Back to Journals » Drug Design, Development and Therapy » Volume 19
Rheumatoid Arthritis Therapy Based on B Cells
Authors Liang Y, Zha M, Liu Q, Lai Z, Li L, Shao Y ![]() , Sun J
, Sun J ![]()
Received 11 March 2025
Accepted for publication 19 August 2025
Published 6 September 2025 Volume 2025:19 Pages 7837—7852
DOI https://doi.org/10.2147/DDDT.S527687
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Manfred Ogris
Yongqi Liang,1,* Menglei Zha,1,* Qifeng Liu,1 Zhifei Lai,1 Lei Li,1 Yiming Shao,2 Jianbo Sun1
1Dongguan Key Laboratory of Chronic Inflammatory Diseases, The First Dongguan Affiliated Hospital, Guangdong Medical University, Dongguan, Guangdong, People’s Republic of China; 2Dongguan Key Laboratory of Sepsis Translational Medicine, The First Dongguan Affiliated Hospital, Guangdong Medical University, Dongguan, Guangdong, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yiming Shao, Email [email protected] Jianbo Sun, Email [email protected]
Abstract: Rheumatoid arthritis (RA) is a chronic autoimmune disorder characterized by persistent synovial inflammation, joint destruction, and progressive disability. While current therapeutic approaches—including corticosteroids, disease-modifying antirheumatic drugs (DMARDs), nonsteroidal anti-inflammatory drugs (NSAIDs), and biologic agents—provide symptomatic relief, their clinical utility remains constrained by substantial limitations such as systemic toxicity, drug resistance, and cumulative adverse effects. These challenges underscore the critical need for novel therapeutic strategies with improved safety and efficacy profiles. The pathogenesis of RA involves multifaceted immune dysregulation, with emerging evidence highlighting the central role of B lymphocytes in both disease initiation and progression. Although B cell-targeted therapies like rituximab demonstrate clinical efficacy, unanswered questions persist regarding the precise immune functions of B cell subpopulations in RA pathogenesis and their potential as translatable therapeutic targets. This comprehensive review examines the clinical burden of RA, limitations of conventional therapies, and the evolving understanding of B cell pathophysiology. We critically evaluate established B cell-directed interventions—including B cell depletion, B cell functional modulation, and regulatory B cell (Breg) promotion—while exploring innovative nanofabrication technologies that may overcome current therapeutic barriers. By synthesizing recent advances in immunomodulatory research, this analysis aims to inform future directions for targeted RA management.
Keywords: RA, B cells, targeted therapy, immune regulation, antigen presentation
Introduction
Characterization of RA
RA is a chronic autoimmune disorder characterized by synovial inflammation. It predominantly affects women, with a female-to-male ratio ranging from 2:1 to 4:1, and typically presents between the ages of 35 and 50 years. RA is associated with substantial morbidity, a chronic progression, multisystem complications, and elevated rates of disability and mortality. While the precise etiology remains unclear, emerging evidence suggests that genetic, environmental, infectious, immune, and endocrine factors contribute to disease development (Figure 1).1
 |
Figure 1 Pathogenic factors of RA. The pathogenesis of RA involves a multifactorial interplay of genetic susceptibility, environmental triggers, immune dysregulation, hormonal fluctuations, aging processes, and lifestyle influences, with immune system aberrations constituting the central pathogenic driver. |
RA presents with heterogeneous clinical manifestations, typically developing insidious with symmetrical inflammatory polyarthritis, predilection for the hands, wrists, and feet. Patients often experience morning stiffness, fatigue, low-grade pyrexia, myalgia, and weight loss. Although rare, fulminant disease onset with rapid joint destruction may occur.2 Once joint damage is established, it is largely irreversible. Without appropriate treatment, RA results in a 70% cumulative disability incidence within three years, with functional limitations progressively worsening over time. Beyond the joints, RA can involve pathogenesis of multiple organ systems such as pericarditis, lung-related complications, renal complications, Sjögren’s syndrome, hematological abnormalities, neurological issues, and other systemic complications.
RA remains incurable, with current management emphasizing early standardized treatment and continuous monitoring. The therapeutic paradigm prioritizes achieving clinical remission or sustaining low disease activity to attenuate disability progression and enhance quality of life. Contemporary interventions comprise pharmacological therapies (central to disease control) and surgical options (Table 1). DMARDs, including methotrexate, leflunomide, and sulfasalazine, constitute first-line therapy to retard disease progression. Combination DMARD regimens are advocated for treatment-refractory cases. Common adverse effects encompass bone marrow suppression, gastrointestinal intolerance, and hepatobiliary toxicity. NSAIDs (eg, ibuprofen, celecoxib) offer symptomatic analgesia but lack disease-modifying properties. Their gastrointestinal ulcerogenic potential and cardiovascular thrombotic risks necessitate cautious drug selection. Glucocorticoids (eg, triamcinolone) serve as short-term anti-inflammatory agents but carry risks of metabolic disturbances and osteoporosis. Phytotherapeutic compounds such as tripterygium glycosides demonstrate therapeutic efficacy yet require rigorous toxicity monitoring. TNF-α inhibitors (eg, infliximab, adalimumab) demonstrate efficacy in DMARD-refractory disease, while CD20-targeted biologics (eg, rituximab) underscore the therapeutic value of B cell-depleting strategies.
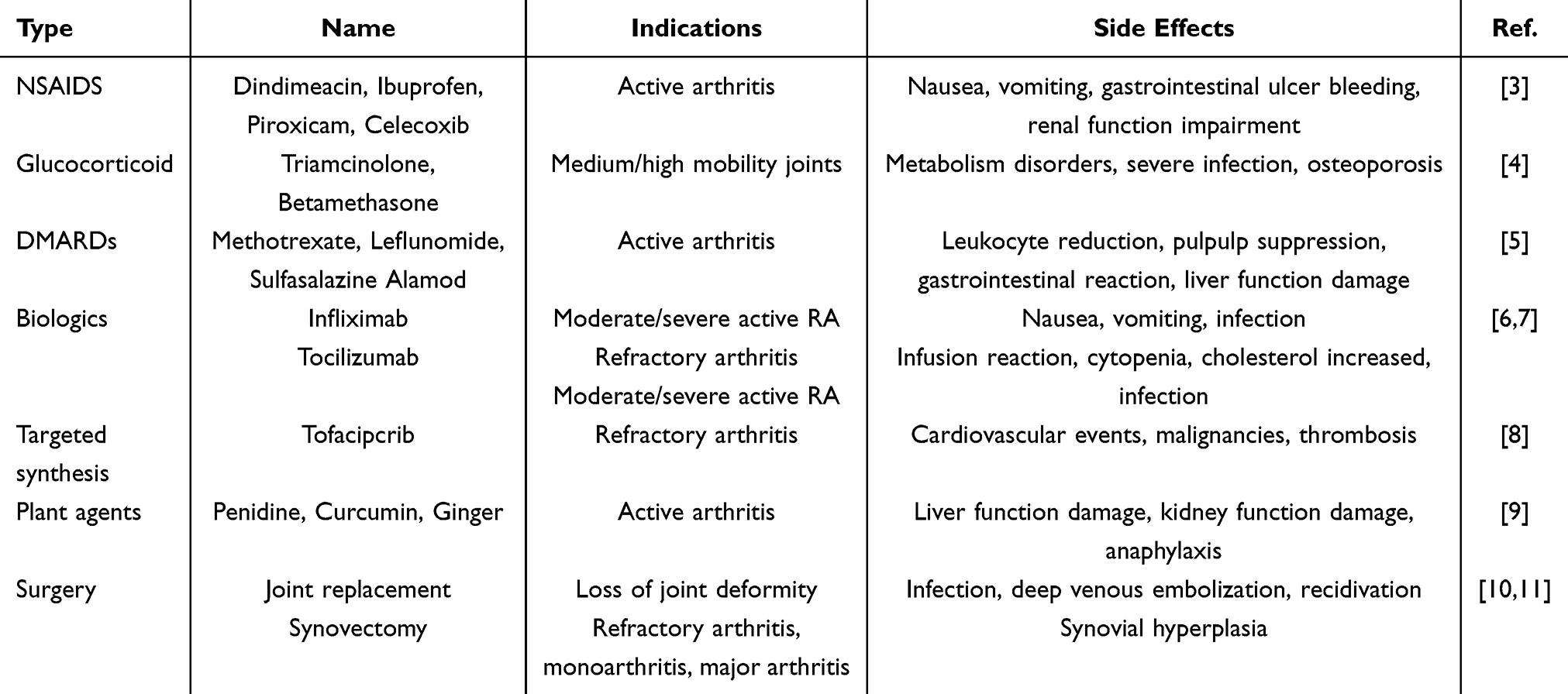 |
Table 1 General Clinical Treatments and Methods for RA |
In summary, while the precise etiopathogenesis of RA remains incompletely elucidated, B cell-directed therapies have emerged as a promising therapeutic paradigm. Current disease-modifying agents lack curative potential, requiring lifelong administration with significant adverse effect profiles. Biologic agents, particularly B cell-depleting modalities, demonstrate enhanced clinical efficacy, highlighting the therapeutic promise of mechanism-driven immunomodulatory strategies in this chronic immune-mediated disorder.
Role and Mechanism of B Cells in RA Development
B lymphocytes, central cellular components of adaptive immunity, fulfill critical immunological roles including high-affinity immunoglobulin production immune memory establishment, antigen presentation capacity, pleiotropic cytokine secretion (IL-6, IFN-γ, TNF-α, GM-CSF, IL-10, TGF-β1, IL-35). Memory B cell subsets and long-lived plasma cells mediate pathogenic antibody synthesis (IgG/IgM/IgE). In the pathogenesis of RA, B cells not only initiate immune responses but also directly contribute to disease progression and joint damage. They perform multifaceted roles, including autoantibody production, secretion of proinflammatory cytokines, and interactions with other immune cells (eg, T cells, dendritic cells) (Figure 2). Auto-antibody generation (rheumatoid factor [RF]/anti-citrullinated protein antibodies [ACPA]) serving as diagnostic/prognostic biomarkers while directly contributing to synovitis.12 T cell activation (MHC-II/TLR-dependent and cytokine-driven pathways) sustaining inflammatory cascades.13,14 Immune complex deposition and proinflammatory cytokine networks. Functional impairment of regulatory lymphocytes (Treg/Breg suppression).15 Thus, understanding the complex interplay between dysregulated B cell function and RA pathogenesis not only enhances our comprehension of disease mechanisms but also paves the way for the development of novel therapeutic strategies targeting B cells for RA management.
 |
Figure 2 Role of B cells in RA. In RA pathogenesis, B lymphocytes orchestrate inflammatory cascades through antigen presentation, immune cell activation, and direct cytokine secretion. Pathologically hyperactivated autoreactive B cells drive excessive autoantibody production, concomitant with Breg depletion, collectively propelling RA initiation and progression. |
Abnormalities of B Cell Function Exacerbated RA
Dysregulated B cell activation constitutes a central pathogenic mechanism in RA initiation and progression. Physiologically, B cell responses are tightly controlled through foreign antigen-specific immune surveillance. In RA, breakdown of self-tolerance drives autoantigen-directed immune responses through: aberrant self-antigen recognition, dysfunctional costimulatory signaling, BCR hyperactivation, pathogenic immune complex formation, Proinflammatory cytokine milieu disruption. Genome-wide association studies implicate HLA-DRβ1 shared epitope alleles in enhanced antigen-presenting cell (APC) function, facilitating citrullinated antigen presentation to autoreactive B cells.16 Complete B cell activation requires both BCR engagement and costimulatory signals. RA synovium exhibits upregulated CD40/CD80-CD86 expression, enhancing B-T cell crosstalk via CD40-CD40L interactions and CD28-mediated costimulation. These signaling axes drive autoreactive B cell expansion, ACPA/RF overproduction, and feed-forward inflammatory loops characterizing RA’s chronic destructive arthritis.17
The development of lymphoid follicle-like architectures within RA synovium pathognomonically reflects sustained immune system overactivation, B lymphocyte clustering, and proinflammatory cytokine dysregulation. These ectopic tertiary lymphoid structures (TLS) mimic secondary lymphoid organ organization, establishing specialized microenvironments that drive pathogenic B cell activation/differentiation while sustaining autoimmune amplification loops.18 Molecular profiling reveals marked upregulation of NF-κB-inducing kinase (NIK) and non-canonical NF-κB pathway components in TLS-positive synovium versus TLS-negative tissue.19 NIK-mediated signaling and high endothelial venule (HEV) neogenesis—induced through lymphotoxin-β (LTβ) secretion from infiltrating immune cells—constitute pivotal mechanisms in TLS ontogeny. Proinflammatory cytokines (TNF-α/IL-6 dominant) orchestrate disease-driving immune responses by stimulating B cell maturation, clonal expansion, and autoantibody secretion.20 Activated B cells undergo terminal differentiation into long-lived plasma cells, perpetuating synovial inflammation through sustained autoantibody production and osteoclastogenic cytokine release.21
Interaction of B Cells with Other Immune Cells in RA
The immunopathogenesis of RA involves multifactorial mechanisms where B lymphocytes orchestrate disease progression through autonomous activation, autoantibody production, and bidirectional crosstalk with T cells, dendritic cells (DCs), macrophages, and natural killer (NK) cells (Figure 3). Antigen-presenting B cells engage CD4+ T cells via MHC-II/antigen complexes, sustaining autoimmune activation through co-stimulatory molecule interactions (CD80/CD86-CD28) and proinflammatory cytokine secretion (IL-6/TNF-α) amplifying chronic synovitis.22 DCs potentiate humoral immunity by enhancing B cell maturation and immunoglobulin class-switching, exacerbating articular immune injury. B cell-macrophage interactions via TNF-α/IL-6 signaling establish feed-forward inflammatory cycles, while Fcγ receptor-bound immune complexes trigger macrophage-mediated B cell hyperactivation.23–25 NK cell cytotoxicity, primed by B cell-derived IL-15, synergizes with IFN-γ-mediated immunomodulation to augment autoimmune responses.26 RA synovial fibroblasts (RA-FLS) exhibit CD40/BAFF-driven MMP secretion when stimulated by B cell contact, directing cartilage/bone matrix degradation.27
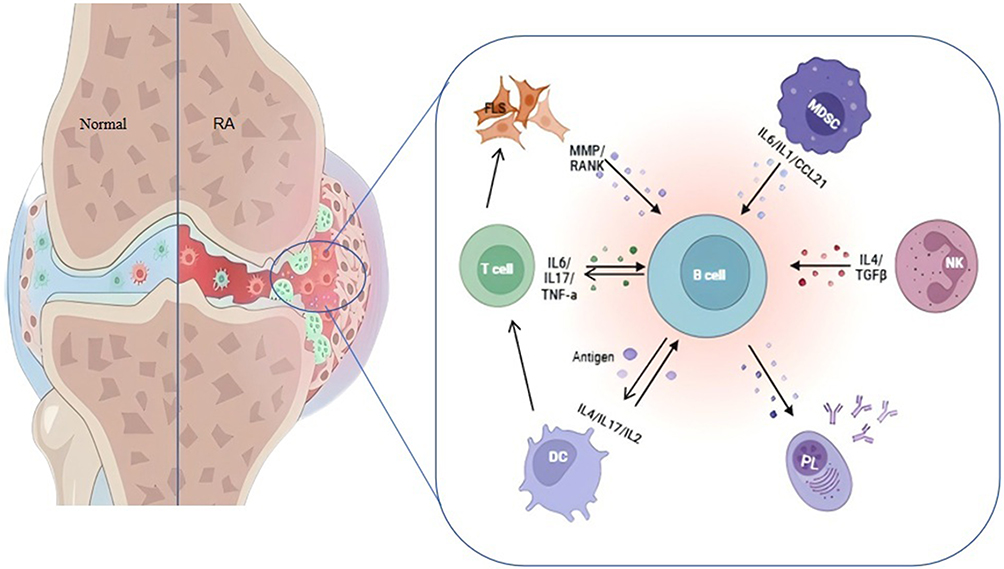 |
Figure 3 Role and immune imbalance between B cells and other immune cells in RA. B lymphocytes exert multifaceted roles in RA pathogenesis, not only generating pathogenic autoantibodies including RF and ACPA directly implicated in immune dysregulation, but also engaging in dynamic crosstalk with T lymphocytes, dendritic cells, macrophages, and other immune effectors. These cellular interactions perpetuate inflammatory cascades, sustaining chronic inflammation while paradoxically disrupting immune homeostasis when inflammatory regulation becomes impaired. |
Paradoxically, regulatory B cells (Bregs) counterbalance inflammation via IL-10-mediated Treg expansion.22
Molecular Mechanism of Abnormal Activation of B Cells in RA
B cell dysregulation in RA involves complex signaling cascades that drive clonal expansion, plasmablast differentiation, and autoantibody overproduction via SYK/PI3K/MAPK/NF-κB pathway activation, perpetuating chronic immune-inflammatory pathology. Targeting these pathways represents novel therapeutic strategies for disease modification.
Bruton’s tyrosine kinase (BTK), essential for BCR signaling, emerges as a key therapeutic target with BTK inhibitors demonstrating significant anti-inflammatory efficacy.28 TLR-MyD88 signaling amplifies autoimmune memory through IFN-α/CpG ODN-mediated B cell activation.29 BAFF/BLyS axis hyperactivation sustains pathogenic B cell survival and complement-mediated inflammation.30 IL-6 trans-signaling via JAK/STAT3 phosphorylation promotes synovial damage, while PI3K-AKT pathway orchestrates B cell proliferative responses.31,32 AKT inhibitors (eg, perifosine) attenuate immune cell activation through AKT-mTOR blockade.33 MAPK/NF-κB cascades coordinate inflammatory gene expression via ERK/p38/IκB kinase activation.34,35 JAK-STAT4 axis modulates B cell differentiation, whereas FcγRIIb-CD19 co-targeting (eg, obexelimab) restores immune tolerance by dual inhibition of B cell activation (Figure 4).36,37
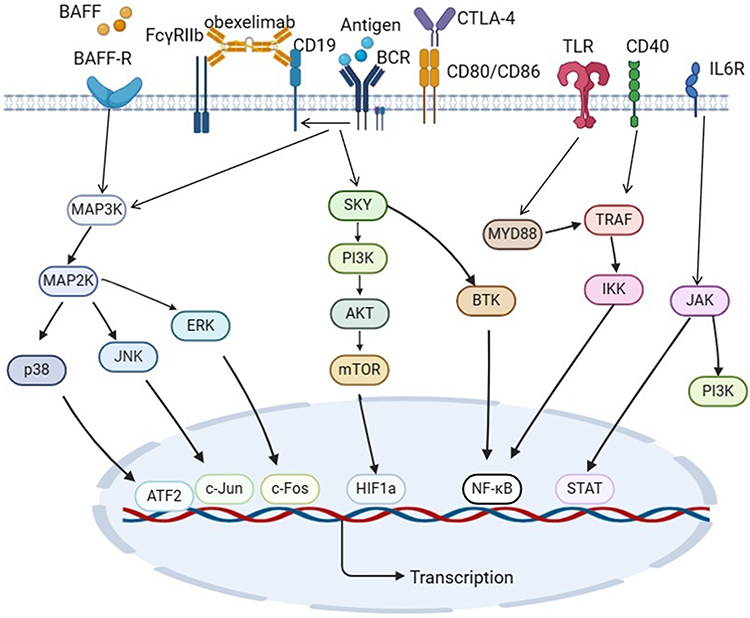 |
Figure 4 Signaling pathways and targets of B cells involved in RA. B lymphocytes orchestrate rheumatoid arthritis pathogenesis through a multi-pathway network involving BTK, TLR-MyD88, BAFF/BLyS, IL-6/IL-6R, PI3K-AKT, MAPK, NF-κB, and FcγR signaling axes. |
In summary, the role of B cells in RA is multifaceted. This intricate signaling network positions B cells as both pathogenic mediators and promising therapeutic targets in RA.
Recent Progress of RA Therapy Based on B Cells
Recent advances in RA therapeutics have yielded refined pharmacological agents with enhanced clinical efficacy and improved safety profiles. Research prioritization has shifted toward B cell-directed strategies, given their multimodal pathogenic contributions—autoantibody production, proinflammatory cytokine secretion, and antigen presentation—collectively driving chronic synovial inflammation. Immunomodulation of B cell effector functions demonstrates therapeutic potential through dampening pathogenic inflammation and ameliorating autoimmunity, thereby mitigating RA symptomatology.
Emerging B cell-targeted therapies are elucidated through three mechanistic categories (Table 2): 1) Surface antigen-directed biologics: CD19/CD20/CD22—key immunoregulatory checkpoints—monoclonal antibodies mediate B cell depletion or functional blockade, suppressing autoantibody titers and synovitis; 2) Pathway-specific inhibitors: BCR-mediated signaling (SYK/BTK), TLR-MyD88 axis, BAFF/APRIL system, IL-6 trans-signaling, PI3K-AKT-mTOR cascade, MAPK/NF-κB pathways, and JAK-STAT network—small-molecule inhibitors modulate cytokine/receptor crosstalk to attenuate B cell hyperactivity; 3) Advanced biotechnological interventions: B cell vaccines, CAR-T (Chimeric Antigen Receptor T-Cell Immunotherapy), mesenchymal stem cell infusions, and Breg augmentation strategically recalibrate B cell homeostasis, ameliorating clinical outcomes.
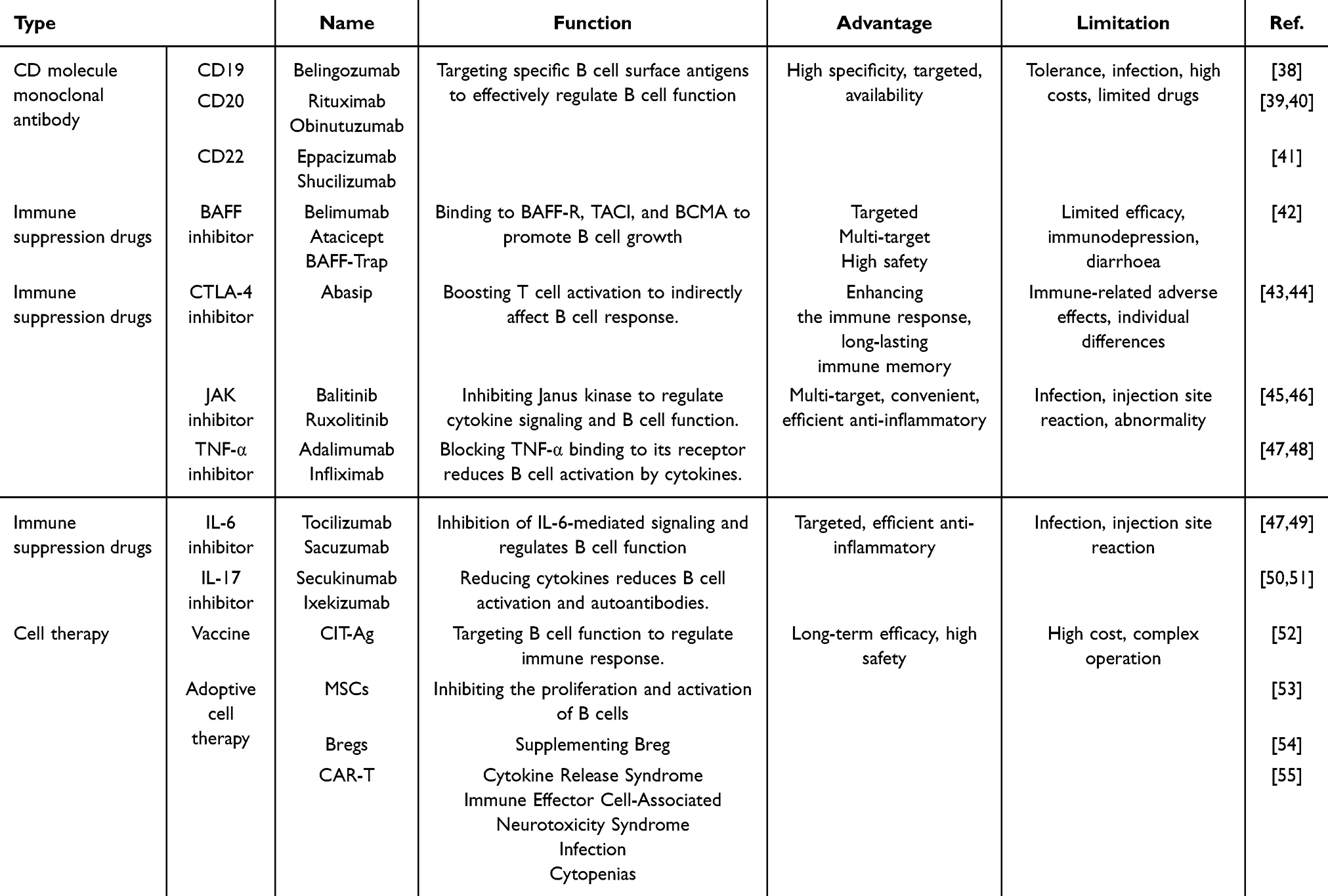 |
Table 2 Regulates the Types and Effects of Drugs and Therapies of B Cells for RA |
These therapeutic interventions stratify into three B cell-centric categories: B cell-depleting modalities, B cell-modulating approaches, and Breg-potentiating strategies, based on their immunomodulatory mechanisms. This chapter delineates these paradigms, with particular emphasis on their mechanistic divergences within B cell-targeted pathways.
Therapy Through B Cell Depletion
In RA pathogenesis, B cell hyperactivation, autoantibody generation, and immune homeostasis disruption constitute central pathogenic drivers. Pharmacological targeting of B cell surface antigens (functional modulation/clonal depletion) represents a cornerstone therapeutic approach. Rituximab, a chimeric anti-CD20 monoclonal antibody, remains the first FDA-approved B cell-depleting agent, mediating selective B cell depletion and demonstrating clinical efficacy in refractory RA management. Emerging agents targeting CD19, BAFF, and BR3 are under active investigation.
Cluster of differentiation (CD) molecules—surface markers dynamically expressed during leukocyte differentiation/activation—serve as critical therapeutic targets. Monoclonal antibodies (mAbs) targeting CD antigens (CD19/CD20/CD22) enable precision B cell modulation in RA through depleting pathogenic B cell clones and disrupting co-receptor signaling. Rituximab (anti-CD20 mAb) remains the clinical cornerstone, while novel CD19-targeted agents demonstrate promise. Georg et al38 developed blinatumomab (CD19×CD3 bispecific T cell engager), inducing T cell-mediated B cell cytotoxicity. In multidrug-refractory RA, low-dose blinatumomab reduced peripheral B cell counts (Figure 5A), reconfigured B cell subsets (Figure 5B and C), lowered autoantibody titers, and ameliorated synovitis (Figure 5D and E). CD22, a BCR co-inhibitory receptor, regulates NF-κB-mediated survival signals. SM03 (recombinant anti-CD22 IgG1) blocks CD22 homotypic interactions, suppressing B cell proliferation via NF-κB inhibition41,56 (Figure 5F).
 |
Figure 5 Targeted CD molecules for treating RA. (A) Temporal dynamics of B cell counts following mAb infusion (18-day observation). (B and C) Comparative analysis of B cell subpopulations pre- vs post-mAb administration. (D) Systemic disease activity quantification pre- and post-intervention. (E) Longitudinal profiles of RF and autoantibody titers. (F) Nano-Glo® luciferase assay (Promega) evaluation of viral transduction efficiency. *p < 0.05, **p < 0.01, ***p < 0.001. Reproduced from Bucci L, Hagen M, Rothe T et al. Bispecific T cell engager therapy for refractory RA. Nat Med. 2024;30(6):1593–1601. Copyright © 2024, The Author(s), under exclusive licence to Springer Nature America, Inc.38 |
CAR-T therapy—a personalized cellular immunotherapy—engineers T cells to express chimeric antigen receptors (CARs), enabling specific recognition and lysis of antigen-expressing cells. Preclinical studies demonstrate CRISPR-Cas9-edited allogeneic CD19-targeting CAR-T cells overcome host immunity with favorable safety/efficacy profiles in refractory autoimmune diseases.57 In RA, pathogenically skewed B cells are eliminated via CD19/CD20/BAFF-R-targeted CAR-T constructs depleting autoreactive B cell clones and mitigation of autoantibody-driven pathology and inflammatory cascades.55 Notably, BAFF-R-specific CAR-T cells achieve dual therapeutic effects by Eradicating BAFF-R B cells and disrupting BAFF/BAFF-R survival signaling. CD19-directed CAR-T therapy—the most clinically advanced approach—clears circulating/tissue-resident B cells, reduces autoantibody/cytokine burden, and attenuates osteoarticular destruction. Szabo et al58 reported a refractory RA case treated with B cell-targeted CAR-T therapy. Diagnosed in 2011, the patient exhibited persistent synovitis despite conventional DMARD/biologic regimens (Figure 6A). Longitudinal analysis demonstrated: declined DAS28-CRP scores, and reduced anti-vimentin/anti-dsDNA autoantibody titers (Figure 6B and C). Following 2023 DLBCL diagnosis, CAR-T therapy (post-chemotherapy failure) yielded: rapid RF decline (48 IU/mL at 3 weeks), sustained serologic remission (13 IU/mL at 1 year), clinical symptom resolution (Figure 6D). This dual therapeutic efficacy highlights CAR-T’s potential beyond hematologic malignancies into immune-mediated disorders.
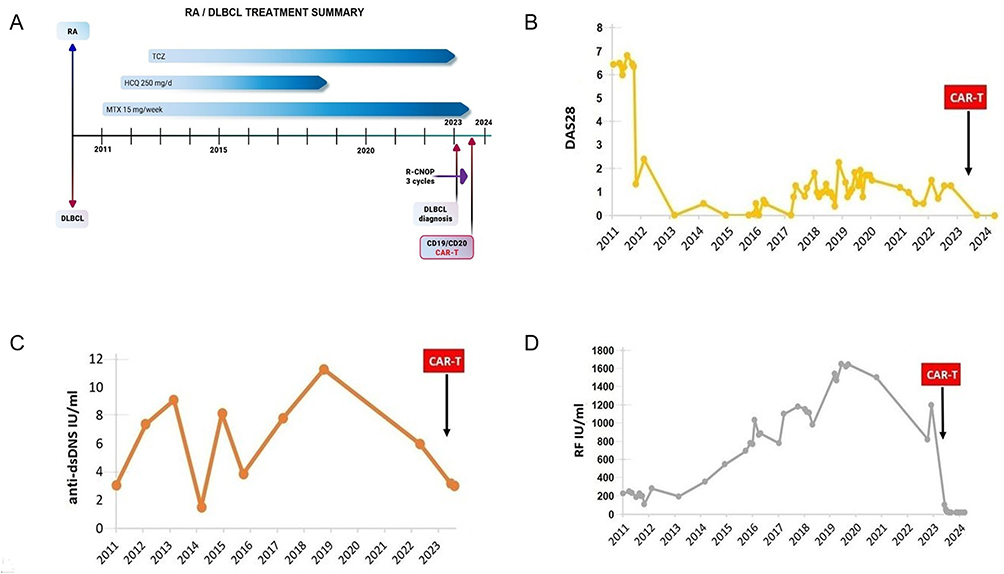 |
Figure 6 CAR-T treatment for refractory RA. (A) Longitudinal treatment history of the RA patient (2011–2024). (B) Temporal evolution of disease activity scores (DAS28), (C) anti-vimentin/anti-dsDNA antibody titers, and (D) RF levels during therapeutic intervention. Reproduced from Szabo D, Balogh A, Gopcsa L et al. Sustained drug-free remission in RA associated with diffuse large B-cell lymphoma following tandem CD20-CD19-directed non-cryopreserved CAR-T cell therapy using zamtocabtagene autoleucel. RMD Open. 2024;10(4): e004727. Copyright © Author(s) (or their employer(s)) 2024. Re-use permitted under CC BY-NC. No commercial re-use. Published by BMJ.58 |
Therapy via Regulating B Cell Function
B lymphocytes orchestrate pivotal pathogenic roles in RA, driving recent research focus on functional reprogramming strategies. Traditional DMARDs exert B cell regulatory effects through pharmacodynamic optimization, while emerging immune checkpoint inhibitors (CTLA-4/BAFFR-targeted) and intracellular signaling antagonists (PI3K/SYK/JAK-STAT/MAPK/BTK) attenuate inflammatory cascades via: Immune cell activity suppression and Synovio-protective effects. B cell-activating factor (BAFF)—a TNF superfamily cytokine predominantly expressed by monocytes, dendritic cells, and T cells—mediates B cell homeostasis through three receptors: BAFFR: Sustains mature B cell survival/response, TACI: Modulates B-T cell crosstalk, BCMA: Regulates humoral immunity. BAFF inhibitors achieve therapeutic efficacy by: competitively binding soluble BAFF, disrupting BAFF-receptor interactions, and normalizing peripheral B cell pools. Sun et al42 engineered BAFF-Trap, a high-affinity BAFF antagonist, demonstrating potent neutralization of soluble BAFF. In collagen-induced arthritis (CIA) models, intraperitoneal BAFF-Trap administration reduced serum BAFF levels (Figure 7A), expanded Breg populations (joint microenvironment modulation) (Figure 7B), suppressed pathogenic autoantibody titers (Figure 7C), and attenuated both CIA and adjuvant-induced arthritis progression. Bruton’s tyrosine kinase (BTK)—a key BCR downstream effector—mediates autoimmune pathogenesis. BTK inhibitors significantly impair: B cell activation thresholds, co-stimulatory molecule upregulation, clonal proliferation (Figure 7D).59 Currie et al60 identified CGI1746, a selective BTK inhibitor blocking enzymatic activation via dual inhibition of autophosphorylation/transphosphorylation. CIA trials revealed reduced autoantibody burden, validating its RA therapeutic potential.
 |
Figure 7 BAFF-Trap and BTi modulate the proliferation and differentiation of B cells. (A) BAFF expression profiles in B lymphocytes following BAFF-Trap administration. (B and C) BAFF-Trap-modulated B cell regulation of IL-10 secretion and antibody titers. (D) BTK inhibitor (BTKi) impact on B cell proliferative capacity, activation thresholds, and co-stimulatory molecule upregulation. *p < 0.05, **p < 0.01, ****p < 0.0001. Zhou B, Zhang H, Su X et al. Therapeutic effects of a novel BAFF blocker on arthritis. Signal Transduct Target Ther. 2019;4:19. © The Author(s) 2019. Creative Commons Attribution 4.0 International License.42 Reproduced from Li R, Tang H, Burns JC, Hopkins BT, Le Coz C, Zhang B, de Barcelos IP, Romberg N, Goldstein AC, Banwell BL, Luning Prak ET, Mingueneau M, Bar-Or A. BTK inhibition limits B-cell-T-cell interaction through modulation of B-cell metabolism: implications for multiple sclerosis therapy. Acta Neuropathol. 2022 ;143(4):505–521. © The Author(s) 2022. Creative Commons Attribution 4.0 International License.59 |
While immune signaling inhibitors demonstrate therapeutic precision in RA through targeted immunomodulation and joint preservation, their clinical application is constrained by mechanism-driven adverse effects including immune-related adverse events (irAEs), infection susceptibility from immunosuppression, treatment refractoriness, and acquired drug resistance. These limitations necessitate risk-stratified patient selection, therapeutic drug monitoring, and personalized therapeutic regimens. Despite these challenges, pathway-specific inhibitors remain a viable therapeutic paradigm when strategically integrated into precision medicine frameworks to optimize risk-benefit profiles.
Therapy with Enhanced Bregs
Bregs serve as critical immunomodulators in RA pathogenesis, suppressing inflammatory cascades and reinstating immune homeostasis through IL-10 secretion and cellular crosstalk with effector lymphocytes. Therapeutic potentiation of Breg functionality represents a promising immunoregulatory strategy; however, translational challenges persist regarding their phenotypic stability, mechanistic elucidation of suppressive networks, and safe clinical translation. Further investigations into Breg plasticity and targeted delivery systems are imperative to harness their full therapeutic potential.
The modulation of B cell populations—either by depleting pathogenic subsets or augmenting immunosuppressive Bregs—holds therapeutic potential to attenuate inflammatory and autoimmune processes in RA, particularly among treatment-refractory patients. RA pathogenesis involves compromised Breg quantity/function, impairing regulatory control over effector B and T lymphocytes, which perpetuates aberrant immune activation. Ex vivo Breg expansion followed by adoptive transfer presents a promising strategy, entailing peripheral blood isolation, in vitro activation/proliferation, and autologous reinfusion to amplify immunoregulatory capacity—an approach demonstrating efficacy in other autoimmune conditions.61 However, clinical translation of Breg-based therapies remains constrained by technical challenges including limited cell yields and suboptimal post-transfer persistence. Korneev et al54 investigated B cell activation strategies to induce immunoregulatory phenotypes, demonstrating that combinatorial stimulation with CD40L, CpG, and IL-21 generates Bregs exhibiting robust immunosuppressive potential (Figure 8A). Notably, combinatorial-treated B cells demonstrated enhanced Treg expansion (Figure 8E) and elevated IL-10 production without concomitant TNF upregulation (Figure 8B and C–F), indicating optimal in vitro differentiation of functionally active Bregs. However, IL-21 inclusion correlated with diminished cellular viability (Figure 8D), highlighting a critical translational barrier requiring protocol refinement.
 |
Figure 8 Therapy with In vitro expanded Bregs. (A) Heatmap depicting expression profiles of EBI3, CD274, IL-10, and TNF in differentially activated B cell subsets. (B) IL-10 and (C) TNF concentrations, with (D) Breg viability rates under therapeutic interventions. (E) Proliferation kinetics of Tregs, activated B cells, and CD4+ T lymphocytes. (F) ELISA quantification of IL-10 in CD4+ T cell co-culture supernatants. *p < 0.05, **p < 0.01, ***p < 0.001. Reproduced from Zheremyan EA, Ustiugova AS, Uvarova AN et al. Differentially activated B cells develop regulatory phenotype and show varying immunosuppressive features: a comparative study. Front Immunol. 2023;14:1178445. Copyright © 2023 Zheremyan, Ustiugova, Uvarova, Karamushka, Stasevich, Gogoleva, Bogolyubova, Mitkin, Kuprash and Korneev.54 |
Short-chain fatty acids (SCFAs)—volatile saturated fatty acids with ≤6 carbons (eg, acetate, propionate, butyrate)—are gut microbiota-derived metabolites from dietary fiber fermentation that modulate Breg function. Valerate enhances Breg immunosuppressive capacity by upregulating IL-10 secretion. Our data demonstrate SCFA-induced Breg expansion under diverse stimuli, including LPS and CD40-activated splenic B cells (Figure 9A), primarily mediated through histone deacetylase (HDAC) inhibition (Figure 9B and C). Butyrate and other HDAC inhibitors further promote B10 cell generation via MAPK pathway activation (Figure 9D). Mauri et al62 identified butyrate-driven elevation of 5-hydroxyindole-3-acetic acid (5-HIAA), a serotonin metabolite that activates aryl hydrocarbon receptor (AhR) signaling to foster Breg differentiation while suppressing germinal center B cells and plasmablasts, ameliorating RA pathology.
 |
Figure 9 SCFAs promote the generation of B10. (A) Flow cytometric analysis assessing IL-10 modulation by LPS/CD40 stimulants. (B) Sodium butyrate-mediated IL-10 regulation with/without signaling pathway inhibitors. (C) Pharmacologic HDAC inhibition impacts IL-10 induction across stimulation conditions. (D) Sodium butyrate’s IL-10 modulation under kinase inhibitor cotreatment. **p < 0.01, ***p < 0.001, ****p < 0.0001. Reproduced from Zou F, Qiu Y, Huang Y et al. Effects of short-chain fatty acids in inhibiting HDAC and activating p38 MAPK are critical for promoting B10 cell generation and function. Cell Death Dis. 2021;12(6):582. Creative Commons Attribution 4.0 International License.61 |
Emerging Nanotherapeutics
Recent advances in nanomedicine offer targeted drug delivery with minimized off-target effects and enhanced therapeutic precision.63 Emerging nanotherapeutic platforms demonstrate clinical potential in RA management through composite nanosystems,64 mRNA/siRNA-based delivery vectors, spleen-targeted mRNA nanoparticles,65 and innovative B cell immunomodulatory strategies.66 B cell-targeted nanotherapies represent a paradigm shift in RA precision medicine. Wang et al40 engineered PEG density- and zeta potential-optimized nanocarriers for in vivo B cell targeting, achieving targeted B cell depletion via CRISPR-Cas9/gBAFFR system delivery—a novel approach for RA intervention. Wang et al67 engineered cationic lipid-assisted PEG-b-PLGA nanoparticles encapsulating BTK-targeting siRNA, effectively suppressing BTK expression in B cells/macrophages while modulating immune cell functionality and alleviating RA symptoms, circumventing high-dose BTK inhibitor toxicity. Yang et al68 developed an exosome-silk fibroin hydrogel composite via in situ photopolymerization, demonstrating potent suppression of T follicular helper (Tfh) cell responses and germinal center B cell-plasma cell differentiation, thereby attenuating synovitis and osteocartilaginous destruction.
FcγRIIB, an inhibitory B cell receptor, suppresses B cell activation via BCR interaction-mediated immunoregulation. Shen et al69 engineered mRNA/LPNs using AMB-POC18 and PEG2k-PLGA2k polymers complexed with mRNA (Figure 10A). Intravenous mRNA/LPNs selectively adsorbed complement C3 within the protein corona, enabling spleen-specific targeting in murine models (Figure 10B and C). mFcγRIIB/LPNs elevated FcγRIIB expression in splenocytes from 54.7% to 62.9%, with >20% increases in B cells and macrophages (Figure 10D and E). Compared to PBS/MTX controls, mFcγRIIB/LPN-treated mice exhibited upregulated FcγRIIB/Lyn/SHP-1 signaling alongside reduced CD19 phosphorylation (Figure 10F), mechanistically inhibiting BCR activation through FcγRIIB-mediated pathways. Furthermore, mFcγRIIB/LPNs attenuated intracellular Ca2+ flux in B cells, effectively curbing pathological hyperactivation and demonstrating therapeutic efficacy in RA (Figure 10G).
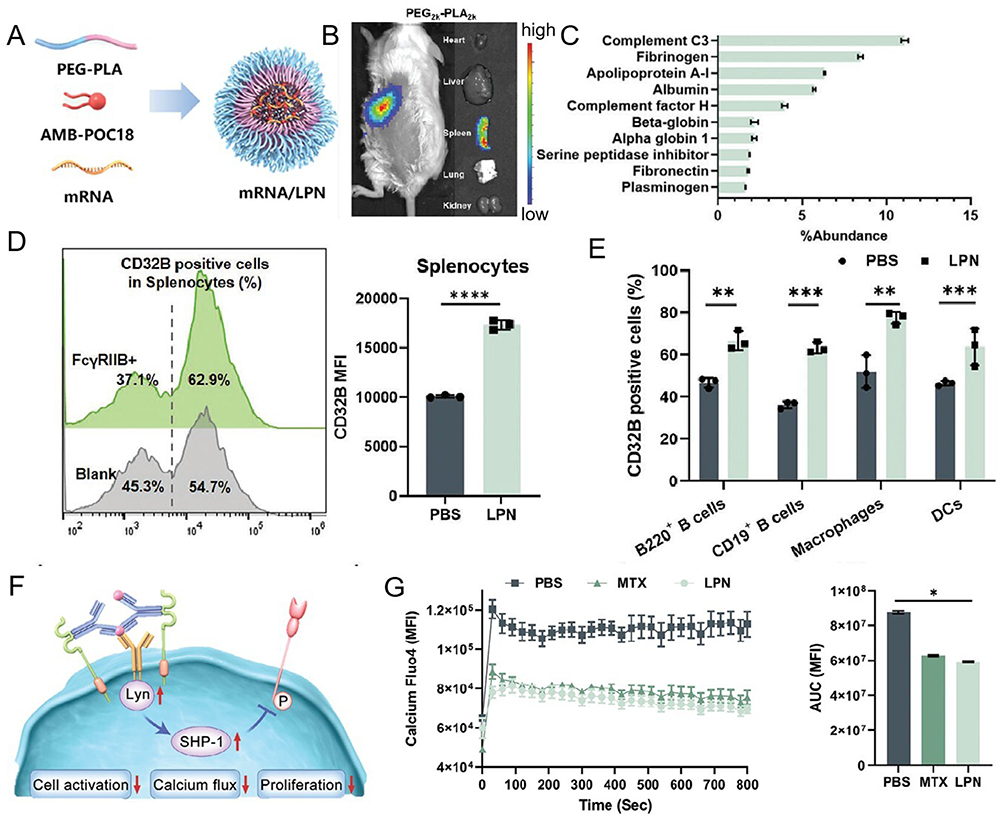 |
Figure 10 Effect and mechanism diagram of LPN on mouse spleen B cells. (A) PEG-PLA polymer formulations with varied molecular weights optimized for LPN-mediated mRNA delivery. (B) Protein Corona composition profiling identifies the top 10 plasma proteins adsorbed onto LPN surfaces. (C) In vivo fluorescence imaging of fluorescein-labeled mRNA/LPN biodistribution following tail vein administration. (D) Splenic transfection efficiency quantification of mFcγRIIB/LPNs. (E) FACS analysis of splenocyte subpopulations post mRNA transfection. (F) Mechanistic schema illustrating FcγRIIB overexpression-mediated BCR signaling suppression via Lyn/SHP-1 cascade. (G) Intracellular Ca2+ flux dynamics in activated B cells. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Reproduced from Yanpeng Liu RZ, Qiu N, Wang S, et al, Spleen-Targeted mRNA Nanoparticles for Modulating B Cell Hyperactivation in RA Therapy. Advanced Function Materials, 2024: 101:1–12. Copyright 2024, Jhon Wiley and Sons.69 |
Discussion and Perspective
RA is a chronic systemic autoimmune disorder characterized by complex pathophysiological mechanisms. Current therapeutic paradigms prioritize achieving clinical remission or maintaining low disease activity through early standardized interventions to mitigate disability progression and enhance quality of life. However, RA management persists with unmet clinical challenges, including incomplete understanding of its multifactorial etiopathogenesis involving dynamic genetic-environmental-immunoendocrine interactions, which complicates precision therapeutic development. Marked interpatient heterogeneity in clinical manifestations and irreversible cumulative joint damage necessitate timely intervention to prevent high disability risks. Conventional pharmacotherapies predominantly provide symptomatic palliation without modifying disease progression, while long-term use carries treatment-related comorbidities. Biologic agents (eg, TNF-α inhibitors, CD20-targeted mAbs) have revolutionized RA management, with emerging evidence supporting obinutuzumab—a glycoengineered anti-CD20 mAb exhibiting augmented antibody-dependent cellular cytotoxicity (ADCC)—originally developed for B cell malignancies. Preliminary trials indicate superior B cell depletion efficacy versus rituximab, suggesting therapeutic potential for refractory RA. However, the safety profile and therapeutic durability of obinutuzumab in RA necessitate further investigation.70 Ruxolitinib, a selective JAK1/2 inhibitor blocking IL-6/IFN-γ signaling, demonstrates clinical potential in RA but warrants additional validation.71 Persistent challenges include primary non-response or acquired resistance to biologics, compounded by economic constraints limiting accessibility. While CD20 antigen loss—a recognized resistance mechanism in B-cell malignancies—is uncommon in RA due to preserved CD20 expression on synovial B cells, alternative pathways contribute to therapeutic refractoriness: chronic B cell activation within RA-specific immune niches, incomplete depletion of synovial tissue-resident B cell reservoirs perpetuating inflammation through anatomical sanctuary effects, and compensatory upregulation of BAFF/APRIL axis signaling post-depletion sustaining residual B cell survival and autoantibody secretion. Prolonged B cell depletion may paradoxically disrupt immunoregulatory balance through Breg attrition, potentially exacerbating inflammatory cascades.
A comprehensive understanding of RA pathogenesis is pivotal for developing targeted therapeutic strategies. The hallmark production of pathogenic autoantibodies—notably RF and ACPA—establishes B lymphocytes as central mediators of RA pathophysiology. B cells perpetuate synovial inflammation and articular destruction through three principal mechanisms: (1) autoantibody synthesis, (2) pro-inflammatory cytokine secretion (IL-6/TNF-α), and (3) antigen presentation-driven T cell activation. This pathogenic triad stems from dysregulated B cell signaling via SYK-PI3K-MAPK-NF-κB axis hyperactivation, creating self-perpetuating immune dyshomeostasis. Therapeutic interventions combining B cell proliferation inhibition with Breg expansion therefore represent promising approaches to restore immunologic equilibrium in RA.
Recent advancements in B cell-targeted therapies have significantly advanced RA management. B cell-depleting modalities, including anti-CD20 monoclonal antibodies (eg, rituximab) and bispecific CD19/CD3 antibodies engaging T cell-mediated cytotoxicity, effectively reduce pathogenic B cell burden. Emerging CAR-T cell therapies targeting B cell surface antigens (eg, CD19/CD20) demonstrate potential in ablating autoreactive B cell populations, though large-scale clinical validation of their safety and efficacy remains imperative. Pharmacologic modulation of B cell activity through BAFF/BTK pathway inhibition attenuates pathogenic B cell survival and signaling, while adjunctive IL-6/TNF-α inhibitors provide complementary immunomodulation. However, therapeutic optimization requires addressing challenges in efficacy durability, adverse event mitigation, and resistance mechanisms.
Enhancement of Breg populations via ex vivo expansion or pharmacologic potentiation represents an emerging immunoregulatory strategy, albeit requiring rigorous clinical validation. Concurrently, nanotherapeutic platforms leveraging targeted delivery systems show promise in enhancing therapeutic precision while minimizing systemic toxicity. Integrative application of single-cell multi-omics, epigenetic profiling, and metabolomics is elucidating B cell heterogeneity and dysregulation in RA, paving the way for personalized therapeutic regimens with optimized safety-efficacy profiles. These multidisciplinary advances hold transformative potential for achieving sustained remission and improved long-term outcomes in RA.
Consent for Publication
Agree to publish any image information.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
The research was supported by National Nature Science Foundation of China (Grant No. 82370969 to J.S.; 82072151 to Y.S.); Guangdong Basic and Applied Basic Research Foundation (Grant No. 2022A1515010678 to J.S.; 2022A1515140101 to L.L.; 2023A1515140177 to Y.S.); Dongguan Science and Technology of Social Development Program (Grant No. 20221800905652to J.S.); Clinical plus Basic Innovation Project of Guangdong Medical University (Grant No. 4SG24012G to J.S.); Talent Development Foundation of The First Dongguan Affiliated Hospital of Guangdong Medical University (No. GCC2022003 to J.S.; GCC2023005 to Y.S.).
Disclosure
All authors declare no competing interests.
References
1. Gibofsky A. Epidemiology, pathophysiology, and diagnosis of rheumatoid arthritis: a synopsis. Am J Manag Care. 2014;20(7 Suppl):S128–S135.
2. Smith MH, Berman JR. What is rheumatoid arthritis? JAMA. 2022;327(12):1194. doi:10.1001/jama.2022.0786
3. Silverstein FE, Faich G, Goldstein JL, et al. Gastrointestinal toxicity with celecoxib vs nonsteroidal anti-inflammatory drugs for osteoarthritis and rheumatoid arthritis: the CLASS study: a randomized controlled trial. Celecoxib long-term arthritis safety study. JAMA. 2000;284(10):1247–1255. doi:10.1001/jama.284.10.1247
4. Boers M, Hartman L, Opris-Belinski D, et al. Low dose, add-on prednisolone in patients with rheumatoid arthritis aged 65+: the pragmatic randomised, double-blind placebo-controlled GLORIA trial. Ann Rheum Dis. 2022;81(7):925–936. doi:10.1136/annrheumdis-2021-221957
5. Nam JL, Emery P. Is there a place for initial treatment with biological DMARDs in the early phase of RA? Best Pract Res Clin Rheumatol. 2013;27(4):537–554. doi:10.1016/j.berh.2013.09.003
6. Kawashiri S-Y, Kawakami A, Ueki Y, et al. Decrement of serum cartilage oligomeric matrix protein (COMP) in rheumatoid arthritis (RA) patients achieving remission after 6 months of etanercept treatment: comparison with CRP, IgM-RF, MMP-3 and anti-CCP Ab. Joint Bone Spine. 2010;77(5):418–420. doi:10.1016/j.jbspin.2010.01.016
7. Iannazzo S, Benucci M, Favalli EG. Tocilizumab after a first-line with anti-TNF in rheumatoid arthritis: a cost-consequence analysis in the Italian setting. Clin Exp Rheumatol. 2018;36(3):479–485.
8. Caporali R, Germinario S, Kacsándi D, Choy E, Szekanecz Z. Start RA treatment - biologics or JAK-inhibitors? Autoimmun Rev. 2024;23(1):103429. doi:10.1016/j.autrev.2023.103429
9. Chopra A, Saluja M, Kianifard T, Chitre D, Venugopalan A. Long term effectiveness of RA-1 as a monotherapy and in combination with disease modifying anti-rheumatic drugs in the treatment of rheumatoid arthritis. J Ayurveda Integr Med. 2018;9(3):201–208. doi:10.1016/j.jaim.2017.07.009
10. Nikiphorou E, Carpenter L, Morris S, et al. Hand and foot surgery rates in rheumatoid arthritis have declined from 1986 to 2011, but large-joint replacement rates remain unchanged: results from two UK inception cohorts. Arthritis Rheumatol. 2014;66(5):1081–1089. doi:10.1002/art.38344
11. Gregorio A, Gambini C, Gerloni V, et al. Lymphoid neogenesis in juvenile idiopathic arthritis correlates with ANA positivity and plasma cells infiltration. Rheumatology. 2007;46(2):308–313. doi:10.1093/rheumatology/kel225
12. Barnas JL, Looney RJ, Anolik JH. B cell targeted therapies in autoimmune disease. Curr Opin Immunol. 2019;61:92–99. doi:10.1016/j.coi.2019.09.004
13. El Shikh MEM, El Sayed RM, Sukumar S, Szakal AK, Tew JG. Activation of B cells by antigens on follicular dendritic cells. Trends Immunol. 2010;31(6):205–211. doi:10.1016/j.it.2010.03.002
14. Manz RA, Hauser AE, Hiepe F, Radbruch A. Maintenance of serum antibody levels. Annu Rev Immunol. 2005;23(1):367–386. doi:10.1146/annurev.immunol.23.021704.115723
15. Hu F, Shi L, Liu X, et al. Proinflammatory phenotype of B10 and B10pro cells elicited by TNF-α in rheumatoid arthritis. Ann Rheum Dis. 2024;83(5):576–588. doi:10.1136/ard-2023-224878
16. Zhang Z, Shao Z, Xu Z, Wang J. Similarities and differences between osteoarthritis and rheumatoid arthritis: insights from Mendelian randomization and transcriptome analysis. J Transl Med. 2024;22(1):851. doi:10.1186/s12967-024-05643-4
17. Lorenzetti R, Janowska I, Smulski CR, et al. Abatacept modulates CD80 and CD86 expression and memory formation in human B-cells. J Autoimmun. 2019;101:145–152. doi:10.1016/j.jaut.2019.04.016
18. Yang M, Zhu L. Osteoimmunology: the crosstalk between T cells, B cells, and osteoclasts in rheumatoid arthritis. Int J Mol Sci. 2024;25(5). doi:10.3390/ijms25052688
19. Noort AR, van Zoest KPM, van Baarsen LG, et al. Tertiary lymphoid structures in rheumatoid arthritis: NF-κB-inducing kinase-positive endothelial cells as central players. Am J Pathol. 2015;185(7):1935–1943. doi:10.1016/j.ajpath.2015.03.012
20. Ye Y, Liang Y, Huang L, Cao X, Xia Z, Liang S. Daptomycin alleviates collagen-induced arthritis via suppressing inflammatory cytokines and NF-κB pathway. Int Immunopharmacol. 2024;144:113648. doi:10.1016/j.intimp.2024.113648
21. Delgado-Arévalo C, Calvet-Mirabent M, Triguero-Martínez A, et al. NLRC4-mediated activation of CD1c+ DC contributes to perpetuation of synovitis in rheumatoid arthritis. JCI Insight. 2022;7(22). doi:10.1172/jci.insight.152886
22. Gautam S, Kumar R, Kumar U, Kumar S, Luthra K, Dada R. Yoga maintains Th17/Treg cell homeostasis and reduces the rate of T cell aging in rheumatoid arthritis: a randomized controlled trial. Sci Rep. 2023;13(1):14924. doi:10.1038/s41598-023-42231-w
23. Burbano C, Villar-Vesga J, Vásquez G, Muñoz-Vahos C, Rojas M, Castaño D. Proinflammatory differentiation of macrophages through microparticles that form immune complexes leads to T- and B-cell activation in systemic autoimmune diseases. Front Immunol. 2019;10:2058. doi:10.3389/fimmu.2019.02058
24. Schmidt T, Najm A, Mussawy H, et al. General synovitis score and immunologic synovitis score reflect clinical disease activity in patients with advanced stage rheumatoid arthritis. Sci Rep. 2019;9(1):8448. doi:10.1038/s41598-019-44895-9
25. Siouti E, Andreakos E. The many facets of macrophages in rheumatoid arthritis. Biochem Pharmacol. 2019;165:152–169. doi:10.1016/j.bcp.2019.03.029
26. Richter J, Capková K, Hříbalová V, et al. Collagen-induced arthritis: severity and immune response attenuation using multivalent N-acetyl glucosamine. Clin Exp Immunol. 2014;177(1):121–133. doi:10.1111/cei.12313
27. Weyand CM, Goronzy JJ. T-cell-targeted therapies in rheumatoid arthritis. Nat Clin Pract Rheumatol. 2006;2(4):201–210. doi:10.1038/ncprheum0142
28. Liu Y-T, Ding H-H, Lin Z-M, et al. A novel tricyclic BTK inhibitor suppresses B cell responses and osteoclastic bone erosion in rheumatoid arthritis. Acta Pharmacol Sin. 2021;42(10):1653–1664. doi:10.1038/s41401-020-00578-0
29. Giordani L, Sanchez M, Libri I, Quaranta MG, Mattioli B, Viora M. IFN-alpha amplifies human naive B cell TLR-9-mediated activation and Ig production. J Leukoc Biol. 2009;86(2):261–271. doi:10.1189/jlb.0908560
30. Daridon C, Burmester GR, Dörner T. Anticytokine therapy impacting on B cells in autoimmune diseases. Curr Opin Rheumatol. 2009;21(3):205–210. doi:10.1097/BOR.0b013e32832a0760
31. Xu J, Jiao W, Wu D-B, et al. Yishen Tongbi decoction attenuates inflammation and bone destruction in rheumatoid arthritis by regulating JAK/STAT3/SOCS3 pathway. Front Immunol. 2024;15:1381802. doi:10.3389/fimmu.2024.1381802
32. He J, Lin X, Wang X, et al. Arecoline hydrobromide suppresses PI3K/AKT pathway in rheumatoid arthritis synovial fibroblasts and relieves collagen-induced arthritis in mice. Int Immunopharmacol. 2023;124(Pt B):110925. doi:10.1016/j.intimp.2023.110925
33. Li Y, Li P, Lin S-H, Zheng Y-Q, Zheng X-X. Paeonol inhibited TNFα-induced GM-CSF expression in fibroblast-like synoviocytes. Int J Clin Pharmacol Ther. 2014;52(11):986–996. doi:10.5414/CP202127
34. Wang X, Chen X, Huang W, et al. Losartan suppresses the inflammatory response in collagen-induced arthritis by inhibiting the MAPK and NF-κB pathways in B and T cells. Inflammopharmacology. 2019;27(3):487–502. doi:10.1007/s10787-018-0545-2
35. Zhang X, Mei D, Wang H, et al. hIgDFc-Ig inhibits B cell function by regulating the BCR-Syk-Btk-NF-κB signalling pathway in mice with collagen-induced arthritis. Pharmacol Res. 2021;173:105873. doi:10.1016/j.phrs.2021.105873
36. Najm A, Masson F-M, Preuss P, et al. MicroRNA-17-5p reduces inflammation and bone erosions in mice with collagen-induced arthritis and directly targets the JAK/STAT pathway in rheumatoid arthritis fibroblast-like synoviocytes. Arthritis Rheumatol. 2020;72(12):2030–2039. doi:10.1002/art.41441
37. Prokopec KE, Rhodiner M, Matt P, Lindqvist U, Kleinau S. Down regulation of Fc and complement receptors on B cells in rheumatoid arthritis. Clin Immunol. 2010;137(3):322–329. doi:10.1016/j.clim.2010.08.006
38. Bucci L, Hagen M, Rothe T, et al. Bispecific T cell engager therapy for refractory rheumatoid arthritis. Nat Med. 2024;30(6):1593–1601. doi:10.1038/s41591-024-02964-1
39. Lee YH, Bae S-C, Song GG. The efficacy and safety of rituximab for the treatment of active rheumatoid arthritis: a systematic review and meta-analysis of randomized controlled trials. Rheumatol Int. 2011;31(11):1493–1499. doi:10.1007/s00296-010-1526-y
40. Palanki R, Han EL, Murray AM, et al. Optimized microfluidic formulation and organic excipients for improved lipid nanoparticle mediated genome editing. Lab Chip. 2024;24(16):3790–3801. doi:10.1039/d4lc00283k
41. Wong KL, Li Z, Ma F, et al. SM03, an anti-CD22 antibody, converts cis-to-trans ligand binding of CD22 against α2,6-linked sialic acid glycans and immunomodulates systemic autoimmune diseases. J Immunol. 2022;208(12):2726–2737. doi:10.4049/jimmunol.2100820
42. Zhou B, Zhang H, Su X, et al. Therapeutic effects of a novel BAFF blocker on arthritis. Signal Transduct Target Ther. 2019;4(1):19. doi:10.1038/s41392-019-0051-z
43. Asai S, Takahashi N, Terabe K, et al. Clinical effectiveness of baricitinib and abatacept in patients with rheumatoid arthritis. Int J Rheum Dis. 2024;27(11):e15414. doi:10.1111/1756-185X.15414
44. Balanean A, Brown-Bickerstaff C, Klink A, et al. Real-world clinical outcomes and rationale for initiating abatacept as a first-line biologic for patients with anticitrullinated protein antibody- and rheumatoid factor-positive rheumatoid arthritis. J Comp Eff Res. 2024;13(12):e230144. doi:10.57264/cer-2023-0144
45. Ekin A, Misirci S, Ildemir S, et al. Efficacy and safety of tofacitinib in rheumatoid arthritis: nine years of real-world data. Clin Transl Sci. 2024;17(11):e70084. doi:10.1111/cts.70084
46. Xu X, Geng L, Xu X, Huang S, Liang J. Cost-effectiveness of upadacitinib vs. Tofacitinib for moderate-to-severe rheumatoid arthritis in China. Immunotherapy. 2024;16(18–19):1141–1151. doi:10.1080/1750743X.2024.2426972
47. Murata K, Uozumi R, Fujii T, et al. Author correction: effects of IL-6, JAK, TNF inhibitors, and CTLA4-Ig on knee symptoms in patients with rheumatoid arthritis. Sci Rep. 2024;14(1):30521. doi:10.1038/s41598-024-81223-2
48. Smolen JS, Landewé R, Bijlsma J, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2016 update. Ann Rheum Dis. 2017;76(6):960–977. doi:10.1136/annrheumdis-2016-210715
49. Jones G, Sebba A, Gu J, et al. Comparison of tocilizumab monotherapy versus methotrexate monotherapy in patients with moderate to severe rheumatoid arthritis: the AMBITION study. Ann Rheum Dis. 2010;69(1):88–96. doi:10.1136/ard.2008.105197
50. Blanco FJ, Möricke R, Dokoupilova E, et al. Secukinumab in active rheumatoid arthritis: a phase III randomized, double-blind, active comparator- and placebo-controlled study. Arthritis Rheumatol. 2017;69(6):1144–1153. doi:10.1002/art.40070
51. Genovese MC, Greenwald M, Cho C-S, et al. A Phase II randomized study of subcutaneous ixekizumab, an anti-interleukin-17 monoclonal antibody, in rheumatoid arthritis patients who were naive to biologic agents or had an inadequate response to tumor necrosis factor inhibitors. Arthritis Rheumatol. 2014;66(7):1693–1704. doi:10.1002/art.38617
52. Jin X, Dong T, Wang Q, et al. A citrullinated antigenic vaccine in treatment of autoimmune arthritis. Sci Bull. 2024;69(18):2920–2929. doi:10.1016/j.scib.2024.02.042
53. Najar M, Raicevic G, Fayyad-Kazan H, Bron D, Toungouz M, Lagneaux L. Mesenchymal stromal cells and immunomodulation: a gathering of regulatory immune cells. Cytotherapy. 2016;18(2):160–171. doi:10.1016/j.jcyt.2015.10.011
54. Zheremyan EA, Ustiugova AS, Uvarova AN, et al. Differentially activated B cells develop regulatory phenotype and show varying immunosuppressive features: a comparative study. Front Immunol. 2023;14:1178445. doi:10.3389/fimmu.2023.1178445
55. Chasov V, Ganeeva I, Zmievskaya E, et al. Cell-based therapy and genome editing as emerging therapeutic approaches to treat rheumatoid arthritis. Cells. 2024;13(15):1282. doi:10.3390/cells13151282
56. Nemazee D. Mechanisms of central tolerance for B cells. Nat Rev Immunol. 2017;17(5):281–294. doi:10.1038/nri.2017.19
57. Wang X, Wu X, Tan B, et al. Allogeneic CD19-targeted CAR-T therapy in patients with severe myositis and systemic sclerosis. Cell. 2024;187(18):4890–4904.e9. doi:10.1016/j.cell.2024.06.027
58. Szabo D, Balogh A, Gopcsa L, et al. Sustained drug-free remission in rheumatoid arthritis associated with diffuse large B-cell lymphoma following tandem CD20-CD19-directed non-cryopreserved CAR-T cell therapy using zamtocabtagene autoleucel. RMD Open. 2024;10(4):e004727. doi:10.1136/rmdopen-2024-004727
59. Li R, Tang H, Burns JC, et al. BTK inhibition limits B-cell-T-cell interaction through modulation of B-cell metabolism: implications for multiple sclerosis therapy. Acta Neuropathol. 2022;143(4):505–521. doi:10.1007/s00401-022-02411-w
60. Di Paolo JA, Huang T, Balazs M, et al. Specific Btk inhibition suppresses B cell- and myeloid cell-mediated arthritis. Nat Chem Biol. 2011;7(1):41–50. doi:10.1038/nchembio.481
61. Zou F, Qiu Y, Huang Y, et al. Effects of short-chain fatty acids in inhibiting HDAC and activating p38 MAPK are critical for promoting B10 cell generation and function. Cell Death Dis. 2021;12(6):582. doi:10.1038/s41419-021-03880-9
62. Rosser EC, Piper CJM, Matei DE, et al. Microbiota-derived metabolites suppress arthritis by amplifying aryl-hydrocarbon receptor activation in regulatory B cells. Cell Metab. 2020;31(4):837–851.e10. doi:10.1016/j.cmet.2020.03.003
63. Ren S, Xu Y, Dong X, et al. Nanotechnology-empowered combination therapy for rheumatoid arthritis: principles, strategies, and challenges. J Nanobiotechnol. 2024;22(1):431. doi:10.1186/s12951-024-02670-7
64. Kim Y-J, Chae SY, Jin C-H, et al. Ionic complex systems based on hyaluronic acid and PEGylated TNF-related apoptosis-inducing ligand for treatment of rheumatoid arthritis. Biomaterials. 2010;31(34):9057–9064. doi:10.1016/j.biomaterials.2010.08.015
65. Luo Z, Lin Y, Meng Y, et al. Spleen-targeted mRNA vaccine doped with manganese adjuvant for robust anticancer immunity in vivo. ACS Nano. 2024;18(44):30701–30715. doi:10.1021/acsnano.4c09902
66. Chu T-W, Zhang R, Yang J, Chao MP, Shami PJ, Kopeček J. A two-step pretargeted nanotherapy for CD20 crosslinking may achieve superior anti-lymphoma efficacy to rituximab. Theranostics. 2015;5(8):834–846. doi:10.7150/thno.12040
67. Zhao G, Liu A, Zhang Y, et al. Nanoparticle-delivered siRNA targeting Bruton’s tyrosine kinase for rheumatoid arthritis therapy. Biomater Sci. 2019;7(11):4698–4707. doi:10.1039/c9bm01025d
68. Rui K, Tang X, Shen Z, et al. Exosome inspired photo-triggered gelation hydrogel composite on modulating immune pathogenesis for treating rheumatoid arthritis. J Nanobiotechnol. 2023;21(1):111. doi:10.1186/s12951-023-01865-8
69. Yanpeng Liu RZ, Qiu N, Wang S, et al. Spleen-targeted mRNA nanoparticles for modulating B cell hyperactivation in rheumatoid arthritis therapy. Adv Func Mater. 2024;2417101:12. doi:10.1002/adfm.202417101
70. Reddy VR, Pepper RJ, Shah K, et al. Disparity in peripheral and renal B-cell depletion with rituximab in systemic lupus erythematosus: an opportunity for obinutuzumab? Rheumatology. 2022;61(7):2894–2904. doi:10.1093/rheumatology/keab827
71. Frede N, Lorenzetti R, Hüppe JM, et al. JAK inhibitors differentially modulate B cell activation, maturation and function: a comparative analysis of five JAK inhibitors in an in-vitro B cell differentiation model and in patients with rheumatoid arthritis. Front Immunol. 2023;14:1087986. doi:10.3389/fimmu.2023.1087986
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.