Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 14
Review on Databases and Bioinformatic Approaches on Pharmacogenomics of Adverse Drug Reactions
Authors Tong H ![]() , Phan NVT
, Phan NVT ![]() , Nguyen TT, Nguyen DV, Vo NS, Le L
, Nguyen TT, Nguyen DV, Vo NS, Le L
Received 6 November 2020
Accepted for publication 26 December 2020
Published 13 January 2021 Volume 2021:14 Pages 61—75
DOI https://doi.org/10.2147/PGPM.S290781
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Hang Tong,1,2 Nga VT Phan,1,2 Thanh T Nguyen,3 Dinh V Nguyen,4,5 Nam S Vo,3 Ly Le1– 3
1School of Biotechnology, International University, Ho Chi Minh City, Vietnam; 2Vietnam National University, Ho Chi Minh City, Vietnam; 3Department of Translational Biomedical Informatics, Vingroup Big Data Institute, Hanoi, Vietnam; 4Department of Respiratory, Allergy and Clinical Immunology, Vinmec International Hospital, Hanoi, Vietnam; 5College of Health Sciences, VinUniversity, Hanoi, Vietnam
Correspondence: Ly Le; Nam S Vo Email [email protected]; [email protected]
Abstract: Pharmacogenomics has been used effectively in studying adverse drug reactions by determining the person-specific genetic factors associated with individual response to a drug. Current approaches have revealed the significant importance of sequencing technologies and sequence analysis strategies for interpreting the contribution of genetic variation in developing adverse reactions. Advance in next generation sequencing and platform brings new opportunities in validating the genetic candidates in certain reactions, and could be used to develop the preemptive tests to predict the outcome of the variation in a personal response to a drug. With the highly accumulated available data recently, the in silico approach with data analysis and modeling plays as other important alternatives which significantly support the final decisions in the transformation from research to clinical applications such as diagnosis and treatments for various types of adverse responses.
Keywords: pharmacogenomics, adverse drug reactions, next generation sequencing, genome-wide association study, candidate gene approach
Adverse Drug Reactions
Adverse drug reactions (ADRs) are defined as adverse events that happen to patients after taking certain drugs in clinical treatment.1,2 ADRs can cause failure to almost every organ, but more frequent targets are skin, blood, and liver.3–16 These reactions have been reported to affect 10–20% inpatients and about 25% outpatients17–19 and becoming the major burden of healthcare globally. Edward and Aronson have characterized ADRs into six types from A-F (Table 1), in which two major causes are determined as pharmacologic (type A) and immunologic effects (type B). Some rare ADRs resulted from complementary drug metabolism and immunogenic responses.20 Type A is the most common ADRs which driven by the pharmacodynamics reactions including drug metabolism and transport. This type of ADRs is dose-dependent and predictable, therefore they can be managed by adjusting drug intake. The incidences of this ADR type depend largely on the manifestations of Phase I and Phase II liver enzymes such as cytochrome P450s and glutathione transferases. The majority of ADR type B is drug hypersensitivity reactions (DHRs) which count for about 20% of total ADR cases and are mostly driven by immune system factors such as human leukocyte antigens. The reactions may happen at a very low amount of drug compared with a normal dose, and are classified based on different sorts of mechanisms. DHRs are more frequently categorized as immediate and delayed reactions regarding the time course of development.21 Even though the other four types of ADRs were also classified based on the involvement of the associated factors and systematic response to the monitoring methods, the most common ADRs observed from literature are type A (on-target or intrinsic) and type B (off-target or idiosyncratic). The ADRs may appear in patients with mild symptoms from dizziness to very severe syndromes or death, causing much uncomfortability during treatment. In some specific conditions, drugs must be withdrawn and the treatments have to be switched to new therapies. Accurate diagnosis therefore becomes vital to save patients and reduce financial tension. The pharmacogenomic studies of gene–drug relationships bring relevant knowledge to add genetic factors as one to be diagnosed prior to using a drug.
 |
Table 1 ADR Classification |
Genetic Predisposition of ADRs
Advances in pharmacogenomics and immunogenomics have revealed the involvement of multiple molecular factors in response to a drug, raising the concept of drug–gene relationship, and moreover, the mechanisms of response. Pharmacogenomics approaches developed to perceive the presentation of genes in a particular group of samples as well as the gene products under certain conditions. Upon exposure to a drug, a particular gene set will express and bring about the products that could be assessed. The completion of the human genome sequencing has brought several advantages to elucidate the relationship between a person’s genome and drug response. Drug metabolizing enzymes cytochrome P450 (CYPs) especially members 1,2 and 3 families play an important role in drug metabolism and toxicity. CYP2D6, CYP2C19 and CYP2C9 involve in the metabolism of about 80% of therapeutic drugs today. The variation in these genes therefore accounts for most of the ADRs type A in literature. In clinical practice, CYP2D6 biomarkers were observed to link with about 18% of ADR cases reported and suggested by the FDA.22 This gene variation is ethnic-specific and required individual validation in a certain population. In drug hypersensitivity, observational studies in patients treated with some drug groups have shown the high association of genes of HLA classes I and II with the incidence of disease states. For example, HLA-B*15:11 is well-known linked with carbamazepine – inducing SCARs in Japanese and Korean people,23,24 while in South East Asian countries, SCARs to the same drug have prevalently appeared in carriers of HLA-B*15:02 alleles.25–28 Antiretroviral abacavir, on the other hand, is more consistently linked with hypersensitivity in patients carrying HLA-B*57:01 across multiple populations of different origins.29–38 The increasing size and abundance of data accumulated from studies make research in data mining now available for applications.39,40 This review presents the features of current approaches and archive, and state the availability of the data obtained for public share.
Pharmacogenomics Approaches to Study ADRs
Studies on Target Genes Candidates: Replication Approach
More than 95 percents of ADR investigations in Asia are replication approaches.41 A similar situation is also seen in other regions. The study is based on case-control design where people using the same drug(s) are chosen for investigation. Those subjects that develop adverse reactions are carefully characterized and set as cases, whereas controls are the ones who can metabolize the same drug normally. Key variants that show strong association parameters in genome-wide association study (GWAS) would be selected for replications. The designated genes or variants might also be chosen from previous investigations in closely related populations.
Replication approach can be considered to assess the prevalence of genes in certain groups of patients in the association study. It can also be used to validate a marker in multiple samples. The replication scheme is therefore designed in a variable approach such as combining different polymerase chain reaction (PCR) – based techniques such as PCR followed with restriction fragment length polymorphism (RFLP); conventional PCR with sequencing; or RT-PCR and in silico techniques.24,42–45 Compared with next generation sequencing, this method is much cheaper and totally affordable for research, and it can be scaled and transformed into an application with little optimization.46,47
Several genetic risk factors responding to drugs were discovered in the gene candidate approach. Abacavir becomes one of the first drugs studied to date to be aware by FDA in patients carrying HLA-B*57:01 alleles (released in 2017). The allele was found more prevalently in abacavir – inducing hypersensitivity patients across continents.30,34–36,48–50 PCR method was repeated simply in several samples using the sequence-specific oligonucleotides followed with sequencing. The consistency of the association between abacavir hypersensitivity and HLA-B*57:01 leads to the general instruction and guideline by FDA to use HLA-B*57:01 test prior to taking this drug. The other example is antiepileptic drug carbamazepine, which has caused highly variable reactions to people in different ethnic groups. Using PCR – based genotyping, HLA-B*15:02 is shown to associate with SJS/TEN in South Asian populations including Han Chinese, Thai, Vietnamese, and Indian,25–28,35,51,52 but in East Asians like Korean and Japanese, the disease is tightly linked with HLA-B*15:11.23,24 In addition to the main allele mentioned above for carbamazepine-induced SCAR, HLA-A*31:01 is also associated with the severe skin disease caused by this drug. A detection test for these alleles has been developed to promisingly use as a diagnostic method in screening the carbamazepine sensitive patients before prescription.53,54 Using PCR in case–control design, several gene-drug associations have been revealed, providing the basis for syndrome – drug linkage using amplification methods (Table 2, Table 3).
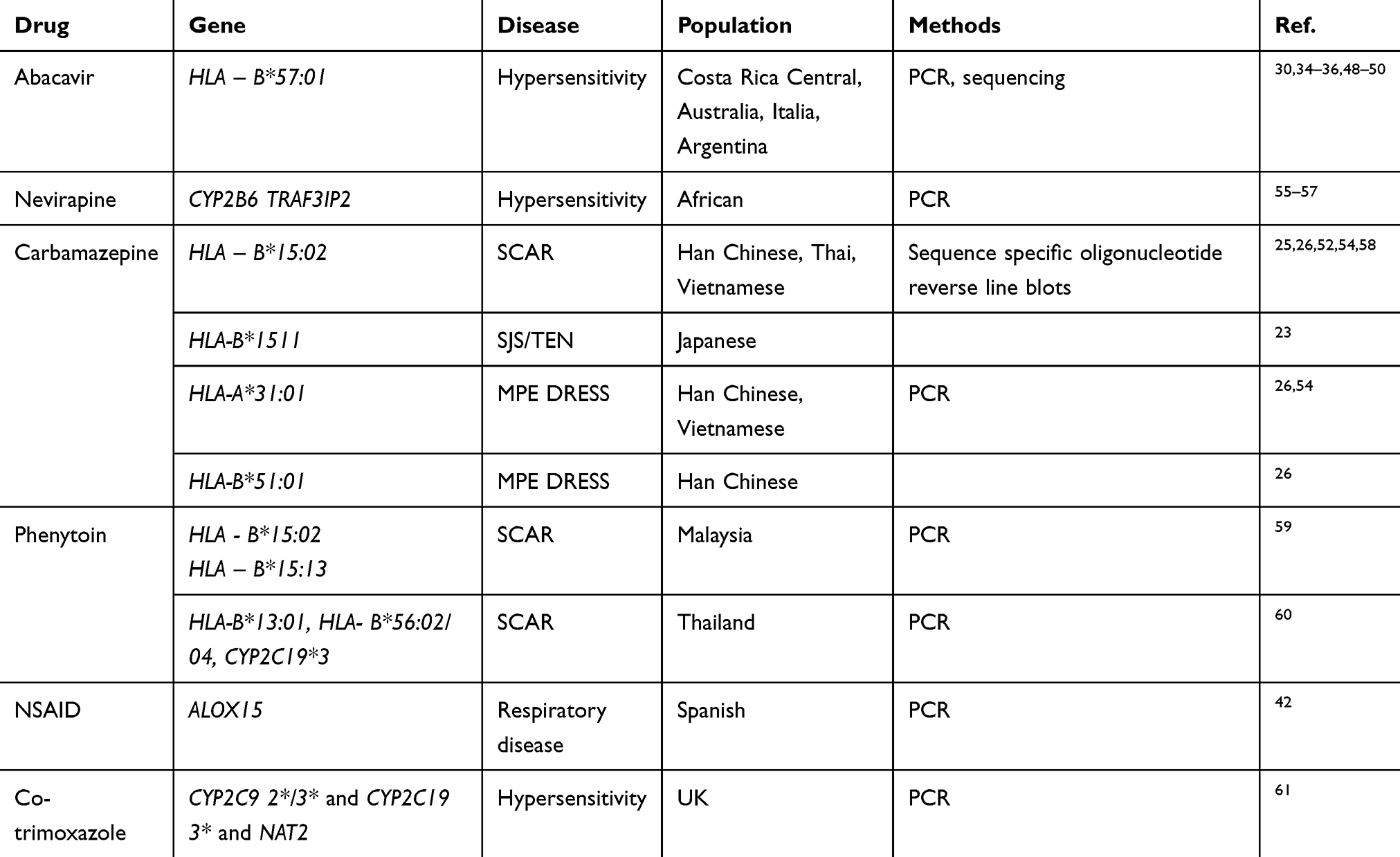 |
Table 2 Selected Genes in Replication Approaches |
Accumulated evidences from individual researches across populations reveal the geographic difference in genetic variability. The association of a gene to a drug response in different populations are varied along with variable allele frequency, haplotype frequency and linkage disequilibrium. Several diseases are multifactorial that require the combinational interpretation of many genes. The meta-analysis of whole exome data from six ethnic populations from different parts of the world shows the consistent findings in drug-related genes. About half of the functional variants of these genes are unique to one of 6 populations studied.62 In addition, amongst the drug-related genes analyzed, CYPs and phase II enzyme have highest difference in cumulative allele probability (CAP), indicating the variation in possibility that a functional variant affects the drug response phenotypes. Although the analysis was based on only the exome data, the major outcome obtained from this result is crucial for each country to build the own pharmacogenomic database. The availability of this data would get us closer to personalized medicine when doctors can more precisely predict the effect of a drug to a patient and choose the best medication for a person.
Genome-Wide Association Studies
Genome-wide association study (GWAS) has shown to be one of the very effective approaches to screen for the risk factors associated with a certain disease.63 To date, there are 4054 publications and almost 140 thousand associations have been published in the GWAS catalog.64 The approaches are designed with a case–control model followed with next generation sequencing and SNP calling against the reference genome. At this present time, the enormous number of SNPs would be obtained and sent for further evaluation of association analysis. Since GWAS evaluated a large number of SNPs, it requires a much larger number of samples to achieve the statistical reliability.65–69 The correlation between the number of SNP and sample size is calculated. It is estimated that testing a single SNP marker requires 248 cases, while testing 500,000 SNPs and 1 million markers requires 1206 cases and 1255 cases, respectively, under the assumption of an odds ratio of 2, 5% disease prevalence, 5% minor allele frequency, complete linkage disequilibrium (LD), 1:1 case/control ratio, and a 5% error rate in an allelic test.70
Recruitment of the large sample in research requires big financial support as well as expertise in data analysis. For this reason, GWAS is preferably used for emerging diseases or complex traits that cannot be simply understood by a single gene phenotype. Specific variants were found in different cohorts providing that risk factors could be thoroughly scanned and validated. GWAS can be designed to integrate with pharmacodynamics to study the drug response in either prospective or retrospective approaches. The suspected drug is administered at common or tested doses and given to individuals. People are recruited based on their response and their genotypes are scanned for all SNPs, and phenotypes are observed through the response of each individual under drug conditions.71 In common design for ADR study, patients are selected with a similar phenotype that demonstrating the characteristics of the same disease in response to a certain drug, compared with the control subjects who take the same prescription and well improved without adverse reactions (drug tolerant). The statistical analysis is then performed to find the association based on genome wide significance, predictive values, et etc.72 The anticonvulsants, antimicrobial and NSAID are among the most causative drugs of ADRs, and were among the first drugs studied and analyzed in GWAS (Table 2).
Several factors have been discussed for the better improvement of the GWAS significance. The sample size is one of the important factors. In the standard approach mentioned, the number of subjects recruited in the study should reach the required quantity in which it could cover the number of possible variants.90 However, in many rare syndromes, recruiting adequate samples seems impossible in a given time. It is therefore requiring consecutive phases of research in which sample collection needs to be completed prior to implementing all other research steps. Multiple attempts have been made to improve the reliability and accuracy of the genome-wide analysis even with a small reference population.91 The findings discussed the adjustment of the model, in which after testing in reference data set of population, both theory and empirical observation from simulation agreed well in the population samples with a high degree of relatedness. These results suggested that higher significance would be obtained in the subjects with the same ethnic origin than in the mixed groups such as meta-analysis. For that, the accuracy and significance at genome-wide scale are still obtained even in the cohort with a small number of subjects (as 50).
Design is another important feature besides sample size added to the success of GWAS. Several samples such as some examples explained above cannot get the genome wide significance assigned by the International Hapmap consortium92 assuming that an OR of 2, MAF 5%, disease prevalence 5% and complete linkage disequilibrium. The outcome might be due to the low allele frequency. The associated p-value suggested 5 x 10−8 is valid for common variants with MAF ≥ 5%. When analyzing variants with lower MAF values such as 1%, 0.5% or 0.1%, the model showed the genome wide significance with p values are 3 × 10−8, 2×10−8 and 1 × 10−8, respectively. The inclusion of LD was indicated not necessary as the model can analyze all variants even they have complete linkage disequilibrium (r2 =1).93
Data Mining and in silico Approach
Although replication approaches have contributed to the mass data accumulation through scanning several cohorts, adding to better understanding the genetics of population, the research design should be utilized with thorough consideration in different aspects.
First, the selected candidate gene should be chosen from the combinational results of genome scanning and/or tested gene of the whole population. The high association of a single gene with the disease might not completely interpret the cause–effect relationship. Pan reported an HLA-B*15:02 negative - case in Taiwan population who developed hypersensitivity in response to carbamazepine,47 suggesting that, the insight about the genetics of a common disease contributes as part of disease development. In the Thai cohort, screening of the HLA genes could help to protect only 22% of SCAR Thai patients (mainly allopurinol - and carbamazepine – inducing) whereas using drug-induced IFN-γ-specific cells scan, approximately 46% of patients with SCAR were detected positive.94 Second, with the complication of the metabolism, approaching a real system to study mechanism sometimes is not possible. The virtual platform may be one of the very good options to try.
The in silico study of carbamazepine and SCAR using pooled data from Asian populations have proved that the polymorphic alleles themselves may not be sufficient to explain the clinical outcomes, instead, the proteins or complex combination of multiple factors could efficiently help to increase the predictive value. For example, when aligning available HLA – B75 protein members on the crystal structure of HLA – B*15:01, all except HLA – B*15:21 imposed and fit on each other. The molecular docking of the chemical structure of carbamazepine to the antigen-presenting sites of tested HLAs could determine the drug binding amino acids on HLA protein. This finding explains for the case reported, in which the HLA – B*15:02 negative but HLA – B*15:21 positive patients still developed SCAR when taking carbamazepine.39 When both alleles belong to the HLA-B75 family, the presence of one and/or another causes hypersensitivity. More investigation combining gene detection and in silico studies have been implemented for dapsone or NSAID.95,96
Analyzing ADRs Using Next Generation Sequencing Data
Data Generation and Collection
In recent years, high-throughput technologies have accelerated genomics/pharmacogenomics studies and resulted in large-scale data. Next-generation sequencing technologies (NGS) such as those provided by Ion Torrent or Illumina platforms are becoming the most common way to get genomics data. Such technologies enabled genomic sequencing on a massive scale at a low-cost and high-quality, which enabled genome-wide studies in large-scale cohorts. Recently, third-generation sequencing technologies (TGS) such as those provided by Pacific Biosciences or Oxford Nanopore Technologies are also exploited in some recent genomics/pharmacogenomics studies.97–101 Such technologies could give read lengths of around tens to even hundreds of thousands of bases although with high-cost and somewhat low-quality compared to second-generation technologies. By generating long reads, TGS has clear advantages in resolving highly repetitive or polymorphic regions which are quite common in pharmacogenomics and immunogenomics studies.
Available Databases for ADRs Studies
Large-scale data from pharmacogenomics studies have been collected and managed under large consortia and networks such as Clinical Pharmacogenetic Consortium (CPIC), Pharmacogenomics Research Network (PGRN), or The South East Asian Pharmacogenomic Network (SEAPHARM). More details of these consortia and networks are shown in Table 3. Some recent work such as The Observational Medical Outcomes Partnership–Common Data Model (OMOP-CDM)102 provides clinical data sources such as electronic health records (EHR) from which ADR-related information can be extracted. Specific databases such as FDA Adverse Event Reporting System (FAERS), Side Effect Resource (SIDER), or Healthcare Cost and Utilization Project (HCUP) provide public datasets that can be used to analyze ADRs and support controlling ADEs.103,104 Currently, many databases have been integrated into larger ones such as PharmGKB.105 PharmGKB also provides a number of datasets and annotations for drugs that have shown adverse reactions, many of them have been pharmacogenetically tested. Some others such as CTD,106 KEGG,107 or SuperTarget108 provide non-clinical data for metabolic pathways, or drug metabolism, interactions in the molecular structures of proteins, or associations of drugs-drugs, drugs-genes. In addition, immunogenomics databases such as HLA-ADR provide allele frequency and haplotype of HLA genes that have been associated with ADRs.109 These databases can be used to help minimize ADRs in drug design or patient treatment.110,111 Some general databases such as dbSNP or the Database of Genomic Variants can be also used as reference for pharmacogenomics studies, eg, to check allele frequency, genotype, and annotation. Figure 1 shows an overview of data, methods and resources for ADR studies.
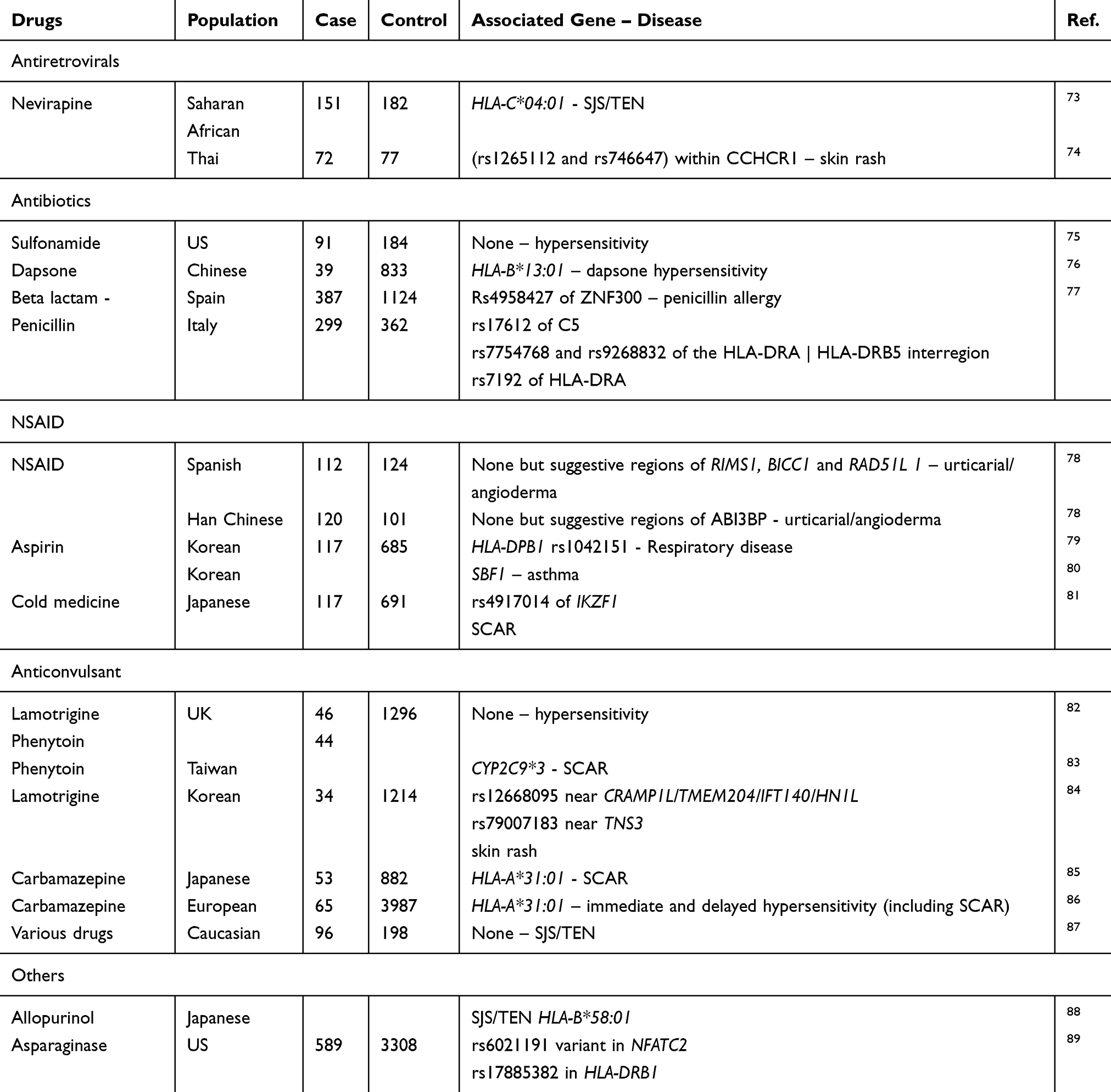 |
Table 2 Approaches of GWAS in Drug Hypersensitivity and Outcomes |
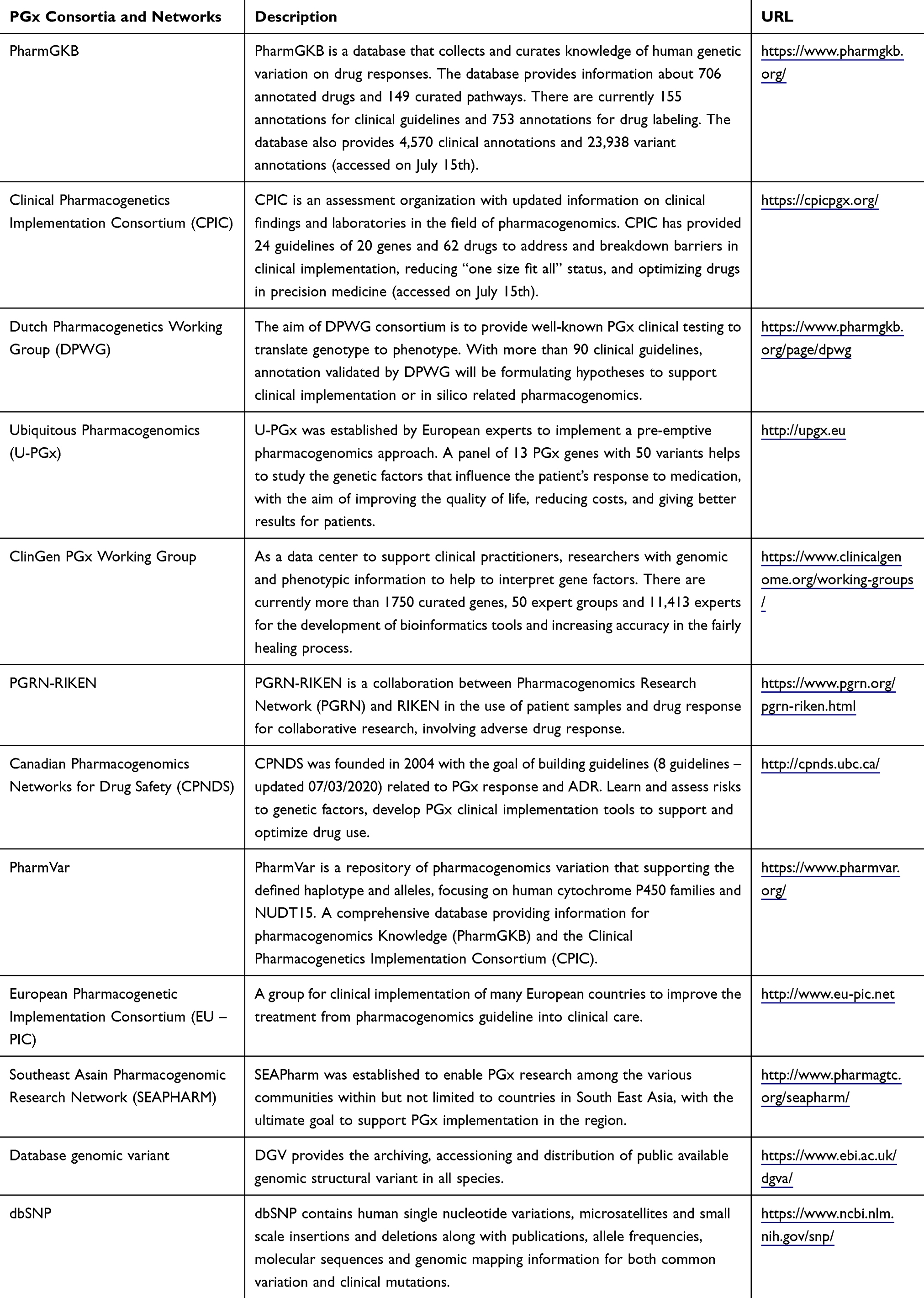 |
Table 3 PGx Consortia and Networks |
 |
Figure 1 Pharmacogenomics for ADRs: networks, data, and pipelines. |
Data Analysis
With the advances in high-performance computing and big data analytics, we can take advantage of large-scale chemical, biological, and biomedical information to discover complex genetic mechanisms of ADRs. Nevertheless, while sequencing data are now easier and cheaper to produce, analyzing such data is still the bottleneck. In the following paragraphs, we will focus on two types of analyses: detecting genomics/pharmacogenomics variants/haplotypes and annotating them for ADR studies. State-of-the-art bioinformatics tools/pipelines for such analyses and emerging machine learning techniques to classify genomic variants into ADRs were summarized in Table 4.
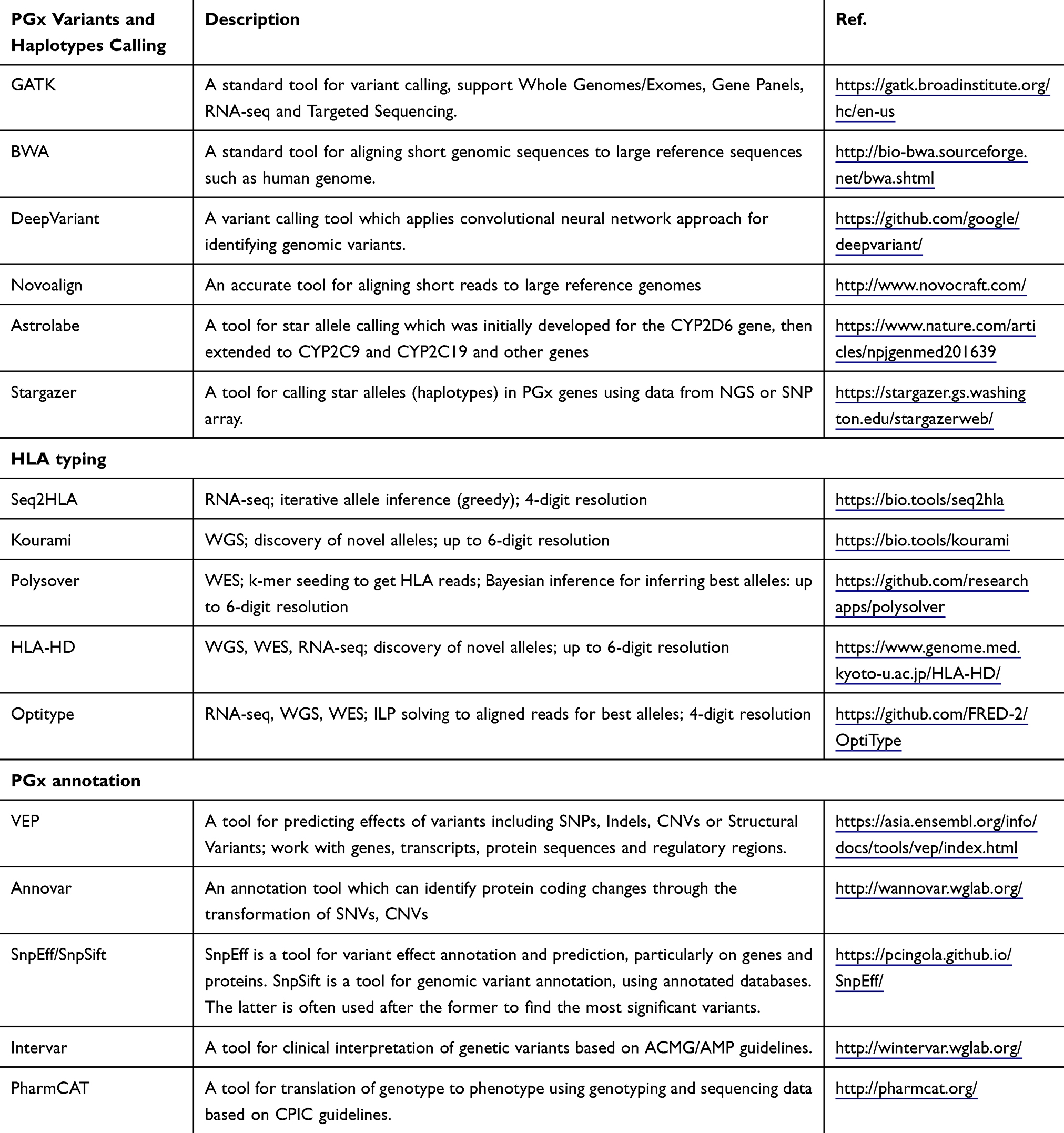 |
Table 4 Common Tools Used for Data Analysis |
Genetic Variant Calling
The GATK best practices112 are often recommended to use for variant calling on NGS data, together with BWA-MEM113 for reading alignment. However, some others such as DeepVariant114 or Novoalign (http://novocraft.com/) can be used as complementary for BWA-MEM or even replacement in some cases to improve performance. This strategy can be widely applied for WGS, WES or sequencing data generated from gene panels. The called variants often serve as a starting point for downstream analyses to study ADRs.
PGx Haplotyping
Due to the complexity of PGx regions, general haplotype callers often have limitations in determining PGx star alleles.101 As a result, specific tools such as Astrolabe,115 Cyrius,116 Aldy,117 or Stargazer118 are often used. However, most of the current PGx haplotyping tools are still limited to detecting star alleles in a subset of PGx genes, eg, Cyrius works on only gene Cytochrome P450 2D6 (CYP2D6), while Stargazer covers only 51 PGx genes. Developing more comprehensive and accurate tools is still an urgent need.
HLA Typing
The complexity and high polymorphisms of HLA regions make HLA typing always challenging, especially for NGS data. HLA typing tools for NGS data such as PHLAT, Polysolver, OptiType, xHLA, or Kourami can get HLA alleles with 4-digits,119,120 6-digits,121,122 up to 8-digits using WGS/WES data. These tools are often limited to detecting known HLA alleles. For RNA-seq data, seq2HLA can be used to get 4-digits resolution.123 Despite a lot of efforts, current tools for HLA typing still suffer from detecting novel or class II HLA alleles.
PGx Annotation
Variant annotation is the process of determining the effects of genetic variants on disease and genes.124 Annotation for PGx variants can be obtained using general tools for annotation of genetic variants such as VEP, SnpEff/SnpSift, Annovar, or Intervar. More informative annotation of PGx genes could be obtained using specific tools such as PharmCAT, which was built based on CPIC’s guidelines. Such guidelines link genotypes to phenotypes and prescribing recommendations based on genotype/phenotype. Recently, PGx guidelines are provided by CPIC, DPWG, CPNDS, and PharmVar are still the gold standard. Due to the limited number of guidelines, such annotation tools can cover only a small subset of PGx genes. The current version of PharmCAT can detect only 12 guidelines out of 64 very important pharmacogenomics (VIPs) of more than 100 known PGx genes.
Classifying Genomic Variants into ADRs
The American College of Medical Genetics and Genomics and Association of Molecular Pathology (ACMG-AMP) has developed guidelines for standardizing and improving disease classification based on genomic variants, which can be used for studying ADRs.125,126 Some tools aim to predict phenotype from genotype using activity score assigned based on allele frequency information from CPIC.127 Some others used hierarchical or k-means clustering to detect the correlation between genotype and phenotype.128 Some recent tools such as Hubble have applied deep learning techniques to predict the functions of PGx alleles.129 Nevertheless, classifying genomic variants to explore the correlation of genes related to ADRs is still challenging and needs significant improvement.
Put-All-Together
A number of data analysis pipelines have been developed to detect and annotate ADR-associated variants and haplotypes. A pipeline for analyzing three genes CYP2C9, CYP2C12, and HLA using WGS/WES/genotyping data have been carried out in.130 They have extracted 39 variants from whole exome sequencing data of 1585 individuals, then haplotype was assigned based on U-PGx translation. Currently the pipeline developed by PGRN seems to be a gold standard for PGx data analysis.131 Meanwhile, some other groups such as RIKEN-Pharmacogenomics Laboratory, SEAPHARM also built their own pipelines for pharmacogenomics data analysis (personal discussion).
Nevertheless, accurate identification of haplotype in pharmacogenomics is still challenging, especially with highly polymorphic regions such as HLA or CYP2D6. Many haplotypes/diplotypes of PGx genes are still unknown. Annotation of PGx genes including novel alleles is still challenging, and there is a need for developing better annotation tools. The gene–drug interaction guidelines are now still very limited, eg, the CPIC database provides only 12 specific guidelines to support optimizing drug therapy. Furthermore, the genotype-phenotype association is still uncertain in many cases due to many factors such as age, gender, the interaction between drug molecules. Developing tools to capture such uncertainty is pretty necessary but very challenging.
Suggestion for Clinical Applications
We have reviewed the typical works in pharmacogenomics to detect adverse drug reactions, a key task in the post-market drug safety surveillance. It is well-known that datadriven131 and data integration approach is essential in solving that problem. Advance in next generation sequencing technology and abundance of genomic data has led to the great outcomes from pharmacogenomic research in drug response to the transformation into clinical applications. A number of drugs have been labeled which concern patients to take the genetic test prior to prescription. Specific programs have also been implemented in particular countries where population genomics have been well analyzed. The US FDA Sentinel Initiative (https://www.sentinelinitiative.org) or The UK CPRD (https://www.cprd.com) are among the programs which have been implemented. These are successful examples for translating research into clinical guidelines for patients or populations who suspect to use specific drugs in the future. For emerging countries, the introduction and improvement of technologies as well as the gradual enlarging of available data bring a number of options of choice. Our review has presented the characteristics, requirements and possible outcomes of some popular and powerful approaches for future applications. Using either one or another depends a lot on the prevalence of current data, design and expectation, however, they can be combined to have the best interpretation of the mechanism of drug response or association of genetic components with a disease, therefore suggest the most effective risk factors for diagnosis or strategic therapy.
Enriching knowledge and attitude towards implementation of pharmacogenomic services among clinicians and administrative staff are crucial for clinical application. In addition, expanding the list of drug-gene pairs covered by national health policy is feasible goal for the near future when sufficient pharmacogenomic and health economic data are generated to aid in the decision-making process. Efforts to implement pharmacogenomics into clinical practice are proceeding at different rates in different countries. It is important to realize the potential benefits of pharmacogenomic implementation on the healthcare system. Effective data and experience sharing at international levels would reduce unnecessary healthcare costs from inefficient or inappropriate drug therapies, maximize the effectiveness of drugs and minimize adverse drug events.
Abbreviations
ADR, adverse drug reaction; DHR, drug hypersensitivity reaction; Ig, immunoglobulin; MPE, maculopapular exanthema; FDE, fixed drug eruption; SCAR, severe cutaneous adverse reaction; DIHS, drug induced hypersensitivity syndrome; DRESS, drug reaction with eosinophilia and systemic symptoms; SJS, Steven Johnson syndrome; TEN, toxic epidermal necrolysis; AGEP, acute generalized exanthematous pustulosis; NSAID, nonsteroidal anti-inflammatory drug; HLA, human leukocyte antigen; GWAS, genome wide association study; WGS, whole genome sequencing; WES, whole exom sequencing; PGx, pharmacogenes; CYP, cytochrome; MAF, minor allele frequency; HWE, Hardy Weinberg equilibrium; OR, odd ratio; LD, linkage disequilibrium; SNP, single nucleotide polymorphism; PCR, polymerase chain reaction; FDA, Food and Drug Administration; RFLP, restriction fragment length polymorphism.
Acknowledgment
This work is partially funded by Vingroup Big Data Institute and Vietnam National University under grant number GEN2019-28-01/HD-KHCN.
Disclosure
The authors declare that they have no conflicts of interest.
References
1. Wheatley LM, Plaut M, Schwaninger JM, et al. Report from the National Institute of Allergy and Infectious Diseases workshop on drug allergy. J Allergy Clin Immunol. 2015;136(2):262–71.e2. doi:10.1016/j.jaci.2015.05.027
2. Uetrecht J. Idiosyncratic drug reactions: current understanding. Annu Rev Pharmacol Toxicol. 2007;47(1):513–539. doi:10.1146/annurev.pharmtox.47.120505.105150
3. Hunziker T, Künzi U-P, Braunschweig S, et al. Comprehensive hospital drug monitoring (CHDM): adverse skin reactions, a 20-year survey. Allergy. 1997;52(4):388–393. doi:10.1111/j.1398-9995.1997.tb01017.x
4. Valeyrie-Allanore L, Sassolas B, Roujeau JC. Drug-induced skin, nail and hair disorders. Drug Saf. 2007;30(11):1011–1030. doi:10.2165/00002018-200730110-00003
5. Sidenius S,PCT,P, Dam M. Lamotrigine-induced rash–worth a rechallenge. Acta Neurol Scand. 2005;111(3):191–194. doi:10.1111/j.1600-0404.2005.00381.x
6. Vonakis BM, Saini SS. New concepts in chronic urticaria. Curr Opin Immunol. 2008;20(6):709–716. doi:10.1016/j.coi.2008.09.005
7. Pereira FA, Mudgil AV, Rosmarin DM. Toxic epidermal necrolysis. J Am Acad Dermatol. 2007;56(2):181–200. doi:10.1016/j.jaad.2006.04.048
8. Downey A, Jackson C, Harun N, et al. Toxic epidermal necrolysis: review of pathogenesis and management. J Am Acad Dermatol. 2012;66(6):995–1003. doi:10.1016/j.jaad.2011.09.029
9. Tang YH, Mockenhaupt M, Henry A, et al. Poor relevance of a lymphocyte proliferation assay in lamotrigine-induced Stevens-Johnson syndrome or toxic epidermal necrolysis. Clin Exp Allergy. 2012;42(2):248–254. doi:10.1111/j.1365-2222.2011.03875.x
10. Wei C-Y, Chung W-H, Huang H-W, et al. Direct interaction between HLA-B and carbamazepine activates T cells in patients with Stevens-Johnson syndrome. J Allergy Clin Immunol. 2012;129(6):1562–9e5. doi:10.1016/j.jaci.2011.12.990
11. Bjornsson E. Review article: drug-induced liver injury in clinical practice. Aliment Pharmacol Ther. 2010;32(1):3–13. doi:10.1111/j.1365-2036.2010.04320.x
12. Clay KD, et al. Brief communication: severe hepatotoxicity of telithromycin: three case reports and literature review. Ann Intern Med. 2006;144(6):415–420. doi:10.7326/0003-4819-144-6-200503210-00121
13. Orman ES, Conjeevaram HS, Vuppalanchi R, et al. Clinical and histopathologic features of fluoroquinolone-induced liver injury. Clin Gastroenterol Hepatol. 2011;9(6):517–523e3. doi:10.1016/j.cgh.2011.02.019
14. Tesfa D, Palmblad J. Late-onset neutropenia following rituximab therapy: incidence, clinical features and possible mechanisms. Expert Rev Hematol. 2011;4(6):619–625. doi:10.1586/ehm.11.62
15. Tesfa D, Keisu M, Palmblad J. Idiosyncratic drug-induced agranulocytosis: possible mechanisms and management. Am J Hematol. 2009;84(7):428–434. doi:10.1002/ajh.21433
16. Aster RH, Curtis BR, McFarland JG, et al. Drug-induced immune thrombocytopenia: pathogenesis, diagnosis, and management. J Thromb Haemost. 2009;7(6):911–918. doi:10.1111/j.1538-7836.2009.03360.x
17. Gandhi TK, Weingart SN, Borus J, et al. Adverse drug events in ambulatory care. N Engl J Med. 2003;348(16):1556–1564. doi:10.1056/NEJMsa020703
18. Gomes ER, Demoly P. Epidemiology of hypersensitivity drug reactions. Curr Opin Allergy Clin Immunol. 2005;5(4):309–316. doi:10.1097/01.all.0000173785.81024.33
19. Lazarou J, Pomeranz BH, Corey PN. Incidence of adverse drug reactions in hospitalized patients: a meta-analysis of prospective studies. JAMA. 1998;279(15):1200–1205. doi:10.1001/jama.279.15.1200
20. Edwards IR, Aronson JK. Adverse drug reactions: definitions, diagnosis, and management. Lancet. 2000;356(9237):1255–1259. doi:10.1016/S0140-6736(00)02799-9
21. Johansson SG, Bieber T, Dahl R, et al. Revised nomenclature for allergy for global use: report of the nomenclature review committee of the World Allergy Organization, October 2003. J Allergy Clin Immunol. 2004;113(5):832–836. doi:10.1016/j.jaci.2003.12.591
22. FDA. Table of Pharmacogenomic Biomarkers in Drug Labeling; 2019.
23. Kaniwa N, Saito Y, Aihara M, et al. HLA-B*1511 is a risk factor for carbamazepine-induced Stevens-Johnson syndrome and toxic epidermal necrolysis in Japanese patients. Epilepsia. 2010;51(12):2461–2465. doi:10.1111/j.1528-1167.2010.02766.x
24. Kim S-H, Lee KW, Song W-J, et al. Carbamazepine-induced severe cutaneous adverse reactions and HLA genotypes in Koreans. Epilepsy Res. 2011;97(1–2):190–197. doi:10.1016/j.eplepsyres.2011.08.010
25. Tassaneeyakul W, Tiamkao S, Jantararoungtong T, et al. Association between HLA-B*1502 and carbamazepine-induced severe cutaneous adverse drug reactions in a Thai population. Epilepsia. 2010;51(5):926–930. doi:10.1111/j.1528-1167.2010.02533.x
26. Hsiao YH, Hui RC-Y, Wu T, et al. Genotype-phenotype association between HLA and carbamazepine-induced hypersensitivity reactions: strength and clinical correlations. J Dermatol Sci. 2014;73(2):101–109. doi:10.1016/j.jdermsci.2013.10.003
27. Yuliwulandari R, Kristin E, Prayuni K, et al. Association of the HLA-B alleles with carbamazepine-induced Stevens-Johnson syndrome/toxic epidermal necrolysis in the Javanese and Sundanese population of Indonesia: the important role of the HLA-B75 serotype. Pharmacogenomics. 2017;18(18):1643–1648. doi:10.2217/pgs-2017-0103
28. Chen P, et al. Carbamazepine-induced toxic effects and HLA-B*1502 screening in Taiwan. N Engl J Med. 2011;364(12):1126–1133. doi:10.1056/NEJMoa1009717
29. Manglani MV, Gabhale YR, Lala MM, et al. HLA- B*5701 Allele in HIV-infected indian children and its association with abacavir hypersensitivity. Indian Pediatr. 2018;55(2):140–141. doi:10.1007/s13312-018-1248-x
30. Mallal S, Nolan D, Witt C, et al. Association between presence of HLA-B*5701, HLA-DR7, and HLA-DQ3 and hypersensitivity to HIV-1 reverse-transcriptase inhibitor abacavir. Lancet. 2002;359(9308):727–732. doi:10.1016/S0140-6736(02)07873-X
31. Agbaji OO, Akanbi M, Otoh I, et al. Absence of human leukocyte antigen-B*57:01 amongst patients on antiretroviral therapy in Nigeria: implications for use of abacavir. Niger Postgrad Med J. 2019;26(4):195–198. doi:10.4103/npmj.npmj_75_19
32. Small CB, Margolis DA, Shaefer MS, et al. HLA-B*57:01 allele prevalence in HIV-infected North American subjects and the impact of allele testing on the incidence of abacavir-associated hypersensitivity reaction in HLA-B*57:01-negative subjects. BMC Infect Dis. 2017;17(1):256. doi:10.1186/s12879-017-2331-y
33. Baniasadi S, et al. Prevalence of HLA-B*5701 and its relationship with abacavir hypersensitivity reaction in Iranian HIV-infected patients. Tanaffos. 2016;15(1):48–52.
34. Martin AM, et al. Predisposition to abacavir hypersensitivity conferred by HLA-B*5701 and a haplotypic Hsp70-Hom variant. Proc Natl Acad Sci U S A. 2004;101(12):4180–4185.
35. Sun HY, Numanagić I, Malikić S, et al. Incidence of abacavir hypersensitivity and its relationship with HLA-B*5701 in HIV-infected patients in Taiwan. J Antimicrob Chemother. 2007;60(3):599–604. doi:10.1093/jac/dkm243
36. Arrieta-Bolanos E, Madrigal JA, Marsh SGE, et al. The frequency of HLA-B(*)57:01 and the risk of abacavir hypersensitivity reactions in the majority population of Costa Rica. Hum Immunol. 2014;75(11):1092–1096. doi:10.1016/j.humimm.2014.09.011
37. Hughes AR, Mosteller M, Bansal AT, et al. Association of genetic variations in HLA-B region with hypersensitivity to abacavir in some, but not all, populations. Pharmacogenomics. 2004;5(2):203–211. doi:10.1517/phgs.5.2.203.27481
38. Nakkam N, et al. HLA pharmacogenetic markers of drug hypersensitivity in a Thai Population. Front Genet. 2018;9:277. doi:10.3389/fgene.2018.00277
39. Jaruthamsophon K, Tipmanee V, Sangiemchoey A, et al. HLA-B*15:21 and carbamazepine-induced Stevens-Johnson syndrome: pooled-data and in silico analysis. Sci Rep. 2017;7(1):45553. doi:10.1038/srep45553
40. Cargnin S, Jommi C, Canonico PL, et al. Diagnostic accuracy of HLA-B*57:01 screening for the prediction of abacavir hypersensitivity and clinical utility of the test: a meta-analytic review. Pharmacogenomics. 2014;15(7):963–976. doi:10.2217/pgs.14.52
41. Ang HX, Chan SL, Sani LL, et al. Pharmacogenomics in Asia: a systematic review on current trends and novel discoveries. Pharmacogenomics. 2017;18(9):891–910. doi:10.2217/pgs-2017-0009
42. Ayuso P, Plaza-Serón MDC, Blanca-López N, et al. Genetic variants in arachidonic acid pathway genes associated with NSAID-exacerbated respiratory disease. Pharmacogenomics. 2015;16(8):825–839. doi:10.2217/pgs.15.43
43. de Oliveira Rodrigues R, Helena Barem Rabenhorst S, Germano de Carvalho P, et al. Association of IL10, IL4, IFNG, and CTLA4 Gene polymorphisms with efavirenz hypersensitivity reaction in patients infected with human immunodeficiency virus. Jpn J Infect Dis. 2017;70(4):430–436. doi:10.7883/yoken.JJID.2016.075
44. Smith RP, Eckalbar WL, Morrissey KM, et al. Genome-wide discovery of drug-dependent human liver regulatory elements. PLoS Genet. 2014;10(10):e1004648. doi:10.1371/journal.pgen.1004648
45. Wadelius M. Point: use of pharmacogenetics in guiding treatment with warfarin. Clin Chem. 2009;55(4):709–711. doi:10.1373/clinchem.2008.115964
46. Nguyen DV, Vidal C, Chu HC, et al. Developing pharmacogenetic screening methods for an emergent country: vietnam. World Allergy Organ J. 2019;12(5):100037. doi:10.1016/j.waojou.2019.100037
47. Pan C-W, Yu C-H, Liao D-L. Carbamazepine-induced hypersensitivity syndrome in chronic schizophrenia. Gen Hosp Psychiatry. 2013;35(5):575 e9–10. doi:10.1016/j.genhosppsych.2012.08.004
48. Dello Russo C, Lisi L, Fabbiani M, et al. Detection of HLA-B*57:01 by real-time PCR: implementation into routine clinical practice and additional validation data. Pharmacogenomics. 2014;15(3):319–327. doi:10.2217/pgs.13.242
49. Moragas M, Belloso WH, Baquedano MS, et al. Prevalence of HLA-B*57:01 allele in Argentinean HIV-1 infected patients. Tissue Antigens. 2015;86(1):28–31. doi:10.1111/tan.12575
50. Dello Russo C, Lisi L, Lofaro A, et al. Novel sensitive, specific and rapid pharmacogenomic test for the prediction of abacavir hypersensitivity reaction: HLA-B*57:01 detection by real-time PCR. Pharmacogenomics. 2011;12(4):567–576. doi:10.2217/pgs.10.208
51. Aggarwal R, Sharma M, Modi M, et al. HLA-B * 1502 is associated with carbamazepine induced Stevens-Johnson syndrome in North Indian population. Hum Immunol. 2014;75(11):1120–1122. doi:10.1016/j.humimm.2014.09.022
52. Nguyen DV, Chu HC, Nguyen DV, et al. HLA-B*1502 and carbamazepine-induced severe cutaneous adverse drug reactions in Vietnamese. Asia Pac Allergy. 2015;5(2):68–77. doi:10.5415/apallergy.2015.5.2.68
53. Mushiroda T, et al. Association of HLA-A*31:01 screening with the incidence of carbamazepine-induced cutaneous adverse reactions in a Japanese population. JAMA Neurol. 2018;75(7):842–849.
54. Nguyen DV, Vidal C, Chi HC, et al. A novel multiplex polymerase chain reaction assay for detection of both HLA-A*31:01/HLA-B*15:02 alleles, which confer susceptibility to carbamazepine-induced severe cutaneous adverse reactions. HLA. 2017;90(6):335–342. doi:10.1111/tan.13143
55. Ciccacci C, et al. Association between CYP2B6 polymorphisms and Nevirapine-induced SJS/TEN: a pharmacogenetics study. Eur J Clin Pharmacol. 2013;69(11):1909–1916. doi:10.1007/s00228-013-1549-x
56. Ciccacci C, Rufini S, Mancinelli S, et al. A pharmacogenetics study in Mozambican patients treated with nevirapine: full resequencing of TRAF3IP2 gene shows a novel association with SJS/TEN susceptibility. Int J Mol Sci. 2015;16(3):5830–5838. doi:10.3390/ijms16035830
57. Carr DF, Chaponda M, Cornejo Castro EM, et al. CYP2B6 c.983T>C polymorphism is associated with nevirapine hypersensitivity in Malawian and Ugandan HIV populations. J Antimicrob Chemother. 2014;69(12):3329–3334. doi:10.1093/jac/dku315
58. Cheung Y-K, Cheng S-H, Chan EJM, et al. HLA-B alleles associated with severe cutaneous reactions to antiepileptic drugs in Han Chinese. Epilepsia. 2013;54(7):1307–1314. doi:10.1111/epi.12217
59. Chang -C-C, Ng -C-C, Too C-L, et al. Association of HLA-B*15:13 and HLA-B*15:02 with phenytoin-induced severe cutaneous adverse reactions in a Malay population. Pharmacogenomics J. 2017;17(2):170–173. doi:10.1038/tpj.2016.10
60. Tassaneeyakul W, Prabmeechai N, Sukasem C, et al. Associations between HLA class I and cytochrome P450 2C9 genetic polymorphisms and phenytoin-related severe cutaneous adverse reactions in a Thai population. Pharmacogenet Genomics. 2016;26(5):225–234. doi:10.1097/FPC.0000000000000211
61. Pirmohamed M, Alfirevic A, Vilar J, et al. Association analysis of drug metabolizing enzyme gene polymorphisms in HIV-positive patients with co-trimoxazole hypersensitivity. Pharmacogenetics. 2000;10(8):705–713. doi:10.1097/00008571-200011000-00005
62. Scharfe CPI, Tremmel R, Schwab M, et al. Genetic variation in human drug-related genes. Genome Med. 2017;9(1):117. doi:10.1186/s13073-017-0502-5
63. Low S-K, Takahashi A, Mushiroda T, et al. Genome-wide association study: a useful tool to identify common genetic variants associated with drug toxicity and efficacy in cancer pharmacogenomics. Clin Cancer Res. 2014;20(10):2541–2552. doi:10.1158/1078-0432.CCR-13-2755
64. Buniello A, et al., The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res, 2019. 47(D1): D1005–D1012.
65. Klein RJ. Power analysis for genome-wide association studies. BMC Genet. 2007;8(1):58. doi:10.1186/1471-2156-8-58
66. Zondervan KT, Cardon LR. Designing candidate gene and genome-wide case-control association studies. Nat Protoc. 2007;2(10):2492–2501. doi:10.1038/nprot.2007.366
67. Spencer CC, et al. Designing genome-wide association studies: sample size, power, imputation, and the choice of genotyping chip. PLoS Genet. 2009;5(5):e1000477.
68. Wu Z, Zhao H, Walsh B. Statistical power of model selection strategies for genome-wide association studies. PLoS Genet. 2009;5(7):e1000582. doi:10.1371/journal.pgen.1000582
69. Park J-H, Wacholder S, Gail MH, et al. Estimation of effect size distribution from genome-wide association studies and implications for future discoveries. Nat Genet. 2010;42(7):570–575. doi:10.1038/ng.610
70. Hong EP, Park JW. Sample size and statistical power calculation in genetic association studies. Genomics Inform. 2012;10(2):117–122. doi:10.5808/GI.2012.10.2.117
71. Wu R, Tong C, Wang Z, et al. A conceptual framework for pharmacodynamic genome-wide association studies in pharmacogenomics. Drug Discov Today. 2011;16(19–20):884–890. doi:10.1016/j.drudis.2011.09.001
72. Hayes B. Overview of statistical methods for Genome-Wide Association Studies (GWAS). Methods Mol Biol. 2013;1019:149–169.
73. Carr DF, Bourgeois S, Chaponda M, et al. Genome-wide association study of nevirapine hypersensitivity in a sub-Saharan African HIV-infected population. J Antimicrob Chemother. 2017;72(4):1152–1162. doi:10.1093/jac/dkw545
74. Chantarangsu S, Mushiroda T, Mahasirimongkol S, et al. Genome-wide association study identifies variations in 6p21.3 associated with nevirapine-induced rash. Clin Infect Dis. 2011;53(4):341–348. doi:10.1093/cid/cir403
75. Reinhart JM, Motsinger-Reif A, Dickey A, et al. Genome-wide association study in immunocompetent patients with delayed hypersensitivity to sulfonamide antimicrobials. PLoS One. 2016;11(6):e0156000. doi:10.1371/journal.pone.0156000
76. Zhang F-R, Liu H, Irwanto A, et al. HLA-B*13:01 and the Dapsone Hypersensitivity Syndrome. N Engl J Med. 2013;369(17):1620–1628. doi:10.1056/NEJMoa1213096
77. Gueant J-L, Romano A, Cornejo-Garcia J-A, et al. HLA-DRA variants predict penicillin allergy in genome-wide fine-mapping genotyping. J Allergy Clin Immunol. 2015;135(1):253–259. doi:10.1016/j.jaci.2014.07.047
78. Cornejo-Garcia JA, Liou L-B, Blanca-López N, et al. Genome-wide association study in NSAID-induced acute urticaria/angioedema in Spanish and Han Chinese populations. Pharmacogenomics. 2013;14(15):1857–1869. doi:10.2217/pgs.13.166
79. Park BL, Kim T-H, Kim J-H, et al. Genome-wide association study of aspirin-exacerbated respiratory disease in a Korean population. Hum Genet. 2013;132(3):313–321. doi:10.1007/s00439-012-1247-2
80. Kim J-H, Park B-L, Cheong HS, et al. Genome-wide and follow-up studies identify CEP68 gene variants associated with risk of aspirin-intolerant asthma. PLoS One. 2010;5(11):e13818. doi:10.1371/journal.pone.0013818
81. Ueta M, Sotozono C, Nakano M, et al. Association between prostaglandin E receptor 3 polymorphisms and Stevens-Johnson syndrome identified by means of a genome-wide association study. J Allergy Clin Immunol. 2010;126(6):1218–25e10. doi:10.1016/j.jaci.2010.08.007
82. McCormack M, Urban TJ, Shianna KV, et al. Genome-wide mapping for clinically relevant predictors of lamotrigine- and phenytoin-induced hypersensitivity reactions. Pharmacogenomics. 2012;13(4):399–405. doi:10.2217/pgs.11.165
83. Su SC, Chen C-B, Chang W-C, et al. HLA Alleles and CYP2C9*3 as predictors of phenytoin hypersensitivity in East Asians. Clin Pharmacol Ther. 2019;105(2):476–485. doi:10.1002/cpt.1190
84. Jang HW, Kim SW, Cho Y-J, et al. GWAS identifies two susceptibility loci for lamotrigine-induced skin rash in patients with epilepsy. Epilepsy Res. 2015;115:88–94. doi:10.1016/j.eplepsyres.2015.05.014
85. Ozeki T, Mushiroda T, Yowang A, et al. Genome-wide association study identifies HLA-A*3101 allele as a genetic risk factor for carbamazepine-induced cutaneous adverse drug reactions in Japanese population. Hum Mol Genet. 2011;20(5):1034–1041. doi:10.1093/hmg/ddq537
86. McCormack M, Alfirevic A, Bourgeois S, et al. HLA-A*3101 and carbamazepine-induced hypersensitivity reactions in Europeans. N Engl J Med. 2011;364(12):1134–1143. doi:10.1056/NEJMoa1013297
87. Shen Y, Nicoletti P, Floratos A, et al. Genome-wide association study of serious blistering skin rash caused by drugs. Pharmacogenomics J. 2012;12(2):96–104. doi:10.1038/tpj.2010.84
88. Tohkin M, et al. A whole-genome association study of major determinants for allopurinol-related Stevens-Johnson syndrome and toxic epidermal necrolysis in Japanese patients. Pharmacogenomics J. 2013;13(1):60–69. doi:10.1038/tpj.2011.41
89. Fernandez CA, Smith C, Yang W, et al. Genome-wide analysis links NFATC2 with asparaginase hypersensitivity. Blood. 2015;126(1):69–75. doi:10.1182/blood-2015-02-628800
90. Nishino J, Ochi H, Kochi Y, et al. Sample size for successful genome-wide association study of major depressive disorder. Front Genet. 2018;9:227. doi:10.3389/fgene.2018.00227
91. Lee SH, Clark S, van der Werf JHJ. Estimation of genomic prediction accuracy from reference populations with varying degrees of relationship. PLoS One. 2017;12(12):e0189775. doi:10.1371/journal.pone.0189775
92. International HapMap C. A haplotype map of the human genome. Nature. 2005;437(7063):1299–1320.
93. Fadista J, Manning AK, Florez JC, et al. The (in)famous GWAS P-value threshold revisited and updated for low-frequency variants. Eur J Hum Genet. 2016;24(8):1202–1205. doi:10.1038/ejhg.2015.269
94. Klaewsongkram J, Sukasem C, Thantiworasit P, et al. Analysis of HLA-B allelic variation and IFN-gamma ELISpot responses in patients with severe cutaneous adverse reactions associated with drugs. J Allergy Clin Immunol Pract. 2019;7(1):219–227e4. doi:10.1016/j.jaip.2018.05.004
95. Watanabe H, Watanabe Y, Tashiro Y, et al. A docking model of dapsone bound to HLA-B*13:01 explains the risk of dapsone hypersensitivity syndrome. J Dermatol Sci. 2017;88(3):320–329. doi:10.1016/j.jdermsci.2017.08.007
96. Schäfer D, Brumen M, Dobovišek A, et al. Dynamic model of eicosanoid production with special reference to non-steroidal anti-inflammatory drug-triggered hypersensitivity. IET Systems Biology. 2015;9(5):204–215. doi:10.1049/iet-syb.2014.0037
97. Ammar R, Paton TA, Torti D, et al. Long read nanopore sequencing for detection of HLA and CYP2D6 variants and haplotypes [version 2; peer review: 2 approved]. F1000Research. 2015;4(17):17. doi:10.12688/f1000research.6037.2
98. Laver TW, et al. Pitfalls of haplotype phasing from amplicon-based long-read sequencing. Sci Rep. 2016;6:21746. doi:10.1038/srep21746
99. Ambardar S, Gowda M. High-resolution full-length HLA typing method using third generation (Pac-Bio SMRT) sequencing technology. Methods Mol Biol. 2018;1802:135–153.
100. Suzuki S, Ranade S, Osaki K, et al. Reference Grade Characterization of Polymorphisms in Full-Length HLA Class I and II genes with short-read sequencing on the ION PGM system and long-reads generated by single molecule, real-time sequencing on the pacbio platform. Front Immunol. 2018;9:2294. doi:10.3389/fimmu.2018.02294
101. Buermans HPJ, Vossen RHAM, Anvar SY, et al. Flexible and scalable full-length CYP2D6 long amplicon PacBio sequencing. Hum Mutat. 2017;38(3):310–316. doi:10.1002/humu.23166
102. Hripcsak G, Ryan PB, Duke JD, et al. Characterizing treatment pathways at scale using the OHDSI network. Proc Natl Acad Sci. 2016;113(27):7329–7336. doi:10.1073/pnas.1510502113
103. Dey S, Luo H, Fokoue A, et al. Predicting adverse drug reactions through interpretable deep learning framework. BMC Bioinform. 2018;19(S21):476. doi:10.1186/s12859-018-2544-0
104. Poudel DR, Acharya P, Ghimire S, et al. Burden of hospitalizations related to adverse drug events in the USA: a retrospective analysis from large inpatient database. Pharmacoepidemiol Drug Saf. 2017;26(6):635–641. doi:10.1002/pds.4184
105. Barbarino JM, Whirl-Carrillo M, Altman RB, et al. PharmGKB: a worldwide resource for pharmacogenomic information. WIREs Syst Biol Med. 2018;10(4):e1417. doi:10.1002/wsbm.1417
106. Davis AP, Grondin CJ, Johnson RJ, et al. The comparative toxicogenomics database: update 2019. Nucleic Acids Res. 2019;47(D1):D948–D954. doi:10.1093/nar/gky868
107. Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30. doi:10.1093/nar/28.1.27
108. Hecker N, et al. SuperTarget goes quantitative: update on drug-target interactions. Nucleic Acids Res. 2012;40(Database issue):D1113–D1117.
109. Ghattaoraya GS, et al. A web resource for mining HLA associations with adverse drug reactions: HLA-ADR. Database. 2016;2016.
110. Cai M-C, Xu Q, Pan Y-J, et al. ADReCS: an ontology database for aiding standardization and hierarchical classification of adverse drug reaction terms. Nucleic Acids Res. 2015;43(Database issue):D907–D913. doi:10.1093/nar/gku1066
111. Ji ZL, Han LY, Yap CW, et al. Drug ADVERSE Reaction Target Database (DART): proteins related to adverse drug reactions. Drug Safety. 2003;26(10):685–690. doi:10.2165/00002018-200326100-00002
112. Van der Auwera GA, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;43(1110):
113. Li H, Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint arXiv:1303.3997, 2013.
114. Poplin R, Chang P-C, Alexander D, et al. A universal SNP and small-indel variant caller using deep neural networks. Nat Biotechnol. 2018;36(10):983–987. doi:10.1038/nbt.4235
115. Twist GP, Gaedigk A, Miller NA, et al. Erratum: constellation: a tool for rapid, automated phenotype assignment of a highly polymorphic pharmacogene, CYP2D6, from whole-genome sequences. Npj Genomic Med. 2017;2(1):16039. doi:10.1038/npjgenmed.2016.39
116. Chen X, et al., Accurate CYP2D6 genotyping using whole genome sequencing data. bioRxiv, 2020: p. 2020.
117. Numanagić I, Malikić S, Ford M, et al. Allelic decomposition and exact genotyping of highly polymorphic and structurally variant genes. Nat Commun. 2018;9(1):828. doi:10.1038/s41467-018-03273-1
118. Lee S-B, Wheeler MM, Thummel KE, et al. Calling star alleles with stargazer in 28 pharmacogenes with whole genome sequences. Clin Pharmacol Ther. 2019;106(6):1328–1337. doi:10.1002/cpt.1552
119. Boegel S, Löwer M, Schäfer M, et al. HLA typing from RNA-Seq sequence reads. Genome Med. 2012;4(12):102. doi:10.1186/gm403
120. Szolek A, Schubert B, Mohr C, et al. OptiType: precision HLA typing from next-generation sequencing data. Bioinformatics (Oxford, England). 2014;30(23):3310–3316. doi:10.1093/bioinformatics/btu548
121. Kawaguchi S, Higasa K, Shimizu M, et al. HLA-HD: an accurate HLA typing algorithm for next-generation sequencing data. Hum Mutat. 2017;38(7):788–797. doi:10.1002/humu.23230
122. Lee H, Kingsford C. Kourami: graph-guided assembly for novel human leukocyte antigen allele discovery. Genome Biol. 2018;19(1):16. doi:10.1186/s13059-018-1388-2
123. Boegel S, et al. In silico HLA typing using standard RNA-Seq sequence reads. In: Bugert P, editor. Molecular Typing of Blood Cell Antigens. New York: Springer New York; 2015:247–258.
124. McCarthy DJ, Humburg P, Kanapin A, et al. Choice of transcripts and software has a large effect on variant annotation. Genome Med. 2014;6(3):26. doi:10.1186/gm543
125. Jarvik GP, Browning BL. Consideration of cosegregation in the pathogenicity classification of genomic variants. Am J Human Genetics. 2016;98(6):1077–1081. doi:10.1016/j.ajhg.2016.04.003
126. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics Med. 2015;17(5):405–424. doi:10.1038/gim.2015.30
127. Gaedigk A, Sangkuhl K, Whirl-Carrillo M, et al. Prediction of CYP2D6 phenotype from genotype across world populations. Genetics Med. 2017;19(1):69–76. doi:10.1038/gim.2016.80
128. Kim E-Y, Shin S-G, Shin J-G. Prediction and visualization of CYP2D6 genotype-based phenotype using clustering algorithms. Transl Clin Pharmacol. 2017;25(3):147–152. doi:10.12793/tcp.2017.25.3.147
129. McInnes G, et al. Transfer Learning Enables Prediction of CYP2D6 Haplotype Function. bioRxiv; 2020:684357.
130. van der Lee M, Allard WG, Bollen S, et al. Repurposing of diagnostic whole exome sequencing data of 1583 individuals for clinical pharmacogenetics. Clin Pharmacol Ther. 2020;107(3):617–627. doi:10.1002/cpt.1665
131. Srinivasan BS, Chen J, Cheng C, et al. Methods for analysis in pharmacogenomics: lessons from the Pharmacogenetics Research Network Analysis Group. Pharmacogenomics. 2009;10(2):243–251. doi:10.2217/14622416.10.2.243
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.