Back to Journals » Neuropsychiatric Disease and Treatment » Volume 14
RIP1 and RIP3 mediate hemin-induced cell death in HT22 hippocampal neuronal cells
Authors Su X, Wang H ![]() , Lin Y, Chen F
, Lin Y, Chen F
Received 22 July 2018
Accepted for publication 27 September 2018
Published 15 November 2018 Volume 2018:14 Pages 3111—3119
DOI https://doi.org/10.2147/NDT.S181074
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yu-Ping Ning
Xingfen Su,1 Handong Wang,2 Yuanxiang Lin,1 Fuxiang Chen1
1Department of Neurosurgery, The First Affiliated Hospital of Fujian Medical University, Fuzhou 350005, Fujian, People’s Republic of China; 2Department of Neurosurgery, Jinling Hospital, School of Medicine, Nanjing University, Nanjing 210002, Jiangsu, People’s Republic of China
Background: Intracerebral hemorrhage (ICH) is a devastating neurological injury associated with significant mortality. Necroptosis is a newly identified type of programmed necrosis initiated by the activation of tumor necrosis factor alpha. Evidences had demonstrated the importance of necroptosis in neuronal cell death. Necrostatin-1 is a specific inhibitor of necroptosis. The present study was carried out to explore whether RIP1/RIP3 pathways participate in hemin induced cell death in HT-22 hippocampal neuronal cells and investigate the potential neuroprotection of necrostatin-1 in hemin induced cell death in HT-22.
Methods: First, different concentrations of hemin (0, 25, 50, 100 µmol/L) were added to HT-22 cells. Propidium iodide (PI) positive cells and cell viability were measured at 24 hours after hemin treatment. Then, necrostatin-1, pan-caspase inhibitor Benzyloxycarbonyl-Val-Ala-Asp(OMe)-fluoromethylketone (z-VAD-fmk) and reactive oxygen species (ROS) scavenger butylated hydroxyanisole (BHA) were applied to hemin-treated HT-22 cells. PI positive cells and cell viability were measured at 24 hours after hemin treatment. MitoSox Red was used to indicate ROS level. Last, the effect of RIP3 in hemin induced HT-22 cell death was explored through RIP3 knockdown using siRNA. PI positive cells, cell viability and ROS lever were measured at 24 h after hemin treatment.
Results: Hemin could induce a dose dependent cell death in HT22 neural cells. RIP1 specific inhibitor necrostatin-1 significantly inhibited cell death induced by hemin in HT-22 cells, greatly reducing PI positive cells, dramatically improving cell viability and decreasing ROS accumulation. BHA could significantly inhibit PI positive cells induced by hemin in HT-22 cells. Furthermore, silencing of RIP3 using siRNA attenuated hemin induced cell death in HT-22 cells, greatly reducing PI positive cells, dramatically improving cell viability and decreasing ROS accumulation.
Conclusion: These data revealed that RIP1/RIP3 might mediate hemin induced cell death in HT-22 cells, and necrostatin-1 played a neuroprotection role in hemin induced cell death in HT-22. RIP1 and RIP3 might represent novel therapeutic targets for ICH.
Keywords: intracerebral hemorrhage, necroptosis, HT22, RIP1, RIP3, necrostatin-1
Introduction
Intracerebral hemorrhage (ICH) accounts for ~10%–15% of all stroke cases and is associated with higher rates of morbidity and mortality than ischemic strokes.1 After the onset of ICH, a lot of changes occur in the brain tissue, including hematoma formation, brain edema, release of cytotoxic blood components, glutamate-induced excitotoxicity, inflammation, and microglia activation.2,3 The synergistic actions of these pathological events lead to neuronal cell death and subsequent neurological dysfunction. However, the molecular mechanisms that promote brain injury after ICH are not fully understood. Hence, exploring these mechanisms may help to identify novel therapeutic targets for ICH.
Hemoglobin degeneration results in sustaining generation of cytotoxic by-products within the parenchyma for weeks after brain hemorrhage, which causes extensive brain injury.4,5 One such product, hemin, accumulates in the hematoma at high micromolar concentrations and is released from lysed red blood cells into the extracellular space of the brain after ICH.4,5 Hemin, oxidized heme, is a lipophilic cytotoxic oxidant and kills brain cells adjacent to the hematoma. Cell culture experiments have shown that hemin is sufficient to kill neurons and astrocytes within 24 hours.6–8 HT22 is an immortalized mouse hippocampal cell line expressing neuron-specific enolase and neurofilament proteins.9
Necrotic cell death is common in a variety of pathological conditions, including ICH.10 Necrotic cell death was thought to be nonregulated. However, recent works showed that some necrotic cell death was highly regulated by certain molecular pathways, specially the RIP1 and RIP3.11–14 This novel type of cell death was designated necroptosis or programmed necrosis.12 Furthermore, a small compound called necrostatin-1 (Nec-1) that could inhibit necroptosis through inhibiting RIP1 was identified.12
A previous study showed that hemin induced necroptotic cell death in a concentration-dependent manner in cortical astrocytes, and this kind of necroptotic cell death could be attenuated by Nec-1 treatment, but not by the pan-caspase inhibitor z-VAD-fmk.5 However, the mechanisms by which hemin caused neurotoxicity in HT22 cells were still not fully understood. The present study explored whether RIP1 and RIP3 pathways participated in hemin-induced cell death in HT22 cells, and the potential therapeutic strategies that could be employed to prevent HT22 cell death.
Materials and methods
Reagents
Nec-1 (Sigma-Aldrich, St Louis, MO, USA) was dissolved in dimethyl sulfoxide (DMSO; the final concentration of DMSO was 0.5%; Sigma-Aldrich) solution. Hemin and butylated hydroxyanisole (BHA) were purchased from Sigma-Aldrich. MitoSox Red was purchased from Thermo Fisher Scientific (Waltham, MA, USA). z-VAD-fmk was purchased from BD Biosciences. DMEM, FBS, and penicillin/streptomycin were bought from Thermo Fisher Scientific.
HT22 cell culture and treatment protocols
HT22 mouse hippocampal cells (ATCC, Manassas, VA, USA) were cultured in DMEM (Thermo Fisher Scientific) containing 10% FBS (Thermo Fisher Scientific) and 1% penicillin/streptomycin (Thermo Fisher Scientific) at 37°C under a humidified atmosphere of 95% air and 5% CO2. To determine the neurotoxicity of hemin, various concentrations (0, 25, 50, and 100 μM) of hemin were added to HT22 cells and treated for 24 hours. Of these concentrations, 50 μM was selected and used in the subsequent experiments. For the neuroprotection studies, HT22 cells were pretreated with Nec-1 (30 μM), z-VAD-fmk (20 μM), and BHA (100 μM) 15 minutes before treatment with 50 μM hemin and were incubated for 24 hours. Control groups were treated with vehicle (DMSO) only. Cell viability and propidium iodide (PI) exclusion assays were performed 24 hours after treatment. Three wells were used for each group. Experiments were repeated three times.
Cell viability and PI exclusion assays
HT22 cells were seeded in 96-well plates at a density of 3,000 cells/well in 100 μL culture medium and fed in the incubator overnight. Then, cells were treated with hemin for 24 hours and cell viability was determined by measuring ATP level. For the cell viability assay, a luminescence-based commercial kit (CellTiter-Glo; Promega) was used. First, 30 μL of the CellTiter-Glo ATP detection reagent was added to each well. After that, we incubated the plates for 10 minutes at room temperature, and then analyzed luminescence using a Wallac Victor II plate reader (PerkinElmer). Cell viability was expressed as a ratio of the DMSO control group. For PI exclusion assay, cells were incubated with PI and Hoechst at a final concentration of 2 μg/mL (Roche) in dark at room temperature for 10 minutes. Then, cells were photographed on a Nikon Eclipse T300 fluorescence microscope (Tokyo, Japan). Three pictures (×200 magnification) per well for PI and Hoechst were randomly taken under the fluorescence microscope and counted by an investigator who was blinded to the treatment scheme. PI-positive cells were expressed as PI+/Hoechst+. Three wells were used for each group. Experiments were repeated three times.
Measurement of mitochondrial ROS
HT22 cells were incubated with 5 μM MitoSox Red and 2 μg/mL Hoechst for 10 minutes at 37°C. Then, cells were washed with PBS and observed under Nikon Eclipse T300 fluorescence microscope. Three pictures (×200 magnification) per well for MitoSox Red and Hoechst were randomly taken under the fluorescence microscope. Three wells were used for each group. Experiments were repeated three times.
RIP3 siRNA knockdown
For the transfection experiments, HT22 cells were seeded in six-well plates. Lipofectamine 2000 (Thermo Fisher Scientific), RIP3 siRNA (On-target Plus Smart Pool with four different sequences; Dharmacon; Thermo Fisher Scientific), and negative nontarget siRNA (nonfunctional mutant RNA; Dharmacon; Thermo Fisher Scientific) were dissolved separately in Optimem I (Thermo Fisher Scientific). After 10 minutes of equilibration at room temperature, each siRNA solution was combined with the respective volume of the Lipofectamine 2000 solution, mixed gently, and allowed to form siRNA liposomes for 20 minutes at room temperature. The transfection mixture was added to the antibiotic-free cell culture medium to a final concentration of 60 nM siRNA and 2 μL/mL Lipofectamine in HT22 cells. After 4 hours of transfection, normal fresh culture medium was added to the transfection medium. After incubation for 48 hours, the cells were digested, and part of them were seeded in 96-well plates at a density of 3,000 cells/well for hemin treatment whereas the other cells were kept in the incubator for 72 hours and then collected to determine the effect of gene silencing by Western blot. Negative nontarget siRNA was used as control.
Western blot
Cells were collected and sonicated on ice in a modified RIPA buffer (50 mmol/L Tris–HCl [pH 7.5], 50 mmol/L NaCl, 4 mol/L urea, 0.5% SDS, 0.5% NP-40, 0.5% Na-deoxycholate, 5 mmol/L phenylmethylsulfonylfluoride, 1 mmol/L EDTA, 5 mmol/L ethyleneglycol tetraacetate [EGTA], 10 mmol/L dithiothreitol) and protease inhibitor cocktail (1:100; Sigma-Aldrich).
Homogenates were cleared by centrifugation at 12,000× g for 15 minutes at 4°C. Protein content of the supernatants was assayed (Bio-Rad Laboratories, Hercules, CA, USA), and aliquots of protein were boiled in denaturing sample buffer (62.5 mmol/L Tris [pH 6.8], 2% SDS, 5 mmol/L EDTA, 10% glycerol, 0.25% 2-mercaptoethanol, 0.01% bromophenol blue). Cell lysate samples were loaded at 100 μg/lane. Denatured proteins were size-fractionated on 12% SDS-PAGE gels (Thermo Fisher Scientific) and blotted onto Immobilon 0.45 mm polyvinylidene difluoride membranes (EMD Millipore, Billerica, MA, USA). Membranes were blocked for 1 hour in 5% milk in Tris-buffered saline (pH 7.4) containing 0.1% Tween 20 (TBST), and then incubated overnight at 4°C with primary antibody anti-RIP3 (1:1,000; Proscience, Poway, CA, USA) or β-actin (1:1,000; Sigma-Aldrich). Membranes were washed in TBST, and then incubated for 1 hour with goat anti-rabbit horseradish peroxidase-conjugated secondary antibody (1:10,000 in TBST) at room temperature. RIP3 or β-actin was detected using the enhanced chemiluminescence Western blotting detection system kit (ECL Plus; Amersham, Little Chalfont, UK) and Hyperfilm (Amersham). Blots were captured on Kodak autoradiographic films. Films were scanned, and densitometric analyses of the bands were performed with ImageJ software.
Statistical analysis
Data were presented as mean ± SEM. GraphPad Prism 5 software was used for the statistical analysis. For comparisons among multiple groups, one-way ANOVA followed by a post hoc (Tukey) test was used to determine significant differences. Statistical significance was set at P<0.05.
Results
Hemin induced a dose-dependent necrosis and neurotoxicity in HT22 cells
To assess whether hemin could induce necrotic cell death in HT22 cells, we treated HT22 cells with various concentrations (0–100 μM) of hemin for 24 hours. As shown in Figure 1A and B, hemin produced a concentration-dependent necrotic cell death (PI+ cells) in HT22 cells. The hemin-induced neurotoxicity was further confirmed by cell viability determined using CellTiter-Glo assay (Figure 1C). Dose-response studies showed that 50 μM hemin efficiently induced necrotic cell death. Therefore, 50 μM hemin was selected and used in the subsequent experiments.
 | Figure 1 Hemin induced dose-dependent necrosis and neurotoxicity in HT22 cells. |
Nec-1 protected against necrotic cell death induced by hemin in HT22 cells
To determine whether hemin could induce necroptosis, HT22 cells were treated with hemin, z-VAD-fmk, and Nec-1. As shown in Figure 2, HT22 cells treated with either z-VAD-fmk or Nec-1 only had the similar number of PI+ cells and cell viability as DMSO group. Hemin at a concentration of 50 μM significantly induced necrotic cell death (as shown by the increased number of PI+ cells) and neurotoxicity. The PI+ cells were greatly inhibited by 30 μM Nec-1 treatment. The quantitative results of protection provided by Nec-1 are shown as number of PI+ cells (Figure 2B) and cell viabilities (Figure 2C). To determine the role of caspase-dependent apoptosis in hemin-induced cell death, a pan-caspase inhibitor z-VAD-fmk was added in the experiment. z-VAD-fmk (20 μM) did not reduce hemin-induced cell death when compared with the hemin group. Nec-1 administration dramatically reduced the cell death of hemin-treated HT22 cells, but additional treatment with z-VAD-fmk did not further reduce cell death (Figure 2B and C). These results indicated that caspase-independent necroptosis might mediate hemin-induced cell death in HT22 cells.
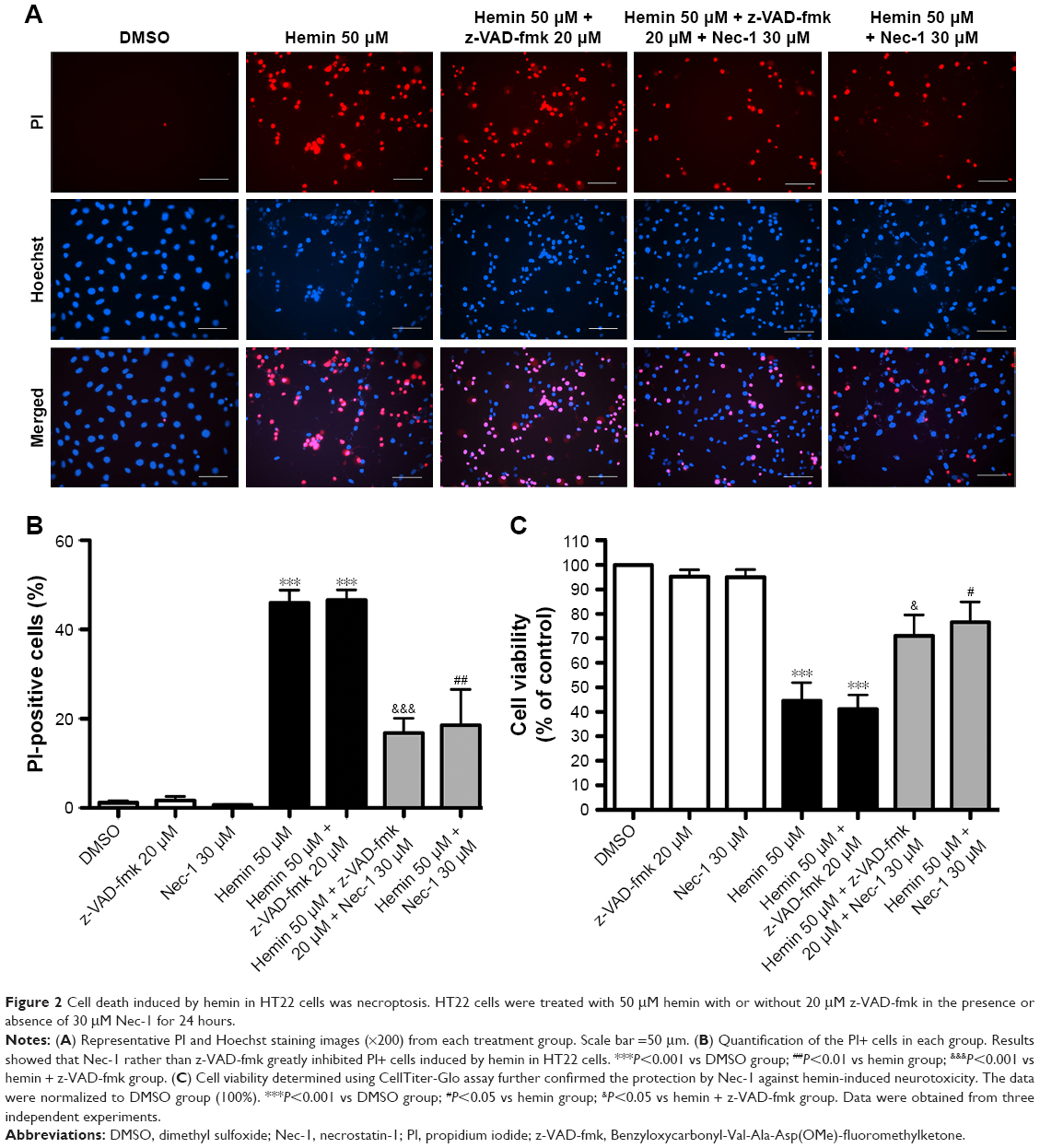 | Figure 2 Cell death induced by hemin in HT22 cells was necroptosis. HT22 cells were treated with 50 μM hemin with or without 20 μM z-VAD-fmk in the presence or absence of 30 μM Nec-1 for 24 hours. |
ROS accumulation induced by hemin mediated necrotic cell death in HT22 cells
It has been reported that accumulation of ROS is critical for necroptosis.14,15 Therefore, we measured mitochondrial ROS using MitoSox Red in HT22 cells treated with hemin. Increased ROS were detected in the HT22 cells treated with hemin, but this response was inhibited by Nec-1 treatment (Figure 3A). Furthermore, the necrotic cell death induced by hemin was abolished in the presence of ROS scavenger BHA (Figure 3B). These results revealed that ROS accumulation might be responsible for necrotic cell death induced by hemin in HT22 cells.
 | Figure 3 ROS accumulation induced by hemin mediated necrotic cell death in HT22 cells. |
RIP3 knockdown by siRNA inhibited necrotic cell death induced by hemin in HT22 cells
It is well known that RIP3 plays an essential role in TNFR-mediated necrosis and multiple necrotic signals converge at RIP3.5,13,14 So, we further investigated the role of RIP3 in hemin-induced necrotic cell death in HT22 cells. RIP3 was efficiently knocked down by RIP3 siRNA when compared with negative siRNA (Figure 4). As shown in Figure 5, hemin caused significant necrotic cell death in negative siRNA group. However, inhibiting RIP3 by siRNA dramatically blocked necrotic cell death induced by hemin in HT22 cells. These results proved that RIP3 might act as a converging signaling molecule in hemin-induced HT22 necrosis.
 | Figure 4 The levels of RIP3 protein expression were determined by Western blotting after siRNA transfection in HT22 cells. |
 | Figure 5 siRNA silencing of RIP3 attenuated hemin-induced necrotic cell death in HT22 cells. |
Discussion
In the present study, we analyzed the mechanisms involved in hemin-induced cell death in HT22 cells. We first demonstrated that RIP1 inhibitor Nec-1 blocked hemin-induced cell death and ROS accumulation was responsible for hemin-induced cell death in HT22 cells. Furthermore, we found that silencing of RIP3 through siRNA attenuated hemin-induced cell death in HT22 cells.
Hemin-induced cell death was involved in the mechanism of brain injury after ICH
The mechanisms of brain injury occurring after ICH are very complex. Acute neuronal death of cells adjacent to the hematoma after ICH is frequently seen after hemorrhagic brain injury. The mechanisms of neuronal cell death are not fully understood. Numerous mechanisms of cell death including caspase-mediated apoptosis have been widely investigated in ICH. Necrotic death of cells adjacent to the hematoma is very common after ICH.10 After ICH, a lot of blood components are released into brain parenchyma. These include glutamate and hemoglobin, which are further broken down into heme or its oxidized form hemin.2 Hemin is a lipophilic oxidant that accumulates in intracranial hematoma at high micromolar concentrations and is toxic to multiple cell types, such as neurons and astrocytes.2,5 Hemin has been used in vitro to study the mechanisms of cell death in cultured neurons and neuronal cell lines.16,17 In the present study, we first demonstrated that hemin induced a concentration-dependent neurotoxicity in HT22 cells and most of the dead cells were PI+ cells (Figure 1). Dose-response studies showed that 50 μM hemin efficiently induced cell death. Therefore, an intermediate dose of 50 μM hemin that caused about 50% cell death was selected and used in the subsequent experiments.
Necroptosis RIP1 pathway mediated ICH-induced cell death in vivo and in vitro
Necrosis was considered as an unregulated and uncontrollable process. However, more recent studies have revealed that necrosis could also occur in a regulated manner. This kind of necrosis was named “necroptosis” or “programmed necrosis”.12,18 Necroptosis is a regulated necrotic cell death that shares the morphological features of necrosis.18 It is regulated by certain molecular pathways, including RIP1 and RIP3 pathways.11,13,14,19 Necroptosis has been reported to participate in many kinds of diseases, including ischemic stroke, myocardial infarction, acute pancreatitis, retinal detachment, traumatic brain injury, subarachnoid hemorrhage, and ICH.12,13,20–25
RIP1, a death domain-containing kinase, is considered as the initiator protein of necroptosis.11,18 During death domain receptor-induced apoptosis, RIP1 is cleaved and inactivated by caspase-8. However, this process is prevented by caspase inhibition and switches cell death from apoptosis to necrosis mode.18 Nec-1 is a specific inhibitor of necroptosis, which does not affect apoptosis, and acts through inhibiting RIP1.11,12 Studies from our group and other groups reported that necroptosis was at least partly involved in ICH in vivo.23,26 They showed that Nec-1 ameliorated cell death, brain edema, neuroinflammation, and neurological score deficits and reduced neurovascular injury after ICH in mice and rats.23,25–28 Genetic inhibition of RIP1 could reduce cell death and neurological deficits after ICH in mice and rats.24,26,29 A previous study further showed that Nec-1 inhibited glutamate-induced cell death in HT22 cells.9 Therefore, we hypothesized that necroptosis also contributed to hemin-induced neurotoxicity in HT22 cells. We investigated whether Nec-1 could block hemin-induced cell death in HT22 cells. Our data showed that hemin-induced cell death in HT22 cells could be inhibited by Nec-1 but not by apoptosis inhibitor z-VAD-fmk (Figure 2), suggesting the reduction of necroptotic cellular injury. The contribution of apoptosis to hemin-induced cell death is still unclear. One study showed the pan-caspase inhibitor z-VAD-fmk failed to reduce cell death of culture astrocytes.5 Our present study got the similar results. Our preliminary unpublished data revealed that Nec-1 had no effect on the expression of caspase-3. These results seemed to exclude apoptosis as a mechanism of hemin-induced cell death and indicated that hemin induced cell death mainly by necroptosis in HT22 cells. Our results were consistent with recent reports from other groups. Shen et al reported that RIP1 mediated oxygen hemoglobin-induced necroptosis in cultured neurons in vitro, which was inhibited by Nec-1.26 Furthermore, a recent study further showed that necroptosis mediated hemin-induced cell death in primary cortical neurons.16 The study also revealed a new kind of neuronal death called ferroptosis following ICH in vitro and in vivo.16 The authors found that ICH shared features of ferroptotic and necroptotic cell death and their inhibitors could rescue over 80% cell death post-ICH in vitro.16 The existence of ferroptotic cell death may explain why Nec-1 cannot inhibit all PI+ cells (Figure 2A and B). However, whether ferroptosis also plays a role in hemin-induced cell death remains to be further investigated.
ROS participated in hemin-induced necroptosis in HT22 cells
ROS have been reported to play a critical role in the execution of necroptosis.14,15,18,19 In our study on hemin neurotoxicity in HT22 cells, ROS were detected in the HT22 cells treated with hemin and were inhibited by Nec-1 treatment (Figure 3A). Furthermore, necrotic cell death induced by hemin could be abolished in the presence of ROS scavenger BHA (Figure 3B). These results indicated that ROS accumulation was at least partially responsible for necrotic cell death induced by hemin in HT22 cells. NADPH oxidase has also been found to participate in TNF-induced ROS production.19,30 After stimulation by TNF, NOXO1 subunit is recruited to RIP1 and initiates ROS production.19,30 RIP3 regulates another source of TNF-induced ROS production, which is responsible for TNF-induced necrosis.14 RIP3 interacts with several metabolic enzymes that are implicated in the metabolism of glutamine and in glycogenolysis, including the cytosolic enzymes PYGL, GLUD1, and GLUL.19 PYGL, GLUD1, and GLUL regulate ROS production by their own pathways.19 However, further studies are needed to understand the precise mechanisms through which ROS execute necroptosis in hemin-treated HT22 cells.
RIP3 was one of the important downstream proteins of necroptosis after ICH
The kinase activity of RIP3, a member of the Ser/Thr kinase family, was originally identified to regulate caspase-dependent and caspase-independent apoptosis and NF-κB activation.31–33 However, RIP3 knockout mice exhibited no remarkable phenotypes and responded normally to apoptosis and NF-κB activation signals.34 Recent studies identified RIP3 as a crucial modulator of necroptosis and that multiple necrotic signals converge at RIP3.13,14,35 RIP3 is recruited to RIP1 and forms a RIP1–RIP3-containing complex through direct interaction between the homotypic RHIM domains of RIP1 and RIP3.13,19,35 This complex has recently been named necrosome.18 The presence of Nec-1 inhibits the formation of the RIP1–RIP3 complex, suggesting that RIP1 kinase activity is involved in necrosome formation.13,19,35 Our previous study showed that the expression of RIP3 was increased after ICH and Nec-1 treatment diminished the interaction of RIP3 with RIP1 after ICH.23 Our present study showed that RIP3 protein was significantly knocked down by siRNA (Figure 4) and RIP3 knockdown greatly inhibited necrotic cell death induced by hemin in HT22 cells (Figure 5). These results implicated that RIP3 might mediate hemin neurotoxicity and necroptosis after ICH. Importantly, a previous study reported that RIP3 knockout significantly reduced necrotic cell death after ICH, which further confirmed that RIP3 mediated necroptosis after ICH.10 Latest studies have added new members to this necroptosis pathway, which are downstream components of RIP3, including MLKL, PGAM5, and Drp1.36,37 However, further work is required to establish whether these components also contribute to hemin-induced cell death in HT22 cells and neurological injury after ICH.
Conclusion
Our study demonstrated that hemin induced programmed necrosis in HT22 hippocampal neurons, which was regulated by RIP1 and RIP3 pathways, and that Nec-1 might have use in preventing hemin-induced cell death. Our data suggest that regulation of necroptosis pathways including RIP1 and RIP3 may represent novel therapeutic targets for ICH, but further studies are needed to explore the underlying mechanisms of necroptosis, ferroptosis, apoptosis, and oxidative stress occurring after ICH in vitro and in vivo.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No 81501012) and the Fujian Province Natural Science Foundation (No 2015J05149).
Disclosure
The authors report no conflicts of interest in this work.
References
Qureshi AI, Tuhrim S, Broderick JP, Batjer HH, Hondo H, Hanley DF. Spontaneous intracerebral hemorrhage. N Engl J Med. 2001;344(19):1450–1460. | ||
Aronowski J, Zhao X. Molecular pathophysiology of cerebral hemorrhage: secondary brain injury. Stroke. 2011;42(6):1781–1786. | ||
Keep RF, Hua Y, Xi G. Intracerebral haemorrhage: mechanisms of injury and therapeutic targets. Lancet Neurol. 2012;11(8):720–731. | ||
Robinson SR, Dang TN, Dringen R, Bishop GM. Hemin toxicity: a preventable source of brain damage following hemorrhagic stroke. Redox Rep. 2009;14(6):228–235. | ||
Laird MD, Wakade C, Alleyne CH, Dhandapani KM. Hemin-induced necroptosis involves glutathione depletion in mouse astrocytes. Free Radic Biol Med. 2008;45(8):1103–1114. | ||
Regan RF, Wang Y, Ma X, Chong A, Guo Y. Activation of extracellular signal-regulated kinases potentiates hemin toxicity in astrocyte cultures. J Neurochem. 2001;79(3):545–555. | ||
Dang TN, Bishop GM, Dringen R, Robinson SR. The metabolism and toxicity of hemin in astrocytes. Glia. 2011;59(10):1540–1550. | ||
Regan RF, Chen J, Benvenisti-Zarom L. Heme oxygenase-2 gene deletion attenuates oxidative stress in neurons exposed to extracellular hemin. BMC Neurosci. 2004;5:34. | ||
Xu X, Chua CC, Kong J, et al. Necrostatin-1 protects against glutamate-induced glutathione depletion and caspase-independent cell death in HT-22 cells. J Neurochem. 2007;103(5):2004–2014. | ||
Zhu X, Tao L, Tejima-Mandeville E, et al. Plasmalemma permeability and necrotic cell death phenotypes after intracerebral hemorrhage in mice. Stroke. 2012;43(2):524–531. | ||
Degterev A, Hitomi J, Germscheid M, et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4(5):313–321. | ||
Degterev A, Huang Z, Boyce M, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1(2):112–119. | ||
He S, Wang L, Miao L, et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137(6):1100–1111. | ||
Zhang DW, Shao J, Lin J, et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325(5938):332–336. | ||
He S, Liang Y, Shao F, Wang X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc Natl Acad Sci U S A. 2011;108(50):20054–20059. | ||
Zille M, Karuppagounder SS, Chen Y, et al. Neuronal death after hemorrhagic stroke in vitro and in vivo shares features of ferroptosis and necroptosis. Stroke. 2017;48(4):1033–1043. | ||
Karuppagounder SS, Alim I, Khim SJ, et al. Therapeutic targeting of oxygen-sensing prolyl hydroxylases abrogates ATF4-dependent neuronal death and improves outcomes after brain hemorrhage in several rodent models. Sci Transl Med. 2016;8(328):ra329. | ||
Vandenabeele P, Galluzzi L, vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11(10):700–714. | ||
Vandenabeele P, Declercq W, van Herreweghe F, vanden Berghe T. The role of the kinases RIP1 and RIP3 in TNF-induced necrosis. Sci Signal. 2010;3(115):re4. | ||
Oerlemans MI, Liu J, Arslan F, et al. Inhibition of RIP1-dependent necrosis prevents adverse cardiac remodeling after myocardial ischemia-reperfusion in vivo. Basic Res Cardiol. 2012;107(4):270. | ||
Trichonas G, Murakami Y, Thanos A, et al. Receptor interacting protein kinases mediate retinal detachment-induced photoreceptor necrosis and compensate for inhibition of apoptosis. Proc Natl Acad Sci U S A. 2010;107(50):21695–21700. | ||
You Z, Savitz SI, Yang J, et al. Necrostatin-1 reduces histopathology and improves functional outcome after controlled cortical impact in mice. J Cereb Blood Flow Metab. 2008;28(9):1564–1573. | ||
Su X, Wang H, Kang D, et al. Necrostatin-1 ameliorates intracerebral hemorrhage-induced brain injury in mice through inhibiting RIP1/RIP3 pathway. Neurochem Res. 2015;40(4):643–650. | ||
Chen F, Su X, Lin Z, et al. Necrostatin-1 attenuates early brain injury after subarachnoid hemorrhage in rats by inhibiting necroptosis. Neuropsychiatr Dis Treat. 2017;13:1771–1782. | ||
Majmundar N, Kim B, Prestigiacomo CJ. Necroptosis pathway in treatment of intracerebral hemorrhage: Novel therapeutic target. World Neurosurg. 2016;89:716–717. | ||
Shen H, Liu C, Zhang D, et al. Role for RIP1 in mediating necroptosis in experimental intracerebral hemorrhage model both in vivo and in vitro. Cell Death Dis. 2017;8(3):e2641. | ||
Chang P, Dong W, Zhang M, et al. Anti-necroptosis chemical necrostatin-1 can also suppress apoptotic and autophagic pathway to exert neuroprotective effect in mice intracerebral hemorrhage model. J Mol Neurosci. 2014;52(2):242–249. | ||
King MD, Whitaker-Lea WA, Campbell JM, Alleyne CH, Dhandapani KM. Necrostatin-1 reduces neurovascular injury after intracerebral hemorrhage. Int J Cell Biol. 2014;(495817):1–10. | ||
Lule S, Wu L, Mcallister LM, et al. Genetic inhibition of receptor interacting protein kinase-1 reduces cell death and improves functional outcome after intracerebral hemorrhage in mice. Stroke. 2017;48(9):2549–2556. | ||
Kim YS, Morgan MJ, Choksi S, Liu ZG. TNF-induced activation of the Nox1 NADPH oxidase and its role in the induction of necrotic cell death. Mol Cell. 2007;26(5):675–687. | ||
Feng S, Yang Y, Mei Y, et al. Cleavage of RIP3 inactivates its caspase-independent apoptosis pathway by removal of kinase domain. Cell Signal. 2007;19(10):2056–2067. | ||
Yu PW, Huang BC, Shen M, et al. Identification of RIP3, a RIP-like kinase that activates apoptosis and NFkappaB. Curr Biol. 1999;9(10):539–542. | ||
Sun X, Lee J, Navas T, Baldwin DT, Stewart TA, Dixit VM. RIP3, a novel apoptosis-inducing kinase. J Biol Chem. 1999;274(24):16871–16875. | ||
Newton K, Sun X, Dixit VM. Kinase RIP3 is dispensable for normal NF-kappa Bs, signaling by the B-cell and T-cell receptors, tumor necrosis factor receptor 1, and Toll-like receptors 2 and 4. Mol Cell Biol. 2004;24(4):1464–1469. | ||
Cho YS, Challa S, Moquin D, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137(6):1112–1123. | ||
Sun L, Wang H, Wang Z, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148(1–2):213–227. | ||
Wang Z, Jiang H, Chen S, Du F, Wang X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell. 2012;148(1–2):228–243. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.