Back to Journals » Drug Design, Development and Therapy » Volume 9
RGD-modified liposomes enhance efficiency of aclacinomycin A delivery: evaluation of their effect in lung cancer
Authors Feng C, Li X, Dong C, Zhang X, Zhang X, Gao Y
Received 2 April 2015
Accepted for publication 5 May 2015
Published 11 August 2015 Volume 2015:9 Pages 4613—4620
DOI https://doi.org/10.2147/DDDT.S85993
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Chan Feng,1,* Xiaoyan Li,2,* Chunyan Dong,1 Xuemei Zhang,1 Xie Zhang,1 Yong Gao1
1Department of Oncology, Shanghai East Hospital, Tongji University, Shanghai, 2Shanghai Tenth People’s Hospital, Tongji University, Shanghai, People’s Republic of China
*These authors contributed equally to this work
Abstract: In this study, long-circulating Arg-Gly-Asp (RGD)-modified aclacinomycin A (ACM) liposomes were prepared by thin film hydration method. Their morphology, particle size, encapsulation efficiency, and in vitro release were investigated. The RGD-ACM liposomes was about 160 nm in size and had the visual appearance of a yellowish suspension. The zeta potential was -22.2 mV and the encapsulation efficiency was more than 93%. The drug-release behavior of the RGD-ACM liposomes showed a biphasic pattern, with an initial burst release and followed by sustained release at a constant rate. After being dissolved in phosphate-buffered saline (pH 7.4) and kept at 4°C for one month, the liposomes did not aggregate and still had the appearance of a milky white colloidal solution. In a pharmacokinetic study, rats treated with RGD-ACM liposomes showed slightly higher plasma concentrations than those treated with ACM liposomes. Maximum plasma concentrations of RGD-ACM liposomes and ACM liposomes were 4,532 and 3,425 ng/mL, respectively. RGD-ACM liposomes had a higher AUC0–∞ (1.54-fold), mean residence time (2.09-fold), and elimination half-life (1.2-fold) when compared with ACM liposomes. In an in vivo study in mice, both types of liposomes inhibited growth of human lung adenocarcinoma (A549) cells and markedly decreased tumor size when compared with the control group. There were no obvious pathological tissue changes in any of the treatment groups. Our results indicate that RGD-modified ACM liposomes have a better antitumor effect in vivo than their unmodified counterparts.
Keywords: RGD, aclacinomycin A, long-circulating liposomes, pharmacokinetic, tumor inhibition
Introduction
RGD is a short tripeptide containing arginine-glycine-aspartic acid, and exists widely in vivo. The extracellular matrix and adhesion proteins in blood, including fibrin (fibrinogen), vitronectin, and collagen, commonly contain the RGD sequence.1 As the recognition sites for integrin and its ligand interactions, RGD peptides had adhesion between mediated cell, extracellular matrix and cells. RGD also has a signaling function. Considerable research attention has been focused on the influence of RGD on the function of platelets,2,3 its application in antithrombotic therapy,4 and its ability to induce regeneration of bone tissue.5
In recent years, treatment targeting angiogenesis had become an important topic in cancer research. It is widely believed that tumor growth, invasion, and metastasis occurs as a result of angiogenesis, and that if the blood vessels feeding the tumor are inadequate, the tumor would become necrotic or apoptotic.6 Meanwhile, research in molecular biology has revealed that integrin, present on the surface of cells, plays an important role in tumor angiogenesis.7 Integrin receptors, in particular ανβ3, are often highly expressed on certain types of tumor cells and vascular endothelial tumor cells, but not in the normal vasculature.8 It has also been shown that exogenous RGD peptides can competitively inhibit ligand binding to integrins, thereby inhibiting angiogenesis and migration of tumor cells. At the same time, tumors can be target marked and anticancer drugs can be target delivered.9–11
Aclacinomycin A (ACM) is an antineoplastic anthracycline drug with relatively strong lipophilic properties that can be taken up rapidly into cells in relatively high concentrations where it acts on the DNA of the cancer cell, inhibiting the synthesis of nucleic acids, in particular RNA. It was cycle non-specific drugs, blocking the cell cycle during the late G1 and late S phases.12 ACM is used in the treatment of acute myelogenous leukemia, acute lymphoblastic leukemia, malignant lymphoma, gastric cancer, lung cancer, breast cancer, ovarian cancer, and other related diseases. However, ACM had relatively high toxicity, which greatly limits its clinical application. To reduce its toxicity and to increase its antitumor efficacy, researchers have prepared long-circulating ACM liposomes and microemulsion,13 with good results for research purposes. However, the therapeutic effect was not significant because of the lack of effective targeting.
In this work, we prepared long-circulating ACM liposomes modified by polyethylene glycol (PEG), and then covalently linked the RGD peptide on the liposome surface. The morphology, particle size, encapsulation efficiency, and in vitro release of these liposomes were examined. In addition, their antitumor activity was investigated in vitro and in an animal model of lung cancer, with the intention of laying a solid foundation for further clinical research.
Materials and methods
Materials
The ACM was gifted from Haikang Pharmaceuticals Co Ltd (Sichuan, People’s Republic of China). Distearoyl-L-a-phosphatidylethanolamine (DSPE)-PEG2000 was provided by the Material Engineering Institute at Tongji University. High purity cholesterol and hydrogenated soybean phosphatidyl choline were purchased by Phospholipid Tech Ltd (Shanghai, People’s Republic of China). RGD peptide was purchased from Biochempartner (Shanghai, People’s Republic of China). All the chemicals and solvents used were of analytical or high-performance liquid chromatography grade. Deionized purified water was prepared in the laboratory.
Animals
The animal experiments were performed using rats weighing 220±20 g and BALB/c nude mice weighing 21±1 g. The animals were kept in cages at a temperature of 25°C±2°C with a 12:12-hour light-dark cycle. Food and water were freely available. All experiments were performed in strict accordance with the Guide for the Care and Use of Laboratory Animals as adopted by the Chinese National Institutes of Health.
Preparation of liposomes
Liposomes composed of hydrogenated soybean phosphatidyl choline, cholesterol, and DSPE-PEG2000 were prepared by thin film hydration as described previously.14,15 Briefly, 20 mg of ACM, 60 mg of hydrogenated soybean phosphatidyl choline, 35 mg of cholesterol, and 5 mg of DSPE-PEG2000 were dissolved in 6.5 mL of chloroform and dried in a rotary evaporator under reduced pressure. The vacuum was maintained overnight to remove any traces of the solvent. The resulting lipid film was hydrated with 5 mL of phosphate-buffered saline (PBS, pH 7.4, containing 110 mg RGD peptide) at 37°C for 30 minutes. The suspension was then subjected to ultrasonic treatment for 2 minutes at 500 W using a high-intensity probe ultrasonicator (JY92-2D; Xinzhi Equipment Instruction, Zhejiang, People’s Republic of China). Control ACM liposomes without RGD peptide was prepared using the same method.
Particle size and zeta potential
The surface and internal morphology of the liposomes were examined using a scanning electron microscope (6010LV, JEOL, Tokyo, Japan). Before analyzing the samples, the freeze-dried liposomes were gold-sputtered in an argon atmosphere. The particle size and zeta potential of the plain liposomes, ACM liposomes, and RGD-ACM liposomes were measured by dynamic light scattering (Nicomp™ 380 ZLS, Particle Sizing Systems, Port Richey, FL, USA). Before analysis, each sample was diluted 20-fold in distilled water (pH 7.4) to obtain the appropriate liposomal concentration.
Encapsulation efficiency
The encapsulation efficiency of the ACM in the liposomes was determined by high-performance liquid chromatography.16 Briefly, 0.5 mL of the RGD-liposomal or liposomal sample was first added into a super filter (UFC501096, Millipore Corporation, Billerica, MA, USA) and centrifuged at 12,000 rpm for 30 minutes. The amount of ultrafiltered ACM was also measured by high-performance liquid chromatography. The encapsulation efficiency of ACM was calculated using the formula: (M1 – M2)/M1 ×100%, where M1 and M2 are the mass of the initially added ACM and non-encapsulated ACM, respectively.
Chromatographic separation was performed on a Diamonsil™ C18 column (250 mm ×4.6 mm, 5 μm; Dikma Technologies Inc, Lake Forest, CA, USA) with the column temperature set at 35°C. A 0.2% N ammonium acetate to acetonitrile to methanol ratio of 32:28:40 (v/v/v, pH 3.0, phosphoric acid) was used as the mobile phase at a flow rate of 1.0 mL per minute. The monitor wavelength was set at 430 nm and the injection volume was 20 μL.
In vitro drug release studies
The in vitro release of RGD-ACM liposomes was measured in PBS (pH 7.4) at 37°C±0.5°C by the dialysis method. RGD-ACM liposomes (containing ~10 mg of ACM) were placed into a dialysis bag and suspended in 250 mL of release medium, and stirred at 50 rpm using the USP paddle method. At predetermined time points (0.25, 0.5, 1, 2, 4, 6, 8, 10, 14, 16, 24, and 48 hours), 2 mL samples were withdrawn with a syringe filter (0.45 μm pore size) from the release medium and replaced by an equal volume of fresh medium to maintain a constant volume. Triplicate tests were conducted and the results were averaged. The controls, ie, ACM liposomes without the RGD peptide and liposomes without ACM, were prepared using the same method.
Pharmacokinetics
The rats were randomized into two groups (n=6 each) to receive either intravenous RGD-ACM liposomes at a dose of 8 mg/kg or the same intravenous dose of ACM liposomes as a control. For the pharmacokinetic studies, 0.5 mL samples of whole blood were collected from both groups at 0.25, 0.5, 1, 1.5, 2, 4, 6, 8, 12, and 24 hours post-dosing and immediately centrifuged at 4,000 rpm for 10 minutes to obtain plasma samples. The supernatant plasma (0.25 mL) was transferred to new tubes and stored at −80°C until analysis. Pharmacokinetic parameters were calculated using Microsoft Office Excel 2013.
Next, 100 μL of the plasma sample was transferred to a 10 mL plastic test tube together with 10 μL of internal standard solution (quercetin, 1 mg/mL). After shaking for 30 seconds using a 5432 vortex mixer (Eppendorf, Hamburg, Germany), 3 mL of ethyl acetate was added and the mixture was vortexed for a further 2 minutes. After centrifugation at 3,000 g for 10 minutes (IEC Micromax, Thermo Scientific, Rockford, IL, USA), the upper organic layer was quantitatively transferred to a 10 mL glass tube and evaporated to dryness using an evaporator at 40°C. The residue was reconstituted in 100 μL of methanol, and then vortex-mixed. After centrifugation at 2,000 g for 5 minutes, a 20 μL aliquot of the solution was injected into the high-performance liquid chromatography system for analysis.
In vivo tumor growth inhibition study
The antitumor effect of RGD-ACM liposomes was evaluated in vivo using BALB/c nude mice (aged 5–6 weeks, weight 20–22 g) that had been inoculated subcutaneously with 2×109 human lung adenocarcinoma (A549) cells. The treatments were started on the day when the tumor volume reached 100–150 mm3, which was designated as day 0.
On day 0, the mice were randomly divided into a negative control (saline) group (n=6), an RGD-ACM liposome group (n=6), and an ACM liposome group (n=6). Samples at a dose of 8 mg/kg were injected intravenously via the tail vein every 3 days, 3 administrations in total. Tumor size and mouse body weight were measured three times per week during the study. On day 10, the mice were euthanized followed by excision and weighing of the tumors. The trial drug administration procedure was as follows: weighing and first injection on day 0; weighing and second injection on day 3; weighing and third injection on day 9; and euthanasia on day 10.
Tumor volume (V) was calculated using the following formula: (W2 × L)/2, where W is the tumor measurement at the widest point and L is the tumor dimension at the longest point. Relative tumor volume (R) was calculated by the following formula: Vi/V0, where V0 is the tumor volume at day 0, and Vi is the tumor volume at the measurement point.
Anti-tumor activity was estimated by the relative tumor inhibitory rate (%), which was calculated by the formula: (1−[R(treatment group)/R(negative control group)]) ×100%.
Statistical analysis
The results are expressed as the mean ± standard deviation. Analysis of variance was used to test the statistical significance of differences among groups. The Student’s t-test was used for single or multiple comparisons of experimental groups. P<0.05 was considered to be statistically significant.
Results and discussion
Preparation of RGD-ACM liposomes
With the use of new technology and equipment, as well as the development and application of excellent carrier materials and accessories, targeted drug delivery technology has been developing rapidly in recent years, and has gradually extended to the treatment of a number of diseases.17–20 Liposomes have been extensively researched as targeted drug delivery carriers, and been found to have sustained-release properties, long-lasting effects, low systemic toxicity, and good biocompatibility, and to be suitable for administration by a number of routes. Traditional liposomes only had a passive targeting role; however, if antibody or ligand molecules targeted to proliferating intimal tissues can be connected to the liposomal membrane to form targeted liposomes, active targeting could be achieved.
In this study, RGD-ACM liposomes were prepared successfully by the film hydration method. They had the visual appearance of a yellowish suspension, and were spherical in shape with a multilamellar nature (Figure 1). The RGD-ACM liposomes and the ACM liposomes were about 160 nm in size, which is ideal for the treatment of cancer according to a report by Gaumet et al.21 The zeta potential of the RGD-ACM liposomes and ACM liposomes was −22.2 mV and −21.4 mV, respectively, and the encapsulation efficiency was more than 93%. The zeta potential results seemed to be rather variable. The colloidal system was stable for 1 week when kept at 4°C. There was no apparent change in size or zeta potential when adding the modified material in the liposomes. Further, the zeta potential of the liposomes indicated that there was a great number of negative charges on their surface. It has been found in previous research that the higher the absolute value of the zeta potential (more than 15 mV), the stronger the electrostatic repulsion between particles, which made these liposome particles more difficult to congregate and more stable in the dispersal system.22 All the indices showed no significant change when adding RGD in the liposomes.
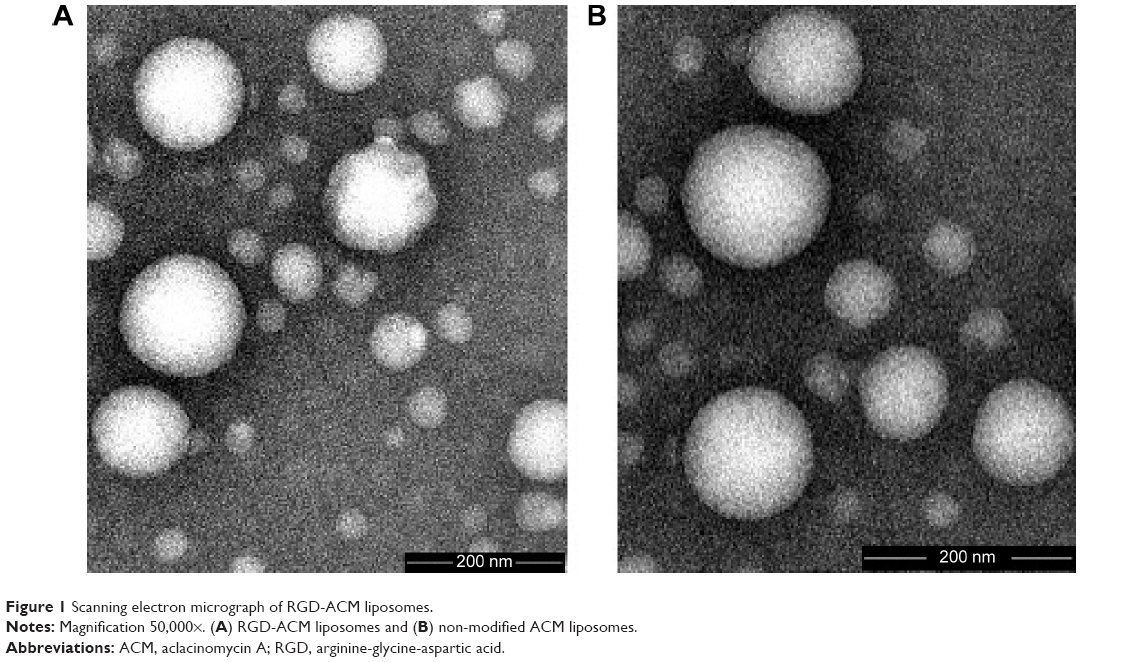 | Figure 1 Scanning electron micrograph of RGD-ACM liposomes. |
In vitro release
The release of ACM from the liposomes and that of the free drug was investigated using the dialysis method. The in vitro cumulative release profiles of ACM-loaded liposomes in PBS are shown in Figure 2. The dissolution rate of the free drug was more rapid than that of both types of liposomes in PBS. The release of ACM from the liposomes and RGD-modified liposomes in PBS over 48 hours (68.3% and 79.2%) was lower than that of the free drug (99%). The drug-release behavior of the RGD-ACM liposomes exhibited a biphasic pattern, ie, an initial burst release followed by sustained release at a constant rate. Burst release occurred for 2 hours after administration. The rate of burst release in 2 hours was 30%, which gradually became slower thereafter. The slow release phase lasted about 46 hours, showing the potential of the liposomes to act as a sustained drug delivery system. The liposomes did not aggregate over time. As shown in Figure 3, after being dissolved in PBS (pH 7.4) and kept in 4°C for one month, the sample dose did not aggregate and still had the appearance of a milky white colloidal solution.
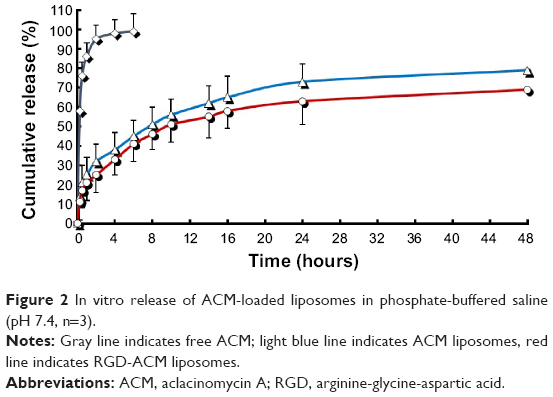 | Figure 2 In vitro release of ACM-loaded liposomes in phosphate-buffered saline (pH 7.4, n=3). |
 | Figure 3 The sample did not aggregate and still had the appearance of a milky white colloidal solution after being dissolved in phosphate-buffered saline (pH 7.4) and kept at 4°C for (A) one month and (B) two months. |
Pharmacokinetics
The pharmacokinetic profiles of ACM in plasma after intravenous injection of an 8 mg/kg dose of ACM liposomes and RGD-ACM liposomes via the tail vein are shown in Figure 4. The pharmacokinetic parameters, including area under the concentration-time curve from time zero to infinity (AUC0–∞), mean residence time, clearance, and biological half-life, are reported in Table 1. As is already known, the liposomal ACM formulation had a much longer systemic circulation time when compared with the solution. The mean residence time was significantly higher than that of the solution. In this study, rats treated with RGD-ACM liposomes had a slightly higher plasma concentration than those treated with ACM liposomes. The plasma profile showed maximum concentration values for RGD-ACM liposomes and ACM liposomes of 4,532 and 3,425 ng/mL, respectively. The RGD-ACM liposomes provided a higher AUC0–∞ (1.54-fold), mean residence time (2.09-fold), and biological half-life (1.2-fold) when compared with the ACM liposomes. However, the RGD-ACM liposomes showed decreased clearance when compared with the ACM liposomes. There was no significant difference in pharmacokinetic parameters between the ACM liposomes and the RGD-ACM liposomes. Little is reported about the pharmacokinetics of RGD-modified liposomes. However, research on the distribution of RGD-modified and nonmodified carriers indicates that the blood concentrations are very close.23 Usually, hydrophilic surface modification of a nanocarrier can prevent phagocytosis via the reticuloendothelial system, produce a long-circulating effect, and change pharmacokinetic behavior. In the present study, hydrophobic RGD was conjugated with ACM liposomes and prolonged the circulation of ACM.
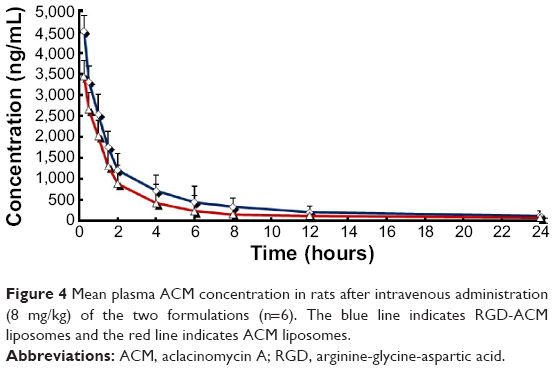 | Figure 4 Mean plasma ACM concentration in rats after intravenous administration (8 mg/kg) of the two formulations (n=6). The blue line indicates RGD-ACM liposomes and the red line indicates ACM liposomes. |
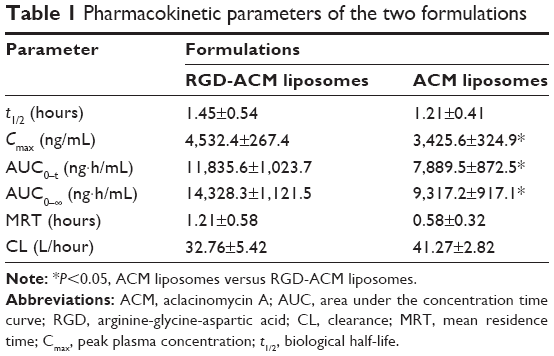 | Table 1 Pharmacokinetic parameters of the two formulations |
In vivo tumor growth inhibition
To test the antitumor activity of RGD-ACM liposomes, mice bearing human lung adenocarcinoma (A549) tumors were administered ACM in liposomes or in RGD-modified liposomes on three occasions over 10 days. As shown in Figure 5, both types of liposomes inhibited growth of human lung adenocarcinoma (A549) cells and decreased tumor size when compared with the control group. However, there was a marked reduction in tumor volume in the group treated with RGD-ACM liposomes and only a slight decrease in tumor volume in the group treated with ACM liposomes, when compared with the control group. The mean tumor weights and volumes were shown in Table 2. The appearance of the tumors was consistent with the statistical analysis of the tumor volume data, ie, when compared with the control group, all treatments significantly (P<0.05) inhibited tumor volume, with the greatest effect seen in the RGD-ACM liposome group. Similarly, the tumor weights were significantly (P<0.05) inhibited by both ACM treatments. Again, the effect was most pronounced in the RGD-ACM liposome group. Interestingly, the data suggest that tumor weight was more sensitive to treatment than tumor volume. Overall, our results indicate that the RGD-modified ACM liposomes had a better antitumor effect in vivo than the nonmodified liposomes. No obvious pathological changes were seen in the tissues of any of the treatment groups (data not shown).
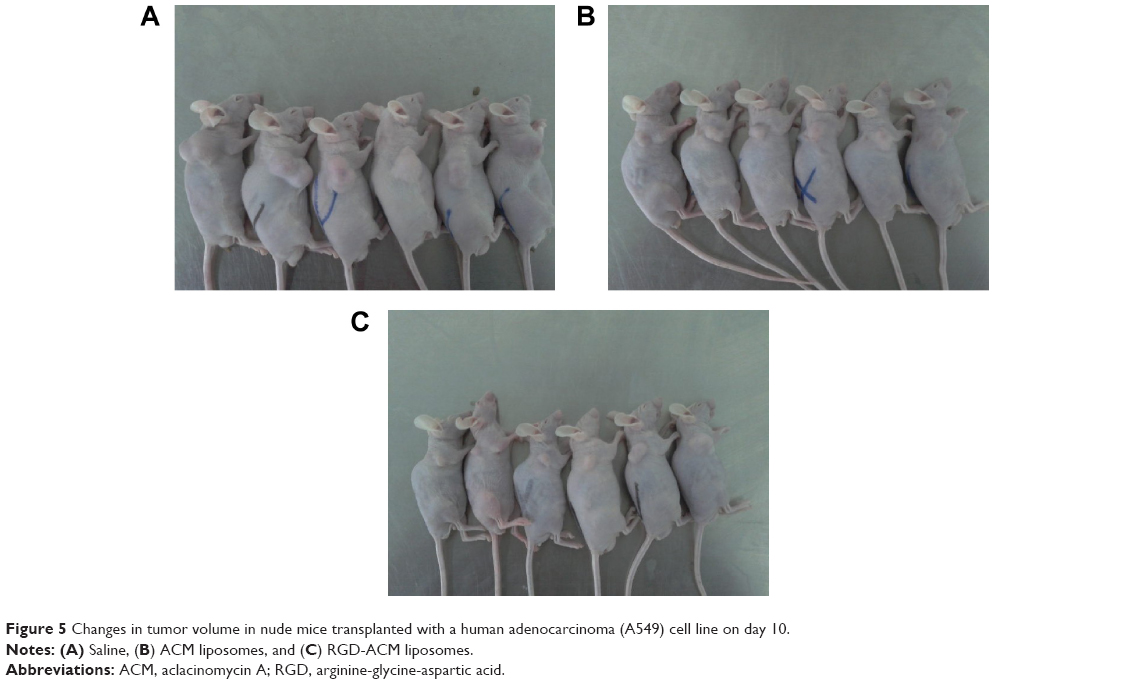 | Figure 5 Changes in tumor volume in nude mice transplanted with a human adenocarcinoma (A549) cell line on day 10. |
 | Table 2 Effect of RGD-ACM liposomes and ACM liposomes on A549 cells in nude mice |
Conclusion
In this study, ACM liposomes were prepared with and without surface modification by RGD. The liposomes were characterized physically and evaluated for ACM release in vitro. The pharmacokinetics and antitumor activity of the liposomes were examined in vivo in A549 tumor-bearing mice. Our results suggest that, compared with ACM liposomes, the tumor-targeting efficiency and subsequent antitumor efficacy of ACM may be improved by administration in the form of RGD-modified liposomes.
Acknowledgments
This research was supported in part by the National Nature Science Foundation of China (81201798), the Project of Shanghai Science and Technology Committee (124119a5100), the Fund of Pudong Health Bureau of Shanghai (pw-12-4), the Fund of Pudong Health Bureau of Shanghai (PWRd2014-01) and the Project of Key Disciplines Group Construction of Pudong Health Bureau of Shanghai (PWZxq2014-04).
Disclosure
The authors report no conflicts of interest in this work.
References
Ruoslahti E. RGD and other recognition sequences for integrins. Annu Rev Cell Dev Biol. 1996;12:697–715. | ||
Bougie DW, Rasmussen M, Zhu J, Aster RH. Antibodies causing thrombocytopenia in patients treated with RGD-mimetic platelet inhibitors recognize ligand-specific conformers of αIIb/β3 integrin. Blood. 2012; 119:6317–6325. | ||
Ma D, Xu X, An S, et al. A novel family of RGD-containing disintegrins (Tablysin-15) from the salivary gland of the horsefly Tabanus yao targets αIIbβ3 or αVβ3 and inhibits platelet aggregation and angiogenesis. Thromb Haemost. 2011;105:1032–1045. | ||
Huang Y, Zhang Y, Wu Y, et al. Expression, purification, and mass spectrometric analysis of 15N, 13C-labeled RGD-hirudin, expressed in Pichia pastoris, for NMR studies. PLoS One. 2012;7:e42207. | ||
Moshaverinia A, Chen C, Xu X, et al. Bone regeneration potential of stem cells derived from periodontal ligament or gingival tissue sources encapsulated in RGD-modified alginate scaffold. Tissue Eng Part A. 2014;20:611–621. | ||
Folkman J. Role of angiogenesis in tumor growth and metastasis. Semin Oncol. 2002;29(6 Suppl 16):15–18. | ||
Pasqualini R, Arap W, McDonald DM. Probing the structural and molecular diversity of tumor vasculature. Trends Mol Med. 2002; 8:563–571. | ||
Zetter BR. On target with tumor blood vessel markers. Nat Biotechnol. 1997;15:1243–1244. | ||
Danhier F, Le Breton A, Préat V. RGD-based strategies to target alpha(v) beta(3) integrin in cancer therapy and diagnosis. Mol Pharm. 2012;9:2961–2973. | ||
Garanger E, Boturyn D, Dumy P. Tumor targeting with RGD peptide ligands – design of new molecular conjugates for imaging and therapy of cancers. Anticancer Agents Med Chem. 2007;7:552–558. | ||
Zitzmann S, Ehemann V, Schwab M. Arginine-glycine-aspartic acid (RGD)-peptide binds to both tumor and tumor-endothelial cells in vivo. Cancer Res. 2002;62:5139–5143. | ||
Nitiss JL, Pourquier P, Pommier Y. Aclacinomycin A stabilizes topoisomerase I covalent complexes. Cancer Res. 1997;57:4564–4569. | ||
Wang J, Maitani Y, Takayama K. Antitumor effects and pharmacokinetics of aclacinomycin A carried by injectable emulsions composed of vitamin E, cholesterol, and PEG-lipid. J Pharm Sci. 2002; 91:1128–1134. | ||
Chen Z, Deng J, Zhao Y, Tao T. Cyclic RGD peptide-modified liposomal drug delivery system: enhanced cellular uptake in vitro and improved pharmacokinetics in rats. Int J Nanomedicine. 2012;7:3803–3811. | ||
Cheng L, Huang FZ, Cheng LF, et al. GE11-modified liposomes for non-small cell lung cancer targeting: preparation, ex vitro and in vivo evaluation. Int J Nanomedicine. 2014;9:921–935. | ||
Jia Y, Ji J, Wang F, Shi L, Yu J, Wang D. Formulation, characterization, and in vitro/vivo studies of aclacinomycin A-loaded solid lipid nanoparticles. Drug Deliv. November 5, 2014. [Epub ahead of print]. | ||
Masaka T, Matsuda T, Li Y, et al. Synthesis of VIP-lipopeptide using a new linker to modify liposomes: Towards the development of a drug delivery system for active targeting. Chem Pharm Bull (Tokyo). 2013;61:1184–1187. | ||
Wang X, Lin Y, Zeng Y, Sun X, Gong T, Zhang Z. Effects of mycophenolic acid-glucosamine conjugates on the base of kidney targeted drug delivery. Int J Pharm. 2013;456:223–234. | ||
Pehlivan SB. Nanotechnology-based drug delivery systems for targeting, imaging and diagnosis of neurodegenerative diseases. Pharm Res. 2013;30:2499–2511. | ||
Arias JL. Liposomes in drug delivery: a patent review (2007 – present). Expert Opin Ther Pat. 2013;23:1399–1414. | ||
Gaumet M, Vargas A, Gurny R, Delie F. Nanoparticles for drug delivery: the need for precision in reporting particle size parameters. Eur J Pharm Biopharm. 2008;69:1–9. | ||
Dong Y, Feng SS. Methoxy poly(ethylene glycol)-poly(lactide) (MPEG-PLA) nanoparticles for controlled delivery of anticancer drugs. Biomaterials. 2004;25:2843–2849. | ||
Xu J, Zhao JH, Liu Y, Feng NP, Zhang YT. RGD-modified poly(D,L-lactic acid) nanoparticles enhance tumor targeting of oridonin. Int J Nanomedicine. 2012;7:211–219. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.