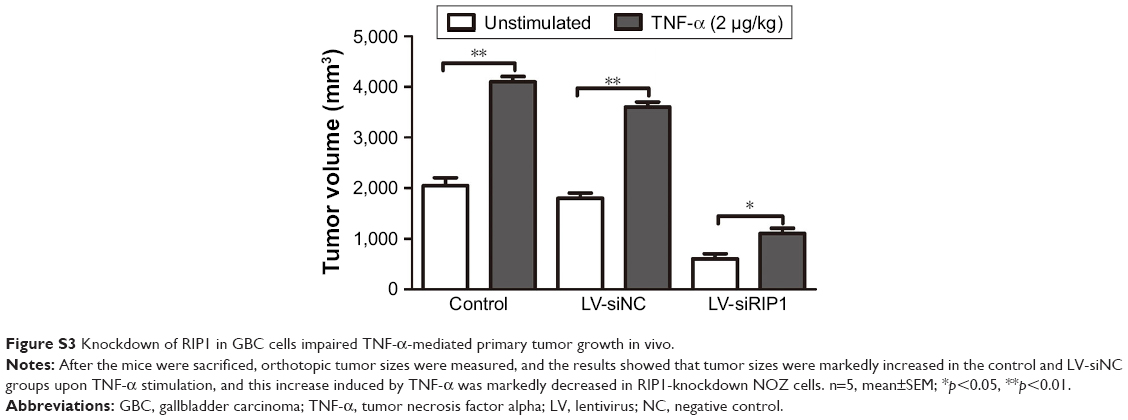 | Figure S3 Knockdown of RIP1 in GBC cells impaired TNF-α-mediated primary tumor growth in vivo. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.