Back to Journals » Therapeutics and Clinical Risk Management » Volume 12
Review of novel therapeutic targets for improving heart failure treatment based on experimental and clinical studies
Authors Osei Bonsu K, Owusu I ![]() , Buabeng K, Reidpath D, Kadirvelu A
, Buabeng K, Reidpath D, Kadirvelu A ![]()
Received 8 February 2016
Accepted for publication 2 April 2016
Published 3 June 2016 Volume 2016:12 Pages 887—906
DOI https://doi.org/10.2147/TCRM.S106065
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
Kwadwo Osei Bonsu,1,2 Isaac Kofi Owusu,3 Kwame Ohene Buabeng,4 Daniel Diamond Reidpath,1 Amudha Kadirvelu1
1School of Medicine and Health Sciences, Monash University Sunway Campus, Jalan Lagoon Selatan, Bandar Sunway, Subang Jaya, Selangor, Malaysia; 2Accident and Emergency Directorate, Komfo Anokye Teaching Hospital, 3Department of Medicine, 4Department of Clinical and Social Pharmacy, College of Health Sciences, Kwame Nkrumah University of Science and Technology, Kumasi, Ghana
Abstract: Heart failure (HF) is a major public health priority due to its epidemiological transition and the world’s aging population. HF is typified by continuous loss of contractile function with reduced, normal, or preserved ejection fraction, elevated vascular resistance, fluid and autonomic imbalance, and ventricular dilatation. Despite considerable advances in the treatment of HF over the past few decades, mortality remains substantial. Pharmacological treatments including β-blockers, angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, and aldosterone antagonists have been proven to prolong the survival of patients with HF. However, there are still instances where patients remain symptomatic, despite optimal use of existing therapeutic agents. This understanding that patients with chronic HF progress into advanced stages despite receiving optimal treatment has increased the quest for alternatives, exploring the roles of additional pathways that contribute to the development and progression of HF. Several pharmacological targets associated with pathogenesis of HF have been identified and novel therapies have emerged. In this work, we review recent evidence from proposed mechanisms to the outcomes of experimental and clinical studies of the novel pharmacological agents that have emerged for the treatment of HF.
Keywords: novel treatment, experimental and clinical studies, therapeutic targets, heart failure
Introduction
Heart failure (HF) is a complex syndrome resulting from disorders in structure and function of the heart associated with a wide variety of cardiovascular diseases and considered a major public health problem owing to its epidemiological transition.1 HF is typified by loss of contractile function with reduced, normal, or preserved ejection fraction (EF), elevated vascular resistance, fluid and autonomic imbalance, and ventricular dilatation.2
Despite considerable gains in the treatment over the past few decades, mortality and morbidity of HF remain substantial. Pharmacological treatments encompassing β-blockers, angiotensin-converting enzyme (ACE) inhibitors, angiotensin receptor blockers (ARBs), and aldosterone antagonists have been proven to significantly reduce mortality and readmissions in HF.1 However, the prognosis is still poor, and a large percentage of these patients progress to advanced HF. Further, treatments for many patients remain unsatisfactory as current therapies often fail to control symptoms and restore quality of life.3 The observation that chronic HF progresses to advanced stages despite optimal treatment has increased the quest for alternatives exploring the roles of additional pathways that contribute to the development and progression of HF.4
Several pharmacological targets associated with pathogenesis of HF have been identified and novel treatments have emerged. The aim of this article was to review emerging therapies, their proposed mechanisms of action, and outcomes of experimental and clinical studies for these new therapies for HF. Figure 1 shows the pathophysiologic mechanisms of HF and novel therapeutic targets of action of pharmacological agents evaluated in this review.
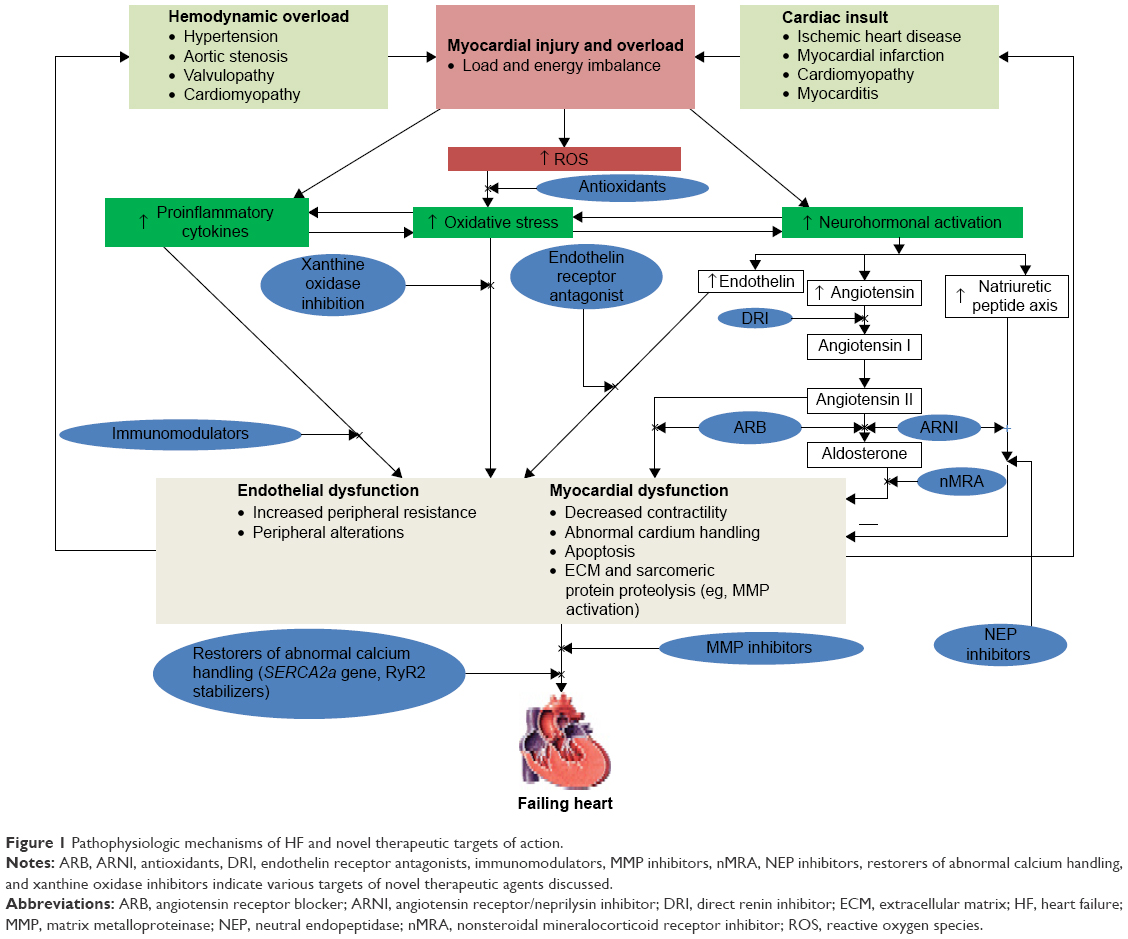 | Figure 1 Pathophysiologic mechanisms of HF and novel therapeutic targets of action. |
Novel approaches to myocardial contractility
Targeting sarcoplasmic–endoplasmic reticulum calcium ATPase 2a to treat HF
Calcium (Ca2+) plays a central role in contractile function of cardiomyocytes. Contractility of cardiomyocytes is regulated by excitation–contraction coupling that occurs through modulation of cytosolic Ca2+ concentration encompassing release of Ca2+ from sarcoplasmic reticulum (SR) through the ryanodine receptor (RyR), then SR Ca2+ reuptake via Ca2+ uptake pump, and Ca2+ removal from myocytes through Na+/Ca2+ exchanger.5 The sarcoplasmic–endoplasmic reticulum calcium ATPase 2a (SERCA2a) is an enzyme responsible for the transfer of Ca2+ from the cytoplasm back into the lumen of the SR, thus shutting off contraction and initiating cardiomyocyte relaxation. Calcium released from the SR into the cytosol during systole activates actin, and myosin coupling accounts for myofilament shortening and the production of contractile force. The rate of myocyte relaxation is controlled by reuptake of calcium during diastole.5,6
Dysregulation of Ca2+ handling/homeostasis in cardiomyocytes plays a critical role in the contractile and relaxation abnormalities that occur in HF.7–10 Deviations from normal Ca2+ handling/homeostasis seen in HF include partial SR Ca2+ depletion, elevated diastolic SR Ca2+ leak, irregular behavior of SR Ca2+ release channels, sarcolemmal Na+/Ca2− exchanger upregulation, and downregulation of SERCA2a.7–10 Thus, approaches aimed at upregulating and restoring SERCA2a activity are being evaluated as a potential therapeutic target for the treatment of HF.
The role of SERCA2a is of particular importance in the war against HF.11 Experimental and human models with HF have demonstrated that downregulating SERCA2a expression and activity is an important element in cardiomyocyte dysfunction.6–8 It has been further shown that even the modest decrease in SERCA2a reduces its activity to the extent that there is a substantial increase in diastolic calcium concentration in homogenates of human heart.12 Abnormal Ca2+ handling/homeostasis by the failing heart is mostly attributed to the reduction in SERCA2a activity, which adversely affects cardiac function and can be corrected by increasing the expression and activity of SERCA2a.
Augmenting SERCA2a expression and activity has shown various favorable effects in HF.6 It has been shown that enhancing SERCA2a activity through a vector-delivered SERCA2a gene improves ventricular metabolic reserve, systolic and diastolic functions, and survival and reduces the probability of developing ventricular arrhythmias during ischemia-induced Ca2+ overload.13–15 Gene transfer therapy provides a novel strategy for targeting abnormalities in cardiac cells that adversely affect cardiac function. Among the nonviral and viral vector systems developed for tissue and organ gene delivery, adeno-associated viruses (AAVs) have emerged as the most valuable gene transfer agents available today. This is because AAVs are preferentially taken up by cardiomyocytes, minimally immunogenic and have the capacity for long-term transgene expression. In addition, AAVs have demonstrated an excellent biosafety record.16 The use of AAVs as viral vector to deliver SERCA2a gene therapy to the heart has shown favorable response in both small17 and large animal models of HF.18–20
The promising outcomes of these experimental studies warranted the initiation of human trials with SERCA2a gene therapy. A Phase I trial with an adeno-associated viral-1 (AAV1) vector used to deliver SERCA2a gene via intracoronary infusion was conducted and subsequently established safety and feasibility of the SERCA2a gene therapy for HF.21 A Phase II trial, the Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID), was then conducted to further evaluate the clinical benefits of SERCA2a gene therapy in HF.22 The CUPID was a double-blind, placebo-controlled trial which enrolled and randomized 39 patients with advanced HF to either AAV1–SERCA2a gene infusions or a matching placebo. The study established the safety of AAV1–SERCA2a gene treatment and exhibited therapeutic response with improved functional status, decreased symptoms of HF, decreased levels of natriuretic peptides (NPs), and attenuated adverse remodeling of the left ventricle in high-dose group. Moreover, a 3-year follow-up study of these patients who received high-dose therapy showed reduction in mortality.23 However, high titers of adenoviral neutralizing antibodies against the AAV1 seen in some patients remain a problem that needs to be addressed.
Despite the promising outcomes of gene transfer therapy for HF, there remains a number of issues to be addressed. Of particular importance is the presence of adenoviral neutralizing antibodies. There are reports of AAV-associated disease that is common in the population, and many develop titers of neutralizing antibodies in response to these infections. The presence of these antibodies adversely affects the efficacy of treatment, thus the therapy is recommended for patients with titers <1.2.
Approximately 60% of >1,500 participants intended to be enrolled in CUPID were excluded due to antibody titers found in serums drawn from them. It is reported that no specific patterns could be detected although significant variability was observed between study participants from the ten countries. However, patients’ age was the only variable that correlated with qualifying AAV1-neutralizing antibody titers.23 Granted that these results are representative, antibody titers seen in serums of patients will be an important obstacle to AAV-based gene transfer therapy in patients with HF. Studies aimed at reducing antibody titers will need to be carried out in order to make this therapy more widely available.
Although currently available AAV vectors have high affinity for cardiac tissues, uptake into other tissues in the body occurs raising the possibility of off-target effects of therapy that could be harmful. More cardiac-specific vectors need to be developed.
RyR stabilization
RyR is an essential tetrameric protein for contraction of myofilaments via regulating the release of Ca2+ from the SR.24 There are three isoforms in mammals. RyR1 is predominant in skeletal muscle,25 and RyR2 is predominant in cardiac muscle.26 RyR3 was first isolated from rabbit brain but known to be more widely distributed in the skeletal muscles, diaphragm, and many other organs.27 Ca2+ enters the cell during the action potential after the voltage-gated L-type calcium channel is activated. The resulting Ca2+ influx causes larger amount of Ca2+ to be released by stimulating the RyR, which is a channel responsible for the release of Ca2+ from the SR membrane. The RyR2 acts as a scaffolding protein which associates with several proteins (protein kinase A, protein phosphatase 1 and 2a, calmodulin, calmodulin kinase II, and phosphodiesterase 4D3 [PDE4D3]) to form a macromolecular complex.28,29 This macromolecular complex is vital to regulating and maintaining the integrity of RyR2 and allows tight control of its function via several phosphorylation and Ca2+ activation and inactivation sites. The RyR2-binding protein, FKBP12.6 (calstabin 2), stabilizes the tetrameric macromolecular complex. The calstabin increases the likelihood of RyR2 channel to maintain a closed state in a resting myocyte. The RyR2 has the strongest affinity for calstabin 2 in the heart, and detaching the macromolecular complex increases channel open probability.9,30 Ca2+/calmodulin-dependent protein kinase II (CaMKII) or protein kinase A phosphorylation of RyR increases its open probability in HF. It has been shown that increased β-adrenergic signaling is closely related to malfunction of SR Ca2+ release via induction of cyclic AMP-dependent PKA hyperphosphorylation of RyR2, thus resulting in calstabin 2 dissociation from the channel complex and as a consequence destabilizing the closed conformation of RyR, while increasing Ca2+ leak.31 These processes lead untimely to the release of Ca2+ during diastole, known as SR Ca2+ leak,9,30 which is characterized by a drastic fall in Ca2+ content of the SR, with less Ca2+ available for release, resulting in weaker muscle contractions. Guo et al32 demonstrated that the activation of CaMKII promotes resting SR Ca2+ release events (Ca2+ sparks and waves) in permeabilized ventricular myocytes via RyR phosphorylation. Furthermore, increased RyR leak during HF has been linked to CaMKII phosphorylation,33 and recent evidence indicates that CaMKII-mediated phosphorylation actually promotes HF development.34–37 It is noteworthy that CaMKII expression and activity is enhanced during chronic β1-adrenergic and angiotensin receptor II stimulation in HF.37 Thus, it is clear that in HF, RyRs become leaky and consequently result in increased frequency of Ca2+ sparks and waves, which promote arrhythmogenesis.38
Approaches to RyR2-Ca2+ leak hypothesis have introduced novel therapeutic strategies for HF.9,30 A number of compounds, collectively called rycals, are known to directly bind and modulate RyR, and 1,4-benzothiazepine derivative, JTV519 (K201), is one of the first agents shown to effectively enhance RyR–calstabin binding and restore abnormal RyR function. These rycals stabilize the closed RyR conformation, but it is unclear whether this action is solely mediated by calstabin 2 binding. An early study in an animal model of HF showed that JTV519-treated hearts restored calstabin 2 binding, inhibited SR Ca2+ leak, and eventually reestablished left ventricular (LV) function.39 An experiment with human myocardium indicated improved diastolic and systolic function under Ca2+ overload conditions with JTV519,40 giving it a great therapeutic promise. Moreover, improved cardiac function could be attributed to the ability of JTV519 to inhibit multiple ion channels.41 A new derivate of JTV519, S107, with a more selective profile has been developed.42 The S107 has been shown to increase the binding of calstabin 2 to RyR, reduce abnormal diastolic Ca2+ release in arrhythmias,42 and attenuate HF progression.43 Further, irregular spontaneous diastolic SR Ca2+ leak resulting from dysfunctional RyR2 is associated with delayed after depolarizations formation, which plausibly accounts for ventricular arrhythmias in HF.44 Thus, in a Phase II, placebo-controlled randomized trial, the antiarrhythmic properties of another RyR-modulating agent, S44121, is being assessed in patients with HF at risk of ventricular arrhythmias (ISCRTN register: 14227980).
Evidence suggests that CaMKII-induced RyR2 phosphorylation accounts for Ca2+ leakage through RYR2 channels.45,46 In HF, the main cardiac isoform – CaMKIIδ – is upregulated in the heart and has been shown to be strongly associated with reduced left ventricular ejection fraction (LVEF). Thus, the administration of CaMKII inhibitors may serve as a novel treatment strategy for HF.45 Specific inhibition of CaMKII by agents such as KN-93 also reduces RyR leak33 and has been shown to improve the force frequency relationship in trabeculae isolated from failing hearts.46 KN-93 was additionally shown to prevent arrhythmia in CaMKIIδ-overexpressing mice.47 Simvastatin increases CAMKK-dependent phosphorylation of AMPK and LKB1 and enhances Rac1 activity in both cultured endothelial cells and mouse aorta.48 These findings identify new points of pharmacological regulation of vascular signaling pathways and uncover new complexities in understanding the pleiotropic effects of statins in the vascular wall and by extension cardiovascular diseases.
Approaches to enhancing neuregulin
Neuregulins (Nrgs) are members of a family of growth factors encoded by four structurally related genes (Nrg-1, 2, 3, and 4) and act through tyrosine kinase receptors (ErbB2 and ErbB4) in the epidermal growth factor receptor family49 to stimulate cellular proliferation, differentiation, and survival in many tissues particularly the heart. Nrg-1 has been shown to play a critical role in cardiac chamber differentiation and trabeculation in the developing embryo.50 Nrg-1 has two isoforms (Nrg-1α and Nrg-1β), which are expressed in the heart; however, β isoforms are more effective in stimulating cardiac myocyte signaling and responses.50 Further, it has been shown that Nrg-1β isoform is required for cardiac development.51 Nrg-1β is expressed in the endothelial cells and stimulates the tyrosine kinase receptors on cardiac myocytes to modulate glucose uptake, growth, and survival.50,52 An experience of Erb-β2-targeted antibody trastuzumab (Herceptin) and its associated cardiotoxicity in patients treated for breast cancer53,54 has led to the quest for better understanding of the physiological roles of Nrg-1 and its receptors in the myocardium. The early stages of HF are characterized by the activation of Nrg-1/ErbB signaling coupled with increased expression of Nrg-1 in the left ventricle. However, Nrg-1 expression and Nrg-1/ErbB signaling are suppressed during the late phase of HF.55
Consequently, experimental and human studies aimed at augmenting Nrg-1 have recently been conducted. A study of recombinant Nrg-1β showed improved ventricular dimensions, function, and parameters of remodeling in rat model.56 Thus, exogenous Nrg-1β agents have been developed and are being evaluated in clinical trials. An early phase study of exogenous Nrg-1β showed improved cardiac contractile function via modulating Ca2+ handling, thus enhancing excitation–contraction coupling and Ca2+ sensitivity.57 Further, recombinant human Nrg-1β2 has been shown to improve cardiac output, structure, and function by 90 days in a dose-ranging study.58
A number of issues have been raised about recombinant NRGs utility in HF since its receptor expression triggers responses in diverse tissues50 and, notably, the potential for tumor growth acceleration. The need for continuous intravenous (IV) administration of these agents over several hours on daily basis limits their use in chronic HF. Persistent nausea and central nervous system effects observed with the administration of these agents in almost all subjects may further limit their use in chronic HF. Nonetheless, despite these caveats, this approach remains a solid and an exciting treatment approach in HF requiring further studies.
Novel approaches to renin–angiotensin aldosterone system blockade and neurohormonal modulation
The inhibition of the renin–angiotensin aldosterone system (RAAS) is important to the treatment of HF with known pharmacological agents – ACE inhibitors, ARBs, and mineralocorticoid receptor (MR) antagonists – already in clinical practice. However, further targets and new pharmacological agents are being discovered and developed to further improve the outcomes in HF.
Direct renin inhibitors
HF is characterized by increased plasma renin activity (PRA).59 PRA is closely associated with key adverse events in patients with HF.60,61 Moreover, PRA has demonstrated to be a powerful independent prognostic predictor of adverse outcomes in HF.62 Direct renin inhibitors (DRIs) block the conversion of angiotensinogen to angiotensin I consistent with the actions and effects of renin. Nguyen63 described a prorenin receptor that has pathophysiological relevance to HF in addition to the action of renin on the RAAS. The activation of prorenin receptor results in prohypertrophic and apoptotic activity through stimulating transforming growth factor-β and several intracellular signaling pathways. Further, it is known that DRI downregulates the RAAS to reduce pro-renin receptor gene expression.63 Several DRIs have been developed; however, their oral bioavailability has been pharmacologically complex and challenging. Aliskiren, the first orally active DRI to be licensed for clinical use, also has very low bioavailability but shows consistent PRA and downstream RAAS inhibition to produce a steady decrease in blood pressure (BP) in hypertension.
Few studies have evaluated DRIs in patients with HF. Neuberg et al64 were the first to investigate renin inhibition in nine patients with chronic HF with an IV enalkiren, which suppressed PRA and lowered BP in the previous studies of patients with hypertension. The acute IV administration of enalkiren (1.0 mg/kg) significantly increases cardiac and stroke volume indexes and decreases left ventricular filling pressure, mean right atrial pressure, heart rate, and systemic vascular resistance.64 Subsequently, the use of IV remikiren was found to be similar to IV enalapril treatment with regard to reducing pulmonary capillary wedge pressure (PCWP) and systemic vascular resistance in chronic HF.65
In recent times, aliskiren – the only orally active DRI – is being studied in chronic HF. The first study randomized 27 patients with HF to ACE inhibitor, ramipril, or aliskiren.66 Aliskiren 300 mg/d given for 7 days to patients with low LVEF significantly reduced PRA and angiotensin II compared to ramipril.66
To confirm the abovementioned claim, ALiskiren Observation of heart Failure Treatment (ALOFT) study evaluated the efficacy of DRIs in chronic HF.67 The ALOFT study randomized 302 patients with chronic HF to aliskiren (150 mg/d) or placebo for 3 months with the change in plasma brain NP (BNP) levels as the primary efficacy endpoint. At the end of the trial, aliskiren significantly reduced the plasma BNP levels compared to placebo. Further, aliskiren significantly increased plasma renin concentration and decreased PRA compared to placebo. Therefore, DRI could provide extra treatment benefits to existing RAAS inhibitors; however, a careful consideration of possible adverse effects will be required in the treatment of HF. Hyperkalemia is particularly common among patients with chronic HF owing to impaired renal perfusion, comorbidities, and concurrent treatments such as β-blockers and RAAS inhibitors. It is worth noting that, in the ALOFT trial,67 hyperkalemia and worsening renal function were slightly higher among patients who received aliskiren than in placebo group. In addition, marginal increase in symptomatic hypotension was seen with aliskiren compared to placebo.
Large-scale trials have been testing aliskiren treatment on major outcomes in HF. The Aliskiren Trial on Acute Heart Failure Outcomes (ASTRONAUT) study68 is among the first trial to evaluate aliskiren on outcomes in HF. The ASTRONAUT study randomized ~1,639 patients hospitalized for acute decompensated HF to aliskiren 150 mg (increased to 300 mg as tolerated) or placebo daily in addition to standard therapy for ~12 months. The initiation of aliskiren in addition to optimal treatment failed to reduce mortality or readmissions in patients hospitalized for HF with reduced EF (HFrEF) at 6 months or 12 months after discharge.69 Another study evaluating aliskiren in HF is the Aliskiren Trial to Minimize OutcomeS in Patients with Heart FailurE (ATMOSPHERE) trial.70 The ATMOSPHERE is currently evaluating the clinical benefit of aliskiren as additional treatment to other RAAS blockers and direct comparison with ACE inhibitor treatment. This clinical trial is expected to clear the doubt about the usefulness of DRIs in chronic HF following the failure of ASTRONAUT and ALTITUDE71 to show clinical benefit with aliskiren in cardiovascular conditions.
Modulation of NPs
NPs are cardiac hormones which are stimulated and produced by the atrium, ventricles, or endothelial cells after myocardial infarction, LV hypertrophy, and HF. Four major isoforms, such as atrial NP, BNP, C-type NP, and dendroaspis NP, have been described to be produced by the heart after injury. These peptides have various favorable hemodynamic, neurohormonal, cardiac, and renal properties, which are believed to further enhance the beneficial RAAS-blocking effects in HF.
A synthetic analog of BNP – nesiritide – has been extensively studied in clinical trials and proven to increase cardiac indexes and reduce cardiac filling pressures and eventually improve the clinical status of patients with acute decompensated HF. Nesiritide is now clinically available for the management of acute decompensated HF although no primary endpoint benefit was reported.72,73 However, adopting this approach to treatment in chronic HF has been problematic because of poor oral bioavailability of NPs.74
Endothelin receptor antagonism
Endothelin activation is considered an integral part of the neurohormonal response in HF.75,76 The vascular endothelial cells produce endothelin, a potent vasoconstrictor, higher plasma levels of which induces adverse ventricular remodeling and worsens HF symptoms via reducing coronary flow and exhibiting direct harmful myocardial effects.75,77 It is well established that higher plasma levels of endothelin-1 is associated with functional impairment and worsening prognosis in HF.76–79 Driven by these pathophysiological insights, bosentan, an orally active nonselective endothelin receptor antagonist which interferes and inhibits endothelin-mediated effects was developed. Experimental studies evaluating the long-term treatment effects of bosentan in HF have reported significant benefits in hemodynamic variables, ventricular remodeling, and survival.80,81 This claim was confirmed in clinical studies where patients with symptomatic HF on standard therapy had short-term benefits of bosentan on hemodynamics reported.82 However, two long-term studies, Endothelin Antagonist Bosentan for Lowering Cardiac Events in HF (ENABLE) and Research on Endothelin Antagonism in Chronic HF (REACH), assessing bosentan were terminated ahead of schedule owing to the increased number of adverse events, with no improvements in clinical status in patients with symptomatic HF.83,84
Neutral endopeptidase inhibition
Neutral endopeptidase (NEP) is an enzyme which degrades NPs, and the inhibition of NEP persistently increases plasma concentration of NPs. Candoxatril has been shown to induce diuresis and natriuresis, improve exercise capacity, and lower PCWP, but produces no effects in PRA. Ecadotril increases plasma concentration of NPs and reduces PRA and PCWP.85 Further, the treatment effects of ecadotril were evaluated in ~280 patients with chronic HF in a multicenter dose-ranging study.86 There were no symptomatic benefits, but rather pancytopenia and deaths were reported among patients treated with ecadotril.
Vasopeptidase inhibition
Among the NEP inhibitors, omapatrilat (a dual antagonist of ACE and neprilysin) is the most widely studied particularly in HF and/or hypertension. The IMPRESS randomized trial87 was the first Phase II evaluation of 40 mg daily omapatrilat versus 20 mg daily lisinopril in 573 New York Heart Association (NYHA) Functional classes II–IV patients with HF with LVEF ≤40% in a head-to-head randomized fashion for 24 weeks. Fewer cardiovascular events were seen in the patients who received omapatrilat compared to the lisinopril group. Omapatrilat treatment significantly reduced composite endpoint of death, admission for HF, or discontinuation of treatment due to worsening symptoms compared to lisinopril. However, both treatment and lisinopril groups were comparable regarding exercise tolerance which was the primary endpoint of the study.
The promising outcomes of these studies prompted the conduct of Phase III study, OVERTURE,88 which evaluated 5,770 patients comparing enalapril (10 mg twice daily) to omapatrilat (40 mg once daily), over a mean duration of 14.5 months. A marginal but nonsignificant decrease in composite endpoint of death or HF hospitalization requiring IV treatment was seen with omapatrilat compared to enalapril treatment. The design of the OVERTURE study is believed to have accounted for the neutral outcomes reported. The twice-daily dosing of enalapril may have led to pharmacokinetic variations than with a daily dosing of omapatrilat which produced significant lowering in BP. In addition, the definition of “hospitalization for HF” as admissions for HF requiring IV diuretics may have underrated the omapatrilat treatment benefits in the OVERTURE study. Finally, angioedema reported in few patients in the IMPRESS and OVERTURE studies are undoubtedly a source of concern and worth investigating.
Angioedema associated with ACE/NEP inhibitor treatment is believed to be due to the accumulation of bradykinin resulting from concurrent ACE and NEP inhibition, both of which play critical roles in the breakdown of bradykinin.89
In addition to single daily dose of omapatrilat failing to show any significant clinical benefit in comparison to twice-daily dosing enalapril, omapatrilat was associated with serious angioedema and hypotension leading to early termination of the OVERTURE study. In contrast to omapatrilat treatment, LCZ696 (sacubitril – neprilysin inhibitor and valsartan – an ARB) has no or minimal effect on ACE and therefore angioedema associated with inhibition of bradykinin breakdown was limited.
Angiotensin receptor blockade and neprilysin inhibition
Driven by the abovementioned pharmacological insights and experiences, newer approaches aimed at inhibiting NEP to enhance RAAS blockade have been developed. The combination of NEP inhibition with ARBs is suggested to be less likely to cause angioedema because the degradation of bradykinin would be lower compared to ACE inhibitors.
LCZ696 is a single molecule consisting of neprilysin inhibitor sacubitril (AHU377) and ARB valsartan designed to curtail the risk of angioedema.90,91 LCZ696 concurrently inhibits RAAS and impairs NP breakdown due to its specific moieties which modulate both pathways.92 Clinical studies have demonstrated that LCZ696 exhibits greater but favorable neurohormonal and hemodynamic effects compared to ARB alone in patients with hypertension or HF with a preserved EF.93,94 These studies established the tolerability and safety in chronic HF.
The PARADIGM-HF is a large trial that evaluated the efficacy and safety of LCZ696 in chronic HF.95,96 This study compared LCZ696 with enalapril in patients who had HF with a reduced LVEF. In this double-blind trial, 8,442 patients with classes II–IV HF and an LVEF ≤40% were randomly assigned to either LCZ696 or enalapril in addition to the recommended therapy. LCZ696 was superior to enalapril in reducing the risks of HF hospitalization and all-cause and cardiovascular mortality after 27 months of follow-up.96 However, higher proportions of patients had hypotension and nonserious angioedema, but lower proportions were seen with renal impairment, hyperkalemia, and cough in the LCZ696 group than the enalapril group.96 Now, LCZ696 is registered as Entresto™ (Novartis International AG, Basel, Switzerland) (sacubitril/valsartan) in the US and has entered the clinic for the treatment of patients with HFrEF and NYHA classes II–IV. Entresto™ is indicated for reducing the risk of HF hospitalizations and cardiovascular mortality and currently being reviewed by health authorities around the world.
NP analogs
Augmenting endogenous NPs is another approach to finding treatment options for HF. The use of chimeric NP (CD-NP) which is a chimeric designer peptide made up of a complete string of C-type NPs fused to the carboxyl-terminal tail of Dendroaspis NP is one of such approaches being evaluated. C-type NP selectively binds to natriuretic peptide receptor-B and results in the reduction in cardiac filling pressures to lessen hypotension. The C-terminus of dendroaspis NP is responsible for a greater percentage of renal activity via promoting natriuresis and diuresis.97 CD-NP has been shown to be safe and improves cardiovascular and renal function without inducing significant levels of hypotension in animal and human studies. This approach is now being evaluated in Phase II trials in HF (NCT01750905 and NCT00620308). Nonetheless, large clinical trials are needed to further validate CD-NP as an effective treatment option for HF.98
Arginine vasopressin receptor antagonism
Arginine vasopressin (AVP) is a neuropeptide hormone synthesized in the nuclei of the hypothalamus in neuronal cell bodies and released from the posterior pituitary into the bloodstream. Circulating AVP levels are significantly elevated in patients with HF compared to healthy controls, and much higher levels found in congestive patients with HF with significant cardiac decompensation and hyponatremia.99 Its actions are linked to free water reabsorption (V2) and arteriolar vasoconstriction (V1A). Novel approaches being developed for the treatment of HF target vasopressin receptor antagonism. Antagonizing V1A receptor produces vasodilation and lessens afterload, while V2 receptor antagonism increases aquaresis with minimal or no sodium loss.4 Two vasopressin antagonists – conivaptan and tolvaptan – have received the approval for the treatment of hyponatremia in HF.4 Tolvaptan, a selective V2 receptor antagonist, promotes diuresis and as a consequence reduces edema, jugular venous pressure, and lessens or relieves congestion in HF. Conivaptan – dual V1A and V2 receptor antagonist – inhibits pressor response, stimulates aquaresis, and significantly reduces PCWP in HF.100–102 Thus, AVP antagonists provide important options for the treatment of patients with volume-overload HF.
Approaches to modulation of catecholamine synthesis and release
Nolomirole is a selective dopamine 2-α2 receptor agonist which inhibits the release of catecholamines from sympathetic nerve endings.4 This inhibition of catecholamines further inhibits the release of tumor necrosis factor (TNF)-α with the improvement of ventricular function. Although nolomirole reduces cardiac hypertrophy and lessens the symptoms of HF in animal models, a large randomized controlled trial in humans did not show clinical benefit in patients with HF.103,104
Further, 5-HT4 receptor antagonist (SB207266) has been found to significantly reduce LV remodeling and improve diastolic function in postinfarction rat model. These favorable effects suggest important contributions of 5-HT4 receptor antagonism in HF.105 A proof concept of double-blind randomized control trial (RCT) showed that the treatment with piboserod (5-HT4 receptor antagonist) 80 mg marginally but significantly improved LVEF in patients with chronic HF in 24 weeks of follow-up.106 However, the study did not show any significant changes in exercise tolerance, neurohormones, and quality of life but reported increases in adverse events. The clinical relevance of piboserod remains unclear since no significant changes in other efficacy parameters was reported.
Novel approaches to aldosterone blockade
Aldosterone is critical to HF progression and MRs are shown to be overexpressed in failing hearts.107 Aldosterone enhances sodium retention, stimulates loss of potassium and magnesium, and is associated with sympathetic activation, parasympathetic inhibition, fibrosis of the myocardium and vascular walls, and impaired arterial compliance.107 ACE inhibitors suppress aldosterone through the inhibition of angiotensin II formation; however, they do not prevent the release of aldosterone from non-ACE-dependent angiotensin II production or activation of aldosterone receptors.107 Thus, aldosterone has become a well-established independent target for the treatments of systolic HF. Pharmacological agents that inhibit MR reduce the mortality in chronic HF. However, clinical use of these approaches is limited by increased risk of hypotension, hypokalemia, and worsened renal function when used together with other RAAS-blocking agents in HF. Several approaches aimed at addressing the concerns of adverse events limiting the clinical use of these agents have been developed.
PF3882845 (Pfizer, Inc., New York, NY, USA)108 and BR-4628 (Bayer AG, Leverkusen, Germany)109 are among several nonsteroidal MR antagonists which have recently been developed. PF3882845 demonstrated greater decreases in BP, albumin, and renal protection compared to eplerenone in an experimental model.108 The BR-4628 is a dihydropyridine (DHP) derivative which was developed as a result of recent discovery that classic dihydropyridine-based L-type calcium channel blockers antagonize MRs in in vitro and in vivo studies.109 BR-4628 is known to prevent or treat renal injury induced by ischemia/reperfusion using Wistar rat models110 and it is currently in clinical trials.111
Another nonsteroidal MR antagonist – BAY 94-8662 – has recently been developed and has been found to be more selective for MR than spironolactone and showed improved affinity for MR compared to eplerenone in experimental models.112 Further, BAY 94-8862 shows a more evident cardiorenal end-organ protection than steroidal MR antagonists in hypertension-driven HF and renal dysfunction model.113
The minerAlocorticoid Receptor Antagonist Tolerability Study (ARTS) was a double-blind randomized, controlled, Phase II trial which evaluated the safety and tolerability of oral BAY 94-8862 in patients with chronic HFrEF and mild or moderate chronic kidney disease (CKD). The ARTS reported that BAY 94-8862 5–10 mg/d was as effective as spironolactone 25 mg/d or 50 mg/d in decreasing hemodynamic stress indicators and was associated with lower incidences of hyperkalemia and worsening renal function in patients with HFrEF and moderate CKD.113 The promising result of ARTS affords a firm reason for further research into the role of BAY 94-8862 in patients with HFrEF and concomitant CKD. This study further guides dosage selection for Phase III clinical trials.
Blockade of aldosterone synthesis is an alternative strategy to MR antagonism. In a failing heart, local aldosterone production is proportionate to the severity of disease. Aldosterone synthase or angiotensin II induces local aldosterone production in the failing ventricles in a manner comparable to disease severity. A number of novel agents targeting aldosterone synthase inhibition have been developed. One such agent is FAD286, an aldosterone synthase inhibitor, which has been shown to improve hemodynamics and the function of the heart in a rat model of HF.114 A selective aldosterone synthase inhibitor – LCI699 – showed a significant lowering BP similar to that of 50 mg twice-daily dosing of eplerenone and further substantial reductions in 24-hour ambulatory BP monitoring compared to placebo in mild-to-moderate hypertension.115 Thus, inhibiting aldosterone synthase may provide alternative or complementary strategies to MR antagonism in chronic HF.
Novel agents targeting cytokines and inflammation
Increased inflammation resulting from the activation of proinflammatory cytokines, cardiac myocytes, cardiac autoantibodies, cell adhesion molecules, and endothelial cells produced by activated macrophages is common in HF.2 Elevated levels of proinflammatory cytokines – TNF-α, interleukin (IL)-1, IL-6, IL-10, IL-12, IL-17, IL-18, and C-reactive protein – have been linked to adverse outcomes in HF.2 Several chemokines such as monocyte chemoattractant peptide-1, IL-8, and macrophage inflammatory protein-1a are also elevated in HF. Established treatments for HF have shown marginal effects on the cytokine network. Consequently, a number of immunomodulatory agents have been developed as novel treatments due to evidence that attenuated inflammation enhances cardiac function in HF.116,117
TNF-α plays a substantial role in HF progression as elevated plasma levels has been linked to extracellular matrix (ECM) remodeling, myocyte hypertrophy, and increased apoptosis.117 Etanercept (TNF-α receptor antagonist) and infliximab (anti-TNF-α antibody) are two agents known to inhibit TNF-α activity and have been widely studied in various clinical trials.118 However, both agents failed to show outcome benefits in patients with HF and several explanations have been proposed for the failure of anti-TNF-α therapy. One such reason is that low plasma concentrations of TNF-α is essential for the repair of tissues and it is possible that infliximab may have reduced TNF-α to concentrations below optimal levels which may be required for the observed favorable effects. In addition, infliximab may have induced apoptosis and other harmful effects on cardiomyocytes via antibody-dependent cellular toxicity and complement-dependent cytotoxic mechanisms. Further research into anti-TNF-α therapy should clarify the appropriate pharmacological agent, optimal dose, and subgroup of patients likely to benefit from treatment.116,117
The inhibition of TNF-α synthesis via competitively inhibiting the TNF-α-converting enzymes (anti-TACEs) has been explored as a therapeutic target in HF. TACEs are important enzymes required for the conversion of pro-TNF-α into matured form. Studies have shown that macrophage inflammatory proteins and aprotinin decrease the processing of TNF-α via nonselective inhibition of TACE. Two selective TACE inhibitors – DPH-067517 and GM 6001– are being assessed as novel therapeutic agents for the treatment of HF.119
TNF-α activates the p38 MAPK pathway to suppress contractility and promote inflammation and ECM remodeling.120 P38-mediated proinflammatory properties via cytokines (eg, IL-1β and IL-6) and TNF-α have been shown to play significant roles in the progression of HF.121 SB203580 and FR167653 are p38 inhibitors which have shown significant decreases in fibrosis, apoptosis, LV hypertrophy, and dilatation as well as improvement in EF and contractility in experimental models.121 Despite being promising agents for the treatment of HF, these agents have not been tested in clinical studies.120
Pentoxifylline (PTX) is a phosphodiesterase inhibitor which downregulates TNF-α synthesis through suppressing gene transcription of TNF-α. Furthermore, PTX is known to have TNF-α-independent immunomodulatory properties and prevents apoptosis to protect the myocardium and subsequently slowing down the progression of HF. A number of trials of PTX therapy have reported improved clinical symptoms, whereas others fail to show any benefits in patients with HF.122 Lysophosphatidic acid acyl transferase inhibitor – lysofylline – is another pharmacological agent which acts by decreasing lipopolysaccharide-induced TNF-α synthesis. Lysofylline is being evaluated for the treatment of sepsis, thus establishing its contribution to HF treatment may be required.119
Myocardial dysfunction,123 dilated and ischemic cardiomyopathy, and the severity of HF are associated with elevated levels of IL-18.124 The IL-18 is shown to be upregulated in the heart and consequently increasing the production of proinflammatory mediators (IL-1β, IL-8, TNF-α, and inducible nitric oxide synthase) in HF. IL-18 coupled with these mediators is associated with reduced contractility of the myocardium, increased β-adrenergic signaling, ECM remodeling, and apoptosis.124 Thus, inhibiting IL-18 may serve as a potential target for the treatment of HF. Recently, trials evaluating the safety and efficacy of the recombinant human IL-18BP (r-hIL-18BP) and agents blocking IL-18 antibody have been designed and initiated in healthy volunteers and patients with various conditions.125–127 Favorable safety profiles with mild-to-moderate adverse effects were observed in healthy volunteers and patients with moderate-to-severe rheumatoid arthritis or plaque psoriasis with r-hIL-18BP treatment.125–127 The longer elimination half-life of IL-18 neutralizing/blocking antibody is a primary advantage which may allow monthly or quarterly administration.
IL-converting enzyme inhibitor has been shown to attenuate inflammation and ultimately cardiac dysfunction in experimental diabetic cardiomyopathy.85 Moreover, a selective IL-converting enzyme inhibitor, pralnacasan, has demonstrated favorable effects in diabetic cardiomyopathy.128 This may present a novel therapeutic approach to HF.
IV immunoglobulin treatment lowers plasma levels of inflammatory mediators (eg, IL-8 and IL-1), upregulates anti-inflammatory mediators (eg, IL-10 and IL-1Ra), and ultimately improves EF and hemodynamic variables and exercise capacity in HF.129 However, IV immunoglobulin therapy did not reduce LV remodeling compared to placebo when 62 patients with myocardial dysfunction during hospitalization after acute myocardial infarction were randomized in a double-blinded fashion for 26 weeks.130
Celacade immune modulation therapy downregulates proinflammatory cytokines and upregulates anti-inflammatory cytokines by inducing apoptosis in human blood sample exposed to oxidative stress. The outcomes of animal and human studies evaluating celacade in HF and other conditions have so far been encouraging. Subsequently, celacade has been approved for use in the European Union for the treatment of HF in all NYHA class II patients and in classes II, III, and IV patients with no prior history of myocardial infarction.131,132
Novel approaches to targeting myocardial fibrosis and remodeling
Fibrosis of the myocardium is an important pathology in cardiac dysfunction. Matrix metalloproteinases (MMPs) play critical roles in myocardial fibrosis and remodeling by directly breaking down matrix components and subsequent release of biologically active factors from the ECM. Further, enhanced TNF-α and IL-1β expression is shown to be related to improved MMPs activity. Thus, altering the activities of MMPs by direct inhibition or anti-cytokine treatment could modulate myocardial remodeling and function.133,134 Several MMP inhibitors including batimastat, ilomastat, marimastat, and prinomastat have been developed for the treatment of HF.133 Treatment with batismastat reduces collagen synthesis and denaturation, prevents cardiac hypertrophy and dysfunction, and improves the survival of TNF1.6 HF mice.135 Moreover, a selective inhibitor of MMP – PG-53072 – was found to attenuate LV dysfunction and cardiac remodeling in the HF model.133 From these promising findings, further research to determine the efficacy of MMP inhibitors in humans with HF may be required.
TGF-β1 is critical to immune system suppression and tissue repair as well as inducing fibroblasts to produce and remodel ECM. Thus, broadly targeting TGF-β signaling may improve outcomes in HF.134 Experimental studies have shown that local angiotensin II generation correlates with expression and synthesis of TGF-β. Moreover, inducing TGF-β via stimulating angiotensin II type 1 receptor (AT1R) is important to developing myocardial fibrosis.134 Thus, ACE inhibitors may have suppressed TGF-β induction to improve outcomes in HF. Thalidomide reduces post-infarction myocardial remodeling and fibrosis via mechanisms that could plausibly be due to its direct anti-fibrotic effects on the expression of myocardial TGF-h1 gene leading to a sudden decrease in major stimuli for ECM remodeling. In addition, thalidomide may have directly affected the proliferation and survival of fibroblasts.136 These anti-fibrotic effects could present a potential treatment approach which needs to be harnessed to improve outcomes in HF.
Novel approaches to modulating metabolism
Myocardial metabolism may be normal in the early stages of HF but as disease advances, oxidative metabolism reduces, and glycolysis increases and as a consequence downregulating glucose and fatty acid oxidation.137 Fatty acid oxidation remains a dominant source of energy production in LV dysfunction. However, increased fatty acid oxidation inhibits glucose oxidation via the Randle cycle phenomenon which ultimately results in proton overload and intracellular acidosis and further decreases cardiac efficiency.137 Studies have shown that a shift of metabolism from fatty acid toward glucose oxidation improves myocardial contractility, thus slowing down the progression and improving the prognosis of HF.138 The growing evidence of substrate metabolism in HF has provided a strong impetus for thorough studies into optimizing myocardial energy metabolism as novel therapeutic targets.137,139 Several novel agents that modulate myocardial substrate metabolic pathways to optimize myocardial energy production have been developed. These agents include carnitine palmitoyl transferase (CPT) inhibitors (perhexiline maleate, etomoxir), malonyl CoA decarboxylase (MCD) inhibitors, 3-ketoacyl coenzyme A thiolase (3-KAT) inhibitors (trimetazidine [TMZ], ranolazine), pyruvate dehydrogenase kinase (PDK) inhibitors (dichloroacetate, DCA), incretins, and thiazolidinediones.
TMZ is a piperazine compound which is a clinically effective antianginal agent and selectively inhibits long-chain 3-KAT enzyme, which plays a critical role in the β-oxidation pathway. A number of RCTs have shown outcome benefits in both short- and long-term evaluation of TMZ in HF.140–144 These clinical studies show that TMZ improves cardiac function, clinical status, and survival in HF. Recent meta-analyses have indicated that TMZ improves clinical symptoms and cardiac function and reduces cardiovascular hospitalization and mortality in patients with HF.145–147 The meta-analyses revealed a compelling evidence for outcome benefits of TMZ in HFrEF since all RCTs included in the analysis enrolled patients with EF >50%.
Ranolazine is a piperazine derivative which is similar but less potent compared to TMZ. The cardioprotective properties of ranolazine are suggested to be mediated via stimulating glucose oxidation and partially blocking fatty acid oxidation through the inhibition of 3-KAT.137 In addition, there are suggestions that the observed beneficial effects with ranolazine are partly due to its ability to inhibit the late inward sodium current and thus averting the sodium-dependent Ca2+ overload in ischemic HF.137,148
CPT-1 catalyzes an important step in the mitochondrial uptake of long-chain fatty acids during fatty acid metabolism. CPT-1 inhibitors (etomoxir, oxfenicine, and perhexiline) suppress fatty acid oxidation while promoting glucose oxidation. Perhexiline maleate is an antianginal agent which inhibits CPT-1 and CPT-2 enzymes,149 to shift the use of muscle substrate from free fatty acids toward carbohydrates.150 A perhexiline-induced shift in metabolism improves cardiac function, exercise capacity, skeletal energetics, and quality of life in HF.151 However, perhexiline inhibition of CPT-1 leads to the accumulation of phospholipids, resulting in peripheral neuropathy and hepatotoxicity, which could be lessened by sustaining constant therapeutic concentrations.137,152 Etomoxir that was previously introduced as a potential hypoglycemic agent acts by inhibiting the enzyme CPT-1 which favors the oxidation of glucose over fatty acid during metabolism.137 Etomoxir improves the oxidation of glucose and reduces oxygen consumption in the myocardium while preserving contractile function in animal models.137 In a clinical study, etomoxir improved the LVEF and clinical status of patients with HF.153 However, this study had a major limitation of the lack of placebo as control. Subsequently, the Etomoxir for Recovery of Glucose Oxidation (ERGO) study randomized 347 patients with chronic HF to etomoxir (80 mg and 40 mg) or a matching placebo for 6 months. The ERGO study did not report any significant changes in exercise tolerance, ventricular dimensions, and quality-of-life assessment scores between treatment and placebo groups. However, ERGO was stopped prematurely, because investigators detected an unacceptably high liver transaminase levels in four patients in the treatment group.154
DCA inhibits pyruvate dehydrogenase (PDH) kinase, an enzyme which deactivates PDH in the mitochondrion. The PDH activation enhances carbohydrate oxidation partly by improving glycolysis and glucose oxidation coupling and as a result lessening acidosis and contractile dysfunction.4 Conversely, studies evaluating DCA failed to improve exercise tolerance and LVEF in patients with HF.155,156
MCD regulates fatty acid oxidation in the myocardium by altering intracellular levels of malonyl CoA. Agents inhibiting MCD increases the levels of malonyl CoA which in turn inhibit CPT-1, thus slowing down fatty acid oxidation.148 CBM-301940 inhibits MCD to speed up glucose oxidation and decelerate fatty acid oxidation.157 Moreover, a direct inhibitor of fatty acid oxidation – CVT-4325 – has been shown to improve cardiac work without increasing cardiac oxygen consumption, thus increasing myocardial efficiency.158
Novel approaches to oxidative stress reduction
Oxidative stress is a state characterized by excess production of reactive oxygen species (ROS) relative to antioxidant defense and plays important roles in the pathophysiology of LV remodeling and HF.159,160 Cyclooxygenase, cytochrome c oxidase, lipoxygenase, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, uncoupled nitric oxide synthase, and xanthine oxidase (XO) are likely ROS sources in HF.160 Excessive production of ROS causes DNA damage and protein and lipid peroxidation, which results in permanent cell damage or death, and these detrimental effects have been implicated in various cardiovascular conditions.160 In addition, ROS impairs contractile function by modulating proteins responsible for excitation–contraction pairing. Excess ROS cause apoptosis via activating diverse hypertrophy signaling kinases and transcription factors. Elevated ROS production leads to ECM remodeling by stimulating proliferation of cardiac fibroblasts and activating the MMPs.159,160 These cellular events play critical roles in the HF progression. Enzymatic (superoxide dismutase and catalase) and nonenzymatic antioxidants such as β-carotene, lipoic acid, vitamins C and E, and ubiquinone (coenzyme Q10) minimize the deleterious effects of oxidative stress.160 However, studies evaluating the proposed benefits of typical antioxidants such as vitamins C and E have been disappointing.161
Ubiquinone is depleted in HF and has been shown to independently predict mortality.160 Treatment with ubiquinone has marginally but significantly improved LV function in a few clinical studies.161 Further, long-term ubiquinone treatment has been found to reduce cardiovascular events in patients with moderate-to-severe HF who were randomized to ubiquinone 100 mg thrice daily or a matching placebo on optimal treatment for 2 years of follow-up.162 Edaravone (3-methyl-1-phenyl-2-pyrazolin-5-one) is a free radical scavenger, which was found to produce significant treatment benefits in various disease conditions, but its proposed beneficial effects in HF are less clear and yet to be evaluated.160,161
XO inhibitors – allopurinol and oxypurinol – suppress XO activity to improve myocardial function.161,163 XO inhibitors reduce ROS levels and decrease Na+/Ca2+ ion exchange but enhance SERCA2a production to increase myocardial contractility.163 Further, XO inhibition produces anti-inflammatory effects.160 Allopurinol treatment (acute and chronic) shows variable outcome benefits in HF. Acute IV administration improves LV function and ameliorates endothelial dysfunction, whereas chronic oral treatment did not improve exercise capacity.164 Thus, large clinical trials were required to evaluate the beneficial effect of XO inhibition in HF.161
Cingolani et al were the first to evaluate the inhibition of XO in congestive HF. This study was a single-center, double-blind, placebo-controlled, randomized trial of 60 patients with NYHA classes II–III congestive HF, comparing 600 mg daily oxypurinol with a matching placebo, in addition to optimal treatment for 1 month.165 The trial reported that XO inhibition by oxypurinol in congestive HF decreases serum uric acid (SUA) and improves LVEF in patients with LVEF ≤40% after 1 month of treatment.165
The Oxypurinol Compared to Placebo for classes III–IV NYHA Congestive HF (OPT-CHF) study is a proof-of-concept trial which randomized 405 patients with NYHA classes III–IV HF due to systolic dysfunction on optimal medical therapy to oxypurinol (600 mg/d) or a matching placebo for 24 weeks.166 Oxypurinol did not show any clinical benefit after 24 weeks of follow-up in moderate-to-severe HF. Nonetheless, further analysis suggests that oxypurinol shows outcome benefits in patients with higher SUA in a manner that correlates with the scale of SUA reduction.166
In a trial conducted by Nasr and Maurice,167 allopurinol (300 mg/d) was evaluated in comparison to a matching placebo in 59 patients with NYHA classes III–IV HF on standard treatment after 36 weeks of follow-up. In agreement with the earlier trial, allopurinol did not show any significant outcome benefits in patients with HF.167 However, further analysis suggest that allopurinol will be beneficial in patients with elevated SUA in a manner correlating with the degree of SUA reduction and thus SUA may serve as a valuable biomarker to target HF therapy.167
A more recent trial, the Effects of Xanthine Oxidase Inhibition in Hyperuricemic Heart Failure Patients (EXACT-HF) study tested the hypothesis that patients with HF with hyperuricemia may have composite clinical benefits with high-dose allopurinol treatment.168,169 The EXACT-HF, which is a multicenter, randomized (1:1), double-blind, placebo-controlled trial enrolled and randomly assigned 253 patients with symptomatic HF, LVEF ≤40%, and SUA levels ≥9.5 mg/dL to allopurinol 600 mg daily or placebo for 24 weeks. The primary composite endpoint was based on survival, worsening HF, and patient global assessment, whereas secondary endpoints were change in LVEF, exercise capacity, and quality of life at 24 weeks. SUA levels were significantly reduced with allopurinol treatment in comparison to placebo at 12 weeks and 24 weeks (both P<0.0001). However, allopurinol did not show any improvement in the major endpoints at 24 weeks in high-risk HFrEF and elevated SUA levels.169
Studies have shown that a cholesterol-lowering agent, probucol, reduces oxidative stress and myocardial remodeling and improves LV function.170–172 Despite the promising effects of probucol, there are no clinical trials evaluating its proposed beneficial properties in HF. ACE inhibitors, ARBs, and β-adrenoceptor blockers particularly β1 blockers (carvedilol, nebivolol) as well as statins have been proven to reduce oxidative stress and may have contributed to their improved outcomes in patients with HF.
Hydroxymethylglutaryl-coezyme A reductase inhibitor (statins)
Statins have demonstrated significant decrease in adverse outcomes in both primary173 and secondary174 prevention studies and remain the mainstay treatment for preventing HF as well as other cardiovascular conditions. Recent meta-analyses showed significant reductions in cardiovascular events with statin use notwithstanding of patients’ lipid profiles. These studies further demonstrated that the treatment benefit of statins exceeds identified risks.175,176
While the benefit of statin therapy in prevention is well established, their role in the treatment of “established” HF is less clear. A number of non-randomized studies reported that statin therapy improved clinical outcomes in patients with HF177–181 and benefits were also shown in several small RCTs identifying improved surrogate (cardiac function and inflammation) and mortality outcomes in HF.182–185 In contrast, two recent large RCTs – the Controlled Rosuvastatin Multinational Study in HF (CORONA)186 and Gruppo Italiano per lo Studio della Sopravvivenza nell’Insufficienza cardiaca Heart Failure (GISSI-HF)187 – did not show statistically significant outcome benefits in primary endpoints compared to placebo. Nonetheless, the CORONA trial reported a significant reduction in hospitalizations for worsening HF. Furthermore, secondary analyses of CORONA data showed improved survival in patients with low galectin-3188 and N-terminal prohormone brain natuiretic peptide (NT-proBNP)189 levels with rosuvastatin therapy.
Two meta-analyses of RCTs,190,191 which evaluated statin versus placebo, did not report any significant outcome benefit in HF. The outcomes of the two large trials together with these two meta-analyses appear to weigh against the evidence for statin treatment in HF. Nonetheless, a careful scrutiny of the RCTs in the meta-analyses show that the researchers did not compare effects of statin types utilized in each trial but treated them as a uniform class of drugs. Within the statins, there are two different types which can be identified by their hydrophilicity or lipophilicity. Rosuvastatin (hydrophilic statin) evaluated in CORONA and GISSI-HF is suggested to have played a part in the neutral outcome in both large RCTs and possibly skewed both meta-analyses toward the results of these two large trials.
Following CORONA and GISSI-HF, several studies still show statins to improve outcomes and prolong life in patients with HF.192–194 These studies, although non-randomized, again suggest that lipophilic statins may be better than hydrophilic statins in improving clinical outcomes in HF.2 Moreover, a meta-analysis that did not show improved overall survival with statin treatment demonstrated that, in subgroup analysis, lipophilic statins may improve survival in HF.190 Pharmacological properties inherent to the two statin types195,196 may have accounted for the differences in clinical outcomes seen in patients with HF. A closer observation of these data attributes the debatable outcomes obtained in HF trials to the hydrophilicity or lipophilicity of statins. Indeed, lipophilic statins have been shown to improve cardiac function and reduce inflammation better than hydrophilic statins in an indirect adjusted comparison meta-analysis involving ~6200 patients with HF from 19 RCTs.197,198 Given that the evidence from the comparisons favors lipophilic statin therapy, it is plausible that their superior pleiotropic effects coupled with cholesterol-lowering properties may have accounted for better clinical outcomes seen with many trials that evaluated lipophilic statins in HF and studies subsequent to CORONA and GISSI-HF.197,198 More recently, Bonsu et al199 have shown in another study involving ~11,000 patients in 13 RCTs that lipophilic statin treatment significantly decreases hospitalizations for worsening HF and all-cause and cardiovascular mortality compared to rosuvastatin treatment.
In addition to well-known effects, statins have shown important modulation of a number of proposed pathways of novel approaches being evaluated for HF treatments. Several experimental studies have shown that, aside well-established pleiotropic properties, statins modulate novel pathways in the pathogenesis of HF.200–204 Statins have earlier been proven to improve cardiac function via increased expression of SERCA and RyR2 gene and protein in rat hearts.205 Further, rosuvastatin therapy attenuated both structural and functional deterioration of decompensated heart in the spontaneously hypertensive rat models. These actions of rosuvastatin occurred together with decreases in collagen deposition but without changes in BP, LDL-C concentrations, or action potential prolongation, to prevent HF progression.203 Moreover, simvastatin treatment increased CaMKKβ-dependent phosphorylation of AMPK and LKB1 and enhanced Rac1 activity in vascular walls, establishing an important role for statin in activation Rac-1-dependent signaling to possibly attenuate arterial stiffness in HF with a preserved EF.48
In another spontaneously hypertensive rat model study, atorvastatin preserved cardiac dysfunction by positively altering calcium regulatory proteins and upregulating expression and activity of SERCA2a. Thus, the favorable effects of statins on calcium regulatory proteins may, in part, explain the mechanisms underlying their beneficial effect on cardiac function.206 Further, rosuvastatin has been shown to be effective in preventing LV remodeling and cardiac dysfunction via normalizing pSer16-phospholamban (PLB) levels, expression and activity of SERCA2a and SERCA, respectively, as well as elevating IL-10 levels and depleting IL-6 levels, independent of the lipid-lowering effect.207 Simvastatin was shown to prevent decreased SERCA2a activity as it inhibited sympathetic activity via β-adrenergic signaling. This occurred primarily by inhibiting sympathetic outflow and preventing reduced β1-Adrenoreceptor density and intracellular cAMP levels in nonischemic HF rabbit models.208
Recently, a number of studies have further demonstrated diverse mechanisms by which statins improve cardiac function in various HF models. Rosuvastatin therapy exerts cardioprotective effects in end-stage hypertension via dephosphorylation of protein kinase C (PKC)α/β2, causing resultant dephosphorylation of PPI-1 and augmented CaMKII expression. As a consequence, these mechanisms increases the expression of SERCA2a, RyR2, and Na+/Ca2− exchanger but decreases PLB expression to exhibit the beneficial effects seen with statin treatment in HF with hypertension.201
Rosuvastatin significantly reduces hypertrophy via AT1R-PKCβ2/α-ERK-c-fos pathway and prevents apoptosis of the myocardium via Akt-FOXO1, Bcl-2 family, and survivin pathways, consequently suppressing caspase-3 activity in the end-stage hypertensive heart disease.202 Statin impairs LV remodeling and improves cardiac function of diabetic rats via mechanisms which are directly linked to repair of Ca2+ dysregulation and recovery of repolarizing K+ currents resulting from the inhibition of RhoA signaling pathway, independent of cholesterol-lowering effects.209 Simvastatin has been proven to prevent the progression of LV hypertrophy in nonischemic HF model through the inhibition of the RhoA–PPARγ–NF-κB signaling axis associated with blockade of RhoA and Rho GTPase activity in the heart.210
In a more recent study, pitavastatin was found to ameliorate the cardiac dysfunction in mice with dilated cardiomyopathy by reversing the changes in the ratios of phosphorylated PLB to total PLB, SERCA2a, and RyR2 via reducing the plasma AngII concentration and the expressions of myocardium AT1R and PKCβ2. The possible underlying mechanism was proposed to be the regulation of myocardial AT1R-PKCβ2-Ca2+ handling proteins.200
In clinical studies, the debate continues about statin treatment in HF since their role remains controversial following neutral outcomes of the two large RCTs – CORONA and GISSI-HF – which evaluated low-dose rosuvastatin. Several studies subsequent to these large trials have shown improved outcomes in HF and thus have raised issues about the design and the type of statins evaluated in CORONA and GISSI-HF.2
A recent meta-analysis of 15 prospective studies (n=45,110) assessing statin treatment in both HF with a preserved EF and HFrEF211 showed a significant reduction in all-cause mortality (risk ratio [RR] 0.71, 95% confidence interval (CI) 0.61–0.83) and rehospitalization rate for HF (RR 0.84, 95% CI 0.74–0.96) with statin treatment.211 The study reported that atorvastatin treatment appeared to reduce all-cause mortality (RR 0.61, P=0.05) and rehospitalization for HF (RR 0.44, P=0.04) compared to other statin treatment.211 Moreover, statin treatment has recently been reported to be associated with the reduced risk of HF hospitalization, all-cause, and cardiovascular mortality in a nationwide population of patients with HF (n=21,864) from the Swedish HF Registry.212
Statin therapy is known to be clinically safe in HF contrary to earlier assertions and thus remains better alternative to be properly evaluated.175,176 Work on novel agents is worth commendation; however, these agents may not be clinically and commercially available until several years. In addition, new molecules require ample time to study and fully understand their safety in clinical use, thus making statins a better novel approach if they indeed modulate both conventional and several novel pathways to HF pathogenesis. Overall, statins have shown outcome benefits that require recommendation to treatment guidelines while we wait for novel agents that have shown huge promise in the treatment of HF to become clinically and commercially available. Statins may have significant outcome benefits for subgroups of patients with HF, if not all. Thus, a prospective study into whether statins improve outcomes in patients with HF and comorbid conditions such as diabetes where β-blocker treatment is contraindicated may be necessary. Further, the evaluation of statins on outcomes in categories of patients who are known to be underrepresented in major clinical trials such as patients of Black African descent whom the efficacy of mainstay treatments – ACE inhibitors/ARBs and β-blockers – remain controversial may be considered.
Disclosure
The authors report no conflicts of interest in this work.
References
McMurray JJ, Adamopoulos S, Anker SD, et al; ESC Committee for Practice Guidelines. ESC guidelines for the diagnosis and treatment of acute and chronic heart failure 2012: The Task Force for the diagnosis and treatment of acute and chronic heart failure 2012 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail. 2012;14(8):803–869. | ||
Bonsu KO, Kadirvelu A, Reidpath DD. Statins in heart failure: do we need another trial? Vasc Health Risk Manag. 2013;9:303–319. | ||
McMurray JJ. Clinical practice. Systolic heart failure. N Engl J Med. 2010;362(3):228–238. | ||
Patel C, Deoghare S. Heart failure: novel therapeutic approaches. J Postgrad Med. 2015;61(2):101–108. | ||
Mudd JO, Kass DA. Tackling heart failure in the twenty-first century. Nature. 2008;451(7181):919–928. | ||
Lipskaia L, Chemaly ER, Hadri L, Lompre A-M, Hajjar RJ. Sarcoplasmic reticulum Ca(2+) ATPase as a therapeutic target for heart failure. Expert Opin Biol Ther. 2010;10(1):29–41. | ||
Meyer M, Schillinger W, Pieske B, et al. Alterations of sarcoplasmic reticulum proteins in failing human dilated cardiomyopathy. Circulation. 1995;92(4):778–784. | ||
Chen Y, Escoubet B, Prunier F, et al. Constitutive cardiac overexpression of sarcoplasmic/endoplasmic reticulum Ca2+-ATPase delays myocardial failure after myocardial infarction in rats at a cost of increased acute arrhythmias. Circulation. 2004;109(15):1898–1903. | ||
Kushnir A, Marks AR. The ryanodine receptor in cardiac physiology and disease. Adv Pharmacol. 2010;59:1–30. | ||
Hasenfuss G, Teerlink JR. Cardiac inotropes: current agents and future directions. Eur Heart J. 2011;32(15):1838–1845. | ||
Braunwald E. The war against heart failure: the Lancet lecture. Lancet. 2015;385(9970):812–824. | ||
Greenberg B. Gene therapy for heart failure. J Cardiol. 2015;66(3):195–200. | ||
del Monte F, Lebeche D, Guerrero JL, et al. Abrogation of ventricular arrhythmias in a model of ischemia and reperfusion by targeting myocardial calcium cycling. Proc Natl Acad Sci U S A. 2004;101(15):5622–5627. | ||
Kiriazis H, Kranias EG. Genetically engineered models with alterations in cardiac membrane calcium-handling proteins. Annu Rev Physiol. 2000;62:321–351. | ||
Periasamy M, Huke S. SERCA pump level is a critical determinant of Ca(2+)homeostasis and cardiac contractility. J Mol Cell Cardiol. 2001;33(6):1053–1063. | ||
Tilemann L, Ishikawa K, Weber T, Hajjar RJ. Gene therapy for heart failure. Circ Res. 2012;110(5):777–793. | ||
Miyamoto MI, del Monte F, Schmidt U, et al. Adenoviral gene transfer of SERCA2a improves left-ventricular function in aortic-banded rats in transition to heart failure. Proc Natl Acad Sci U S A. 2000;97(2):793–798. | ||
Kawase Y, Ly HQ, Prunier F, et al. Reversal of cardiac dysfunction after long-term expression of SERCA2a by gene transfer in a pre-clinical model of heart failure. J Am Coll Cardiol. 2008;51(11):1112–1119. | ||
Beeri R, Chaput M, Guerrero JL, et al. Gene delivery of sarcoplasmic reticulum calcium ATPase inhibits ventricular remodeling in ischemic mitral regurgitation. Circ Heart Fail. 2010;3(5):627–634. | ||
Byrne MJ, Power JM, Preovolos A, Mariani JA, Hajjar RJ, Kaye DM. Recirculating cardiac delivery of AAV2/1SERCA2a improves myocardial function in an experimental model of heart failure in large animals. Gene Ther. 2008;15(23):1550–1557. | ||
Jaski BE, Jessup ML, Mancini DM, et al; Calcium Up-Regulation by Percutaneous Administration of Gene Therapy In Cardiac Disease (CUPID) Trial Investigators. Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID Trial), a first-in-human phase 1/2 clinical trial. J Card Fail. 2009;15(3):171–181. | ||
Jessup M, Greenberg B, Mancini D, et al; Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID) Investigators. Calcium upregulation by percutaneous administration of gene Therapy in cardiac disease (CUPID): a phase 2 trial of intracoronary gene therapy of sarcoplasmic reticulum Ca2+-ATPase in patients with advanced heart failure. Circulation. 2011;124(3):304–313. | ||
Zsebo K, Yaroshinsky A, Rudy JJ, et al. Long-term effects of AAV1/SERCA2a gene transfer in patients with severe heart failure: analysis of recurrent cardiovascular events and mortality. Circ Res. 2014;114(1):101–108. | ||
Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415(6868):198–205. | ||
Marks AR, Tempst P, Hwang KS, et al. Molecular cloning and characterization of the ryanodine receptor/junctional channel complex cDNA from skeletal muscle sarcoplasmic reticulum. Proc Natl Acad Sci U S A. 1989;86(22):8683–8687. | ||
Nakai J, Imagawa T, Hakamat Y, Shigekawa M, Takeshima H, Numa S. Primary structure and functional expression from cDNA of the cardiac ryanodine receptor/calcium release channel. FEBS Lett. 1990;271(1–2):169–177. | ||
Hakamata Y, Nakai J, Takeshima H, Imoto K. Primary structure and distribution of a novel ryanodine receptor/calcium release channel from rabbit brain. FEBS Lett. 1992;312(2–3):229–235. | ||
Zalk R, Lehnart SE, Marks AR. Modulation of the ryanodine receptor and intracellular calcium. Annu Rev Biochem. 2007;76:367–385. | ||
Bers DM. Macromolecular complexes regulating cardiac ryanodine receptor function. J Mol Cell Cardiol. 2004;37(2):417–429. | ||
Andersson DC, Marks AR. Fixing ryanodine receptor Ca leak – a novel therapeutic strategy for contractile failure in heart and skeletal muscle. Drug Discov Today Dis Mech. 2010;7(2):e151–e157. | ||
Marx SO, Reiken S, Hisamatsu Y, et al. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101(4):365–376. | ||
Guo T, Zhang T, Mestril R, Bers DM. Ca2+/Calmodulin-dependent protein kinase II phosphorylation of ryanodine receptor does affect calcium sparks in mouse ventricular myocytes. Circ Res. 2006;99(4):398–406. | ||
Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ Res. 2005;97(12):1314–1322. | ||
Ling H, Zhang T, Pereira L, et al. Requirement for Ca2+/calmodulin-dependent kinase II in the transition from pressure overload-induced cardiac hypertrophy to heart failure in mice. J Clin Invest. 2009;119(5):1230–1240. | ||
van Oort RJ, Respress JL, Li N, et al. Accelerated development of pressure overload-induced cardiac hypertrophy and dysfunction in an RyR2-R176Q knockin mouse model. Hypertension. 2010;55(4):932–938. | ||
Respress JL, van Oort RJ, Li N, et al. Role of RyR2 phosphorylation at S2814 during heart failure progression. Circ Res. 2012;110(11):1474–1483. | ||
Anderson ME. CaMKII and a failing strategy for growth in heart. J Clin Invest. 2009;119(5):1082–1085. | ||
Ter Keurs HE, Boyden PA. Calcium and arrhythmogenesis. Physiol Rev. 2007;87(2):457–506. | ||
Yano M, Kobayashi S, Kohno M, et al. FKBP12.6-mediated stabilization of calcium-release channel (ryanodine receptor) as a novel therapeutic strategy against heart failure. Circulation. 2003;107(3):477–484. | ||
Toischer K, Lehnart SE, Tenderich G, et al. K201 improves aspects of the contractile performance of human failing myocardium via reduction in Ca2+ leak from the sarcoplasmic reticulum. Basic Res Cardiol. 2010;105(2):279–287. | ||
Kimura J, Kawahara M, Sakai E, Yatabe J, Nakanishi H. Effects of a novel cardioprotective drug, JTV-519, on membrane currents of guinea pig ventricular myocytes. Jpn J Pharmacol. 1999;79(3):275–281. | ||
Lehnart SE, Mongillo M, Bellinger A, et al. Leaky Ca2+ release channel/ryanodine receptor 2 causes seizures and sudden cardiac death in mice. J Clin Invest. 2008;118(6):2230–2245. | ||
Shan J, Betzenhauser MJ, Kushnir A, et al. Role of chronic ryanodine receptor phosphorylation in heart failure and beta-adrenergic receptor blockade in mice. J Clin Invest. 2010;120(12):4375–4387. | ||
Blayney LM, Lai FA. Ryanodine receptor-mediated arrhythmias and sudden cardiac death. Pharmacol Ther. 2009;123(2):151–177. | ||
Bers DM. CaMKII inhibition in heart failure makes jump to human. Circ Res. 2010;107(9):1044–1046. | ||
Sossalla S, Fluschnik N, Schotola H, et al. Inhibition of elevated Ca2+/calmodulin-dependent protein kinase II improves contractility in human failing myocardium. Circ Res. 2010;107(9):1150–1161. | ||
Bristow MR, Ginsburg R, Minobe W, et al. Decreased catecholamine sensitivity and beta-adrenergic-receptor density in failing human hearts. N Engl J Med. 1982;307(4):205–211. | ||
Kou R, Sartoretto J, Michel T. Regulation of Rac1 by simvastatin in endothelial cells: differential roles of AMP-activated protein kinase and calmodulin-dependent kinase kinase-beta. J Biol Chem. 2009;284(22):14734–14743. | ||
Bublil EM, Yarden Y. The EGF receptor family: spearheading a merger of signaling and therapeutics. Curr Opin Cell Biol. 2007;19(2):124–134. | ||
Sawyer DB, Caggiano A. Neuregulin-1beta for the treatment of systolic heart failure. J Mol Cell Cardiol. 2011;51(4):501–505. | ||
Meyer D, Birchmeier C. Multiple essential functions of neuregulin in development. Nature. 1995;378(6555):386–390. | ||
Zhao YY, Sawyer DR, Baliga RR, et al. Neuregulins promote survival and growth of cardiac myocytes. Persistence of ErbB2 and ErbB4 expression in neonatal and adult ventricular myocytes. J Biol Chem. 1998;273(17):10261–10269. | ||
Guglin M, Cutro R, Mishkin JD. Trastuzumab-induced cardiomyopathy. J Card Fail. 2008;14(5):437–444. | ||
Slamon DJ, Leyland-Jones B, Shak S, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344(11):783–792. | ||
Lemmens K, Segers VF, Demolder M, De Keulenaer GW. Role of neuregulin-1/ErbB2 signaling in endothelium-cardiomyocyte cross-talk. J Biol Chem. 2006;281(28):19469–19477. | ||
Liu X, Gu X, Li Z, et al. Neuregulin-1/erbB-activation improves cardiac function and survival in models of ischemic, dilated, and viral cardiomyopathy. J Am Coll Cardiol. 2006;48(7):1438–1447. | ||
Timolati F, Ott D, Pentassuglia L, et al. Neuregulin-1 beta attenuates doxorubicin-induced alterations of excitation-contraction coupling and reduces oxidative stress in adult rat cardiomyocytes. J Mol Cell Cardiol. 2006;41(5):845–854. | ||
Gao R, Zhang J, Cheng L, et al. A Phase II, randomized, double-blind, multicenter, based on standard therapy, placebo-controlled study of the efficacy and safety of recombinant human neuregulin-1 in patients with chronic heart failure. J Am Coll Cardiol. 2010;55(18):1907–1914. | ||
Packer M. Pathophysiology of chronic heart failure. Lancet. 1992;340(8811):88–92. | ||
Latini R, Masson S, Anand I, et al. The comparative prognostic value of plasma neurohormones at baseline in patients with heart failure enrolled in Val-HeFT. Eur Heart J. 2004;25(4):292–299. | ||
Vergaro G, Emdin M, Iervasi A, et al. Prognostic value of plasma renin activity in heart failure. Am J Cardiol. 2011;108(2):246–251. | ||
Krum H. Role of renin in heart failure and therapeutic potential of direct renin inhibition. J Renin Angiotensin Aldosterone Syst. 2008;9(3):177–180. | ||
Nguyen G. The (pro)renin receptor: pathophysiological roles in cardiovascular and renal pathology. Curr Opin Nephrol Hypertens. 2007;16(2):129–133. | ||
Neuberg GW, Kukin ML, Penn J, Medina N, Yushak M, Packer M. Hemodynamic effects of renin inhibition by enalkiren in chronic congestive heart failure. Am J Cardiol. 1991;67(1):63–66. | ||
Kiowski W, Beermann J, Rickenbacher P, et al. Angiotensinergic versus nonangiotensinergic hemodynamic effects of converting enzyme inhibition in patients with chronic heart failure. Assessment by acute renin and converting enzyme inhibition. Circulation. 1994;90(6):2748–2756. | ||
Seed A, Gardner R, McMurray J, et al. Neurohumoral effects of the new orally active renin inhibitor, aliskiren, in chronic heart failure. Eur J Heart Fail. 2007;9(11):1120–1127. | ||
McMurray JJ, Pitt B, Latini R, et al; Aliskiren Observation of Heart Failure Treatment (ALOFT) Investigators. Effects of the oral direct renin inhibitor aliskiren in patients with symptomatic heart failure. Circ Heart Fail. 2008;1(1):17–24. | ||
Gheorghiade M, Albaghdadi M, Zannad F, et al; ASTRONAUT investigators and study coordinators. Rationale and design of the multicentre, randomized, double-blind, placebo-controlled Aliskiren trial on acute heart failure outcomes (ASTRONAUT). Eur J Heart Fail. 2011;13(1):100–106. | ||
Gheorghiade M, Bohm M, Greene SJ, et al; ASTRONAUT Investigators and Coordinators. Effect of aliskiren on postdischarge mortality and heart failure readmissions among patients hospitalized for heart failure: the ASTRONAUT randomized trial. JAMA. 2013;309(11):1125–1135. | ||
Krum H, Massie B, Abraham WT, et al; ATMOSPHERE Investigators. Direct renin inhibition in addition to or as an alternative to angiotensin converting enzyme inhibition in patients with chronic systolic heart failure: rationale and design of the Aliskiren Trial to Minimize OutcomeS in Patients with HEart failuRE (ATMOSPHERE) study. Eur J Heart Fail. 2011;13(1):107–114. | ||
Parving HH, Brenner BM, McMurray JJ, et al. Aliskiren trial in type 2 diabetes using cardio-renal endpoints (ALTITUDE): rationale and study design. Nephrol Dial Transplant. 2009;24(5):1663–1671. | ||
O’Connor CM, Starling RC, Hernandez AF, et al. Effect of nesiritide in patients with acute decompensated heart failure. N Engl J Med. 2011;365(1):32–43. | ||
Bernardo WM, Moreira FT. Does nesiritide reduce mortality and readmission in decompensated heart failure? Rev Assoc Méd Bras. 2012;58(2):133–134. | ||
Wylie JV, Tsao L. Nesiritide for the treatment of decompensated heart failure. Expert Rev Cardiovasc Ther. 2004;2(6):803–813. | ||
Nambi P, Clozel M, Feuerstein G. Endothelin and heart failure. Heart Fail Rev. 2001;6(4):335–340. | ||
Ertl G, Bauersachs J. Endothelin receptor antagonists in heart failure. Drugs. 2004;64(10):1029–1040. | ||
Wei CM, Lerman A, Rodeheffer RJ, et al. Endothelin in human congestive heart failure. Circulation. 1994;89(4):1580–1586. | ||
Pacher R, Bergler-Klein J, Globits S, et al. Plasma big endothelin-1 concentrations in congestive heart failure patients with or without systemic hypertension. Am J Cardiol. 1993;71(15):1293–1299. | ||
Cody RJ, Haas GJ, Binkley PF, Capers Q, Kelley R. Plasma endothelin correlates with the extent of pulmonary hypertension in patients with chronic congestive heart failure. Circulation. 1992;85(2):504–509. | ||
Mulder P, Richard V, Derumeaux G, et al. Role of endogenous endothelin in chronic heart failure: effect of long-term treatment with an endothelin antagonist on survival, hemodynamics, and cardiac remodeling. Circulation. 1997;96(6):1976–1982. | ||
Fraccarollo D, Hu K, Galuppo P, Gaudron P, Ertl G. Chronic endothelin receptor blockade attenuates progressive ventricular dilation and improves cardiac function in rats with myocardial infarction: possible involvement of myocardial endothelin system in ventricular remodeling. Circulation. 1997;96(11):3963–3973. | ||
Sutsch G, Kiowski W, Yan XW, et al. Short-term oral endothelin-receptor antagonist therapy in conventionally treated patients with symptomatic severe chronic heart failure. Circulation. 1998;98(21):2262–2268. | ||
Mylona P, Cleland JG. Update of REACH-1 and MERIT-HF clinical trials in heart failure. Cardio.net Editorial Team. Eur J Heart Fail. 1999;1(2):197–200. | ||
Kalra PR, Moon JC, Coats AJ. Do results of the ENABLE (endothelin antagonist bosentan for lowering cardiac events in heart failure) study spell the end for non-selective endothelin antagonism in heart failure? Int J Cardiol. 2002;85(2–3):195–197. | ||
Balakumar P, Singh M. Recent advances in pharmacotherapy for heart failure: future directions. Trends Med Res. 2007;2:61–71. | ||
Cleland JG, Swedberg K. Lack of efficacy of neutral endopeptidase inhibitor ecadotril in heart failure. The International Ecadotril Multi-centre Dose-ranging Study Investigators. Lancet. 1998;351(9116):1657–1658. | ||
Rouleau JL, Pfeffer MA, Stewart DJ, et al. Comparison of vasopeptidase inhibitor, omapatrilat, and lisinopril on exercise tolerance and morbidity in patients with heart failure: IMPRESS randomised trial. Lancet. 2000;356(9230):615–620. | ||
Packer M, Califf RM, Konstam MA, et al. Comparison of omapatrilat and enalapril in patients with chronic heart failure: the omapatrilat versus enalapril randomized trial of utility in reducing events (OVERTURE). Circulation. 2002;106(8):920–926. | ||
Messerli FH, Nussberger J. Vasopeptidase inhibition and angio-oedema. Lancet. 2000;356(9230):608–609. | ||
Gu J, Noe A, Chandra P, et al. Pharmacokinetics and pharmacodynamics of LCZ696, a novel dual-acting angiotensin receptor-neprilysin inhibitor (ARNi). J Clin Pharmacol. 2010;50(4):401–414. | ||
Hegde LG, Yu C, Renner T, et al. Concomitant angiotensin AT1 receptor antagonism and neprilysin inhibition produces omapatrilat-like antihypertensive effects without promoting tracheal plasma extravasation in the rat. J Cardiovasc Pharmacol. 2011;57(4):495–504. | ||
Waeber B, Feihl F. Blood-pressure reduction with LCZ696. Lancet. 2010;375(9722):1228–1229. | ||
Ruilope LM, Dukat A, Bohm M, Lacourciere Y, Gong J, Lefkowitz MP. Blood-pressure reduction with LCZ696, a novel dual-acting inhibitor of the angiotensin II receptor and neprilysin: a randomised, double-blind, placebo-controlled, active comparator study. Lancet. 2010;375(9722):1255–1266. | ||
Solomon SD, Zile M, Pieske B, et al; Prospective comparison of ARNI with ARB on Management Of heart failUre with preserved ejectioN fracTion (PARAMOUNT) Investigators. The angiotensin receptor neprilysin inhibitor LCZ696 in heart failure with preserved ejection fraction: a phase 2 double-blind randomised controlled trial. Lancet. 2012;380(9851):1387–1395. | ||
Packer M, McMurray JJ, Desai AS, et al; PARADIGM-HF Investigators and Coordinators. Angiotensin receptor neprilysin inhibition compared with enalapril on the risk of clinical progression in surviving patients with heart failure. Circulation. 2015;131(1):54–61. | ||
McMurray JJV, Packer M, Desai AS, et al; PARADIGM-HF Investigators and Committees. Angiotensin–neprilysin inhibition versus enalapril in heart failure. New England Journal of Medicine. 2014;371(11):993–1004. | ||
Lisy O, Huntley BK, McCormick DJ, Kurlansky PA, Burnett JC Jr. Design, synthesis, and actions of a novel chimeric natriuretic peptide: CD-NP. J Am Coll Cardiol. 2008;52(1):60–68. | ||
Rose RA. CD-NP, a chimeric natriuretic peptide for the treatment of heart failure. Curr Opin Investig Drugs. 2010;11(3):349–356. | ||
Chatterjee K. Neurohormonal activation in congestive heart failure and the role of vasopressin. Am J Cardiol. 2005;95(9, suppl 1):8–13. | ||
Mondritzki T, Kolkhof P, Sabbah HN, et al. Differentiation of arginine vasopressin antagonistic effects by selective V2 versus dual V2/V1a receptor blockade in a preclinical heart failure model. Am J Ther. 2011;18(1):31–37. | ||
Veeraveedu PT, Watanabe K, Ma M, et al. Effects of nonpeptide vasopressin V2 antagonist tolvaptan in rats with heart failure. Biochem Pharmacol. 2007;74(10):1466–1475. | ||
Gheorghiade M, Niazi I, Ouyang J, et al; Tolvaptan Investigators. Vasopressin V2-receptor blockade with tolvaptan in patients with chronic heart failure: results from a double-blind, randomized trial. Circulation. 2003;107(21):2690–2696. | ||
Torp-Pedersen C, Kober L, Carlsen JE, et al. A randomised trial of a pre-synaptic stimulator of DA2-dopaminergic and alpha2-adrenergic receptors on morbidity and mortality in patients with heart failure. Eur J Heart Fail. 2008;10(1):89–95. | ||
Pasini E, Cargnioni A, Pastore F, et al. Effect of nolomirole on monocrotaline-induced heart failure. Pharmacol Res. 2004;49(1):1–5. | ||
Birkeland JA, Sjaastad I, Brattelid T, et al. Effects of treatment with a 5-HT4 receptor antagonist in heart failure. Br J Pharmacol. 2007;150(2):143–152. | ||
Kjekshus JK, Torp-Pedersen C, Gullestad L, et al. Effect of piboserod, a 5-HT4 serotonin receptor antagonist, on left ventricular function in patients with symptomatic heart failure. Eur J Heart Fail. 2009;11(8):771–778. | ||
Bramlage P, Turgonyi E, Montalescot G. Aldosterone blockade: current research and future trends. Eur Heart J Suppl. 2011;13(suppl B):B46–B50. | ||
Meyers MJ, Arhancet GB, Hockerman SL, et al. Discovery of (3S,3aR)-2-(3-chloro-4-cyanophenyl)-3-cyclopentyl-3,3a,4,5-tetrahydro-2H-benzo[g]indazole-7-carboxylic Acid (PF-3882845), an orally efficacious mineralocorticoid receptor (MR) antagonist for hypertension and nephropathy. J Med Chem. 2010;53(16):5979–6002. | ||
Fagart J, Hillisch A, Huyet J, et al. A new mode of mineralocorticoid receptor antagonism by a potent and selective nonsteroidal molecule. J Biol Chem. 2010;285(39):29932–29940. | ||
Le Mercier A, Barrera-Chimal J, Prince S, et al. 0278: mineralocorticoid receptor antagonism with BR-4628 protects against renal injury induced by ischemia/reperfusion. Arch Cardiovasc Dis Suppl. 2015;7(2):152. | ||
Kolkhof P, Borden SA. Molecular pharmacology of the mineralocorticoid receptor: prospects for novel therapeutics. Mol Cell Endocrinol. 2012;350(2):310–317. | ||
Barfacker L, Kuhl A, Hillisch A, et al. Discovery of BAY 94-8862: a nonsteroidal antagonist of the mineralocorticoid receptor for the treatment of cardiorenal diseases. ChemMedChem. 2012;7(8):1385–1403. | ||
Pitt B, Kober L, Ponikowski P, et al. Safety and tolerability of the novel non-steroidal mineralocorticoid receptor antagonist BAY 94-8862 in patients with chronic heart failure and mild or moderate chronic kidney disease: a randomized, double-blind trial. Eur Heart J. 2013;34(31):2453–2463. | ||
Mulder P, Mellin V, Favre J, et al. Aldosterone synthase inhibition improves cardiovascular function and structure in rats with heart failure: a comparison with spironolactone. Eur Heart J. 2008;29(17):2171–2179. | ||
Calhoun DA, White WB, Krum H, et al. Effects of a novel aldosterone synthase inhibitor for treatment of primary hypertension: results of a randomized, double-blind, placebo- and active-controlled phase 2 trial. Circulation. 2011;124(18):1945–1955. | ||
Hofmann U, Frantz S. How can we cure a heart “in flame”? A translational view on inflammation in heart failure. Basic Res Cardiol. 2013;108(4):356. | ||
Heymans S, Hirsch E, Anker SD, et al. Inflammation as a therapeutic target in heart failure? A scientific statement from the Translational Research Committee of the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail. 2009;11(2):119–129. | ||
Woolacott N, Bravo Vergel Y, Hawkins N, et al. Etanercept and infliximab for the treatment of psoriatic arthritis: a systematic review and economic evaluation. Health Technol Assess. 2006;10(31):iii–iv, xiii–xvi, 1–239. | ||
Balakumar P, Singh M. Anti-tumour necrosis factor-alpha therapy in heart failure: future directions. Basic Clin Pharmacol Toxicol. 2006;99(6):391–397. | ||
Kerkela R, Force T. p38 mitogen-activated protein kinase: a future target for heart failure therapy? J Am Coll Cardiol. 2006;48(3):556–558. | ||
Bellahcene M, Jacquet S, Cao XB, et al. Activation of p38 mitogen-activated protein kinase contributes to the early cardiodepressant action of tumor necrosis factor. J Am Coll Cardiol. 2006;48(3):545–555. | ||
Shaw SM, Shah MK, Williams SG, Fildes JE. Immunological mechanisms of pentoxifylline in chronic heart failure. Eur J Heart Fail. 2009;11(2):113–118. | ||
Mallat Z, Henry P, Fressonnet R, et al. Increased plasma concentrations of interleukin-18 in acute coronary syndromes. Heart. 2002;88(5):467–469. | ||
Mallat Z, Heymes C, Corbaz A, et al. Evidence for altered interleukin 18 (IL)-18 pathway in human heart failure. FASEB J. 2004;18(14):1752–1754. | ||
O’Brien LC, Mezzaroma E, Van Tassell BW, et al. Interleukin-18 as a therapeutic target in acute myocardial infarction and heart failure. Mol Med. 2014;20:221–229. | ||
Tak PP, Bacchi M, Bertolino M. Pharmacokinetics of IL-18 binding protein in healthy volunteers and subjects with rheumatoid arthritis or plaque psoriasis. Eur J Drug Metab Pharmacokinet. 2006;31(2):109–116. | ||
Marchetti C, Chojnacki J, Toldo S, et al. A novel pharmacologic inhibitor of the NLRP3 inflammasome limits myocardial injury after ischemia-reperfusion in the mouse. J Cardiovasc Pharmacol. 2014;63(4):316–322. | ||
Westermann D, Van Linthout S, Dhayat S, et al. Cardioprotective and anti-inflammatory effects of interleukin converting enzyme inhibition in experimental diabetic cardiomyopathy. Diabetes. 2007;56(7):1834–1841. | ||
Aukrust P, Damas JK, Gullestad L. Immunomodulating therapy: new treatment modality in congestive heart failure. Congest Heart Fail. 2003;9(2):64–69. | ||
Gullestad L, Orn S, Dickstein K, et al. Intravenous immunoglobulin does not reduce left ventricular remodeling in patients with myocardial dysfunction during hospitalization after acute myocardial infarction. Int J Cardiol. 2013;168(1):212–218. | ||
Sporter RJ, Kim JH, Frishman WH. Device-based nonspecific immunomodulation therapy (Celacade), and its potential role in the treatment of chronic heart failure. Cardiol Rev. 2008;16(6):280–287. | ||
Cleland JG, Coletta AP, Clark AL. Clinical trials update from the joint European Society and World Congress of Cardiology meeting: PEP-CHF, ACCLAIM and the HHH study. Eur J Heart Fail. 2006;8(6):658–661. | ||
Kaludercic N, Lindsey ML, Tavazzi B, Lazzarino G, Paolocci N. Inhibiting metalloproteases with PD 166793 in heart failure: impact on cardiac remodeling and beyond. Cardiovasc Ther. 2008;26(1):24–37. | ||
Li YY, McTiernan CF, Feldman AM. Interplay of matrix metalloproteinases, tissue inhibitors of metalloproteinases and their regulators in cardiac matrix remodeling. Cardiovasc Res. 2000;46(2):214–224. | ||
Li YY, Kadokami T, Wang P, McTiernan CF, Feldman AM. MMP inhibition modulates TNF-alpha transgenic mouse phenotype early in the development of heart failure. Am J Physiol Heart Circ Physiol. 2002;282(3):H983–H989. | ||
Yndestad A, Vinge LE, Bjornerheim R, et al. Thalidomide attenuates the development of fibrosis during post-infarction myocardial remodelling in rats. Eur J Heart Fail. 2006;8(8):790–796. | ||
Palaniswamy C, Mellana WM, Selvaraj DR, Mohan D. Metabolic modulation: a new therapeutic target in treatment of heart failure. Am J Ther. 2011;18(6):e197–e201. | ||
Di Napoli P, Barsotti A. Prognostic relevance of metabolic approach in patients with heart failure. Curr Pharm Des. 2009;15(8):883–892. | ||
Lee L, Horowitz J, Frenneaux M. Metabolic manipulation in ischaemic heart disease, a novel approach to treatment. Eur Heart J. 2004;25(8):634–641. | ||
Belardinelli R, Cianci G, Gigli M, Mazzanti M, Lacalaprice F. Effects of trimetazidine on myocardial perfusion and left ventricular systolic function in type 2 diabetic patients with ischemic cardiomyopathy. J Cardiovasc Pharmacol. 2008;51(6):611–615. | ||
El-Kady T, El-Sabban K, Gabaly M, Sabry A, Abdel-Hady S. Effects of trimetazidine on myocardial perfusion and the contractile response of chronically dysfunctional myocardium in ischemic cardiomyopathy: a 24-month study. Am J Cardiovasc Drugs. 2005;5(4):271–278. | ||
Di Napoli P, Di Giovanni P, Gaeta MA, D’Apolito G, Barsotti A. Beneficial effects of trimetazidine treatment on exercise tolerance and B-type natriuretic peptide and troponin T plasma levels in patients with stable ischemic cardiomyopathy. Am Heart J. 2007;154(3):e601–e605. | ||
Fragasso G, Palloshi A, Puccetti P, et al. A randomized clinical trial of trimetazidine, a partial free fatty acid oxidation inhibitor, in patients with heart failure. J Am Coll Cardiol. 2006;48(5):992–998. | ||
Thrainsdottir IS, von Bibra H, Malmberg K, Ryden L. Effects of trimetazidine on left ventricular function in patients with type 2 diabetes and heart failure. J Cardiovasc Pharmacol. 2004;44(1):101–108. | ||
Zhang L, Lu Y, Jiang H, Sun A, Zou Y, Ge J. Additional use of trimetazidine in patients with chronic heart failure: a meta-analysis. J Am Coll Cardiol. 2012;59(10):913–922. | ||
Zhou X, Chen J. Is treatment with trimetazidine beneficial in patients with chronic heart failure? PLoS One. 2014;9(5):e94660. | ||
Gao D, Ning N, Niu X, Hao G, Meng Z. Trimetazidine: a meta-analysis of randomised controlled trials in heart failure. Heart. 2011;97(4):278–286. | ||
Dyck JR, Cheng JF, Stanley WC, et al. Malonyl coenzyme a decarboxylase inhibition protects the ischemic heart by inhibiting fatty acid oxidation and stimulating glucose oxidation. Circ Res. 2004;94(9):e78–e84. | ||
Kennedy JA, Kiosoglous AJ, Murphy GA, Pelle MA, Horowitz JD. Effect of perhexiline and oxfenicine on myocardial function and metabolism during low-flow ischemia/reperfusion in the isolated rat heart. J Cardiovasc Pharmacol. 2000;36(6):794–801. | ||
Jeffrey FM, Alvarez L, Diczku V, Sherry AD, Malloy CR. Direct evidence that perhexiline modifies myocardial substrate utilization from fatty acids to lactate. J Cardiovasc Pharmacol. 1995;25(3):469–472. | ||
Lee L, Campbell R, Scheuermann-Freestone M, et al. Metabolic modulation with perhexiline in chronic heart failure: a randomized, controlled trial of short-term use of a novel treatment. Circulation. 2005;112(21):3280–3288. | ||
Wang P, Fraser H, Lloyd SG, McVeigh JJ, Belardinelli L, Chatham JC. A comparison between ranolazine and CVT-4325, a novel inhibitor of fatty acid oxidation, on cardiac metabolism and left ventricular function in rat isolated perfused heart during ischemia and reperfusion. J Pharmacol Exp Ther. 2007;321(1):213–220. | ||
Schmidt-Schweda S, Holubarsch C. First clinical trial with etomoxir in patients with chronic congestive heart failure. Clin Sci (London). 2000;99(1):27–35. | ||
Holubarsch CJ, Rohrbach M, Karrasch M, et al. A double-blind randomized multicentre clinical trial to evaluate the efficacy and safety of two doses of etomoxir in comparison with placebo in patients with moderate congestive heart failure: the ERGO (etomoxir for the recovery of glucose oxidation) study. Clin Sci (London). 2007;113(4):205–212. | ||
Lewis JF, DaCosta M, Wargowich T, Stacpoole P. Effects of dichloroacetate in patients with congestive heart failure. Clin Cardiol. 1998;21(12):888–892. | ||
Wilson JR, Mancini DM, Ferraro N, Egler J. Effect of dichloroacetate on the exercise performance of patients with heart failure. J Am Coll Cardiol. 1988;12(6):1464–1469. | ||
Cheng JF, Huang Y, Penuliar R, et al. Discovery of potent and orally available malonyl-CoA decarboxylase inhibitors as cardioprotective agents. J Med Chem. 2006;49(14):4055–4058. | ||
Wu L, Belardinelli L, Fraser H. A novel partial fatty acid oxidation inhibitor decreases myocardial oxygen consumption and improves cardiac efficiency in demand-induced ischemic heart. J Cardiovasc Pharmacol. 2008;51(4):372–379. | ||
Tsutsui H, Kinugawa S, Matsushima S. Oxidative stress and heart failure. Am J Physiol Heart Circ Physiol. 2011;301(6):H2181–H2190. | ||
Lakshmi SV, Padmaja G, Kuppusamy P, Kutala VK. Oxidative stress in cardiovascular disease. Indian J Biochem Biophys. 2009;46(6):421–440. | ||
Hajjar RJ, Leopold JA. Xanthine oxidase inhibition and heart failure: novel therapeutic strategy for ventricular dysfunction? Circ Res. 2006;98(2):169–171. | ||
Mortensen SA, Rosenfeldt F, Kumar A, et al; Q-SYMBIO Study Investigators. The effect of coenzyme Q10 on morbidity and mortality in chronic heart failure: results from Q-SYMBIO: a randomized double-blind trial. JACC Heart Fail. 2014;2(6):641–649. | ||
Minhas KM, Saraiva RM, Schuleri KH, et al. Xanthine oxidoreductase inhibition causes reverse remodeling in rats with dilated cardiomyopathy. Circ Res. 2006;98(2):271–279. | ||
Cappola TP, Kass DA, Nelson GS, et al. Allopurinol improves myocardial efficiency in patients with idiopathic dilated cardiomyopathy. Circulation. 2001;104(20):2407–2411. | ||
Cingolani HE, Plastino JA, Escudero EM, Mangal B, Brown J, Pérez NG. The effect of xanthine oxidase inhibition upon ejection fraction in heart failure patients: La Plata Study. J Card Fail. 2006;12(7):491–498. | ||
Hare JM, Mangal B, Brown J, et al; OPT-CHF Investigators. Impact of oxypurinol in patients with symptomatic heart failure. Results of the OPT-CHF study. J Am Coll Cardiol. 2008;51(24):2301–2309. | ||
Nasr G, Maurice C. Allopurinol and global left myocardial function in heart failure patients. J Cardiovasc Dis Res. 2010;1(4):191–195. | ||
Givertz MM, Mann DL, Lee KL, et al. Xanthine oxidase inhibition for hyperuricemic heart failure patients: design and rationale of the EXACT-HF study. Circ Heart Fail. 2013;6(4):862–868. | ||
Givertz MM, Anstrom KJ, Redfield MM, et al; NHLBI Heart Failure Clinical Research Network. Effects of xanthine oxidase inhibition in hyperuricemic heart failure patients: the xanthine oxidase inhibition for hyperuricemic heart failure patients (EXACT-HF) study. Circulation. 2015;131(20):1763–1771. | ||
El-Demerdash E, Awad AS, Taha RM, El-Hady AM, Sayed-Ahmed MM. Probucol attenuates oxidative stress and energy decline in isoproterenol-induced heart failure in rat. Pharmacol Res. 2005;51(4):311–318. | ||
Nakamura R, Egashira K, Machida Y, et al. Probucol attenuates left ventricular dysfunction and remodeling in tachycardia-induced heart failure: roles of oxidative stress and inflammation. Circulation. 2002;106(3):362–367. | ||
Sia YT, Lapointe N, Parker TG, et al. Beneficial effects of long-term use of the antioxidant probucol in heart failure in the rat. Circulation. 2002;105(21):2549–2555. | ||
Kjekshus J, Pedersen TR, Olsson AG, Faergeman O, Pyorala K. The effects of simvastatin on the incidence of heart failure in patients with coronary heart disease. J Card Fail. 1997;3:249–254. | ||
Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. The long-term intervention with pravastatin in ischaemic disease (LIPID) study group. N Engl J Med. 1998;339(19):1349–1357. | ||
Baigent C, Keech A, Kearney PM, et al; Cholesterol Treatment Trialists’ (CTT) Collaborators. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet. 2005;366(9493):1267–1278. | ||
Baigent C, Blackwell L, Emberson J, et al. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet. 2010;376(9753):1670–1681. | ||
Go AS, Lee WY, Yang J, Lo JC, Gurwitz JH. Statin therapy and risks for death and hospitalization in chronic heart failure. JAMA. 2006;296:2105–2111. | ||
Anker SD, Clark AL, Winkler R, et al. Statin use and survival in patients with chronic heart failure: results from two observational studies with 5200 patients. Int J Cardiol. 2006;112:234–242. | ||
Ray JG, Gong Y, Sykora K, Tu JV. Statin use and survival outcomes in elderly patients with heart failure. Arch Intern Med. 2005;165:62–67. | ||
Foody J, Shah R, Galusha D, Masoudi FA, Havranek EP, Krumholz HM. Statin and mortality among elderly patients hospitalized with heart failure. Circulation. 2006;113:1086–1092. | ||
Hognestad A, Dickstein K, Myhre E, Snapinn S, Kjekshus J; OPTIMAAL Investigators. Effect of combined statin and beta-blocker treatment on one-year morbidity and mortality after acute myocardial infarction associated with heart failure. Am J Cardiol. 2004;93(5):603–606. | ||
Yamada T, Node K, Mine T, et al. Long-term effect of atorvastatin on neurohumoral activation and cardiac function in patients with chronic heart failure: a prospective randomized controlled study. Am Heart J. 2007;153(6):1055.e1051–1055.e1055. | ||
Bielecka-Dabrowa A, Goch JH, Mikhailidis DP, Rysz J, Maciejewski M, Banach M. The influence of atorvastatin on parameters of inflammation and function of the left ventricle in patients with dilated cardiomyopathy. Med Sci Monit. 2009;15(12):MS12–MS23. | ||
Sola S, Mir MQ, Rajagopalan S, Helmy T, Tandon N, Khan BV. Statin therapy is associated with improved cardiovascular outcomes and levels of inflammatory markers in patients with heart failure. J Card Fail. 2005;11(8):607–612. | ||
Vrtovec B, Okrajsek R, Golicnik A, et al. Atorvastatin therapy may reduce the incidence of sudden cardiac death in patients with advanced chronic heart failure. J Card Fail. 2008;14(2):140–144. | ||
Kjekshus J, Apetrei E, Barrios V, et al; CORONA Group. Rosuvastatin in older patients with systolic heart failure. N Engl J Med. 2007;357(22):2248–2261. | ||
Tavazzi L, Maggioni AP, Marchioli R, et al; Gissi-HF Investigators. Effect of rosuvastatin in patients with chronic heart failure (the GISSI-HF trial): a randomised, double-blind, placebo-controlled trial. Lancet. 2008;372(9645):1231–1239. | ||
Gullestad L, Ueland T, Kjekshus J, et al; CORONA Study Group. Galectin-3 predicts response to statin therapy in the controlled rosuvastatin multinational trial in heart failure (CORONA). Eur Heart J. 2012;33(18):2290–2296. | ||
Cleland JG, McMurray JJ, Kjekshus J, et al; CORONA Study Group. Plasma concentration of amino-terminal pro-brain natriuretic peptide in chronic heart failure: prediction of cardiovascular events and interaction with the effects of rosuvastatin: a report from CORONA (controlled rosuvastatin multinational trial in heart failure). J Am Coll Cardiol. 2009;54(20):1850–1859. | ||
Lipinski MJ, Cauthen CA, Biondi-Zoccai GG, et al. Meta-analysis of randomized controlled trials of statins versus placebo in patients with heart failure. Am J Cardiol. 2009;104(12):1708–1716. | ||
Zhang S, Zhang L, Sun A, Jiang H, Qian J, Ge J. Efficacy of statin therapy in chronic systolic cardiac insufficiency: a meta-analysis. Eur J Intern Med. 2011;22(5):478–484. | ||
Gastelurrutia P, Lupón J, de Antonio M, et al. Statins in heart failure: the paradox between large randomized clinical trials and real life. Mayo Clin Proc. 2012;87(6):555–560. | ||
Maison P, Desamericq G, Hemery F, et al. Relationship between recommended chronic heart failure treatments and mortality over 8 years in real-world conditions: a pharmacoepidemiological study. Eur J Clin Pharmacol. 2012;69(4):901–908. | ||
Thambidorai SK, Deshmukh AR, Walters RW, et al. Impact of statin use on heart failure mortality. Int J Cardiol. 2011;147(3):438–443. | ||
Mason RP, Walter MF, Day CA, Jacob RF. Intermolecular differences of 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitors contribute to distinct pharmacologic and pleiotropic actions. Am J Cardiol. 2005;96(5A):11F–23F. | ||
Schachter M. Chemical, pharmacokinetic and pharmacodynamic properties of statins: an update. Fundam Clin Pharmacol. 2005;19(1):117–125. | ||
Bonsu KO, Reidpath DD, Kadirvelu A. Effects of statin treatment on inflammation and cardiac function in heart failure: an adjusted indirect comparison meta-analysis of randomised trials. Cardiovasc Ther. 2015;33(6):338–346. | ||
Bonsu KO, Kadirvelu A, Reidpath DD. Lipophilic versus hydrophilic statin therapy for heart failure: a protocol for an adjusted indirect comparison meta-analysis. Syst Rev. 2013;2:22. | ||
Bonsu KO, Reidpath DD, Kadirvelu A. Lipophilic statin versus rosuvastatin (hydrophilic) treatment for heart failure: a meta-analysis and adjusted indirect comparison of randomised trials. Cardiovasc Drugs Ther. Epub 2016 Jan 18. | ||
Hu W, Jiang WB. Pitavastatin-attenuated cardiac dysfunction in mice with dilated cardiomyopathy via regulation of myocardial calcium handling proteins. Acta Pharm. 2014;64(1):105–115. | ||
Qiu Z, Zhang W, Fan F, et al. Rosuvastatin-attenuated heart failure in aged spontaneously hypertensive rats via PKCalpha/beta2 signal pathway. J Cell Mol Med. 2012;16(12):3052–3061. | ||
Zhang WB, Du QJ, Li H, et al. The therapeutic effect of rosuvastatin on cardiac remodelling from hypertrophy to fibrosis during the end-stage hypertension in rats. J Cell Mol Med. 2012;16(9):2227–2237. | ||
Loch D, Chan V, Hoey A, Brown L. Rosuvastatin attenuates heart failure and cardiac remodelling in the ageing spontaneously hypertensive rat. Basic Clin Pharmacol Toxicol. 2009;105(4):262–270. | ||
Chang SA, Kim YJ, Lee HW, et al. Effect of rosuvastatin on cardiac remodeling, function, and progression to heart failure in hypertensive heart with established left ventricular hypertrophy. Hypertension. 2009;54(3):591–597. | ||
Zheng X, Hu SJ. Effects of simvastatin on cardiac performance and expression of sarcoplasmic reticular calcium regulatory proteins in rat heart. Acta Pharmacol Sin. 2005;26(6):696–704. | ||
Yao L, Chen GP, Lu X, Zheng LR, Mou Y, Hu SJ. Effects of atorvastatin on calcium-regulating proteins: a possible mechanism to repair cardiac dysfunction in spontaneously hypertensive rats. Basic Res Cardiol. 2009;104(3):258–268. | ||
Yang Y, Mou Y, Hu SJ, Fu M. Beneficial effect of rosuvastatin on cardiac dysfunction is associated with alterations in calcium-regulatory proteins. Eur J Heart Fail. 2009;11(1):6–13. | ||
Zou C, Liu Z, Qu F, et al. WITHDRAWN: simvastatin prevents decreased SERCA2a activity in non-ischemic heart failure in rabbits via inhibition of beta-adrenergic signaling. Biomed Pharmacother. Epub 2010 Oct 14. | ||
Ozturk N, Yaras N, Ozmen A, Ozdemir S. Long-term administration of rosuvastatin prevents contractile and electrical remodelling of diabetic rat heart. J Bioenerg Biomembr. 2013;45(4):343–352. | ||
Zou C, Qi H, Liu ZH, Han L, Zhao C, Yang X. Simvastatin activates the PPARgamma-dependent pathway to prevent left ventricular hypertrophy associated with inhibition of RhoA signaling. Tex Heart Inst J. 2013;40(2):140–147. | ||
Wang JQ, Wu GR, Wang Z, Dai XP, Li XR. Long-term clinical outcomes of statin use for chronic heart failure: a meta-analysis of 15 prospective studies. Heart Lung Circ. 2014;23(2):105–113. | ||
Alehagen U, Benson L, Edner M, Dahlstrom U, Lund LH. Association between use of statins and outcomes in heart failure with reduced ejection fraction: prospective propensity score matched cohort study of 21 864 patients in the Swedish Heart Failure Registry. Circ Heart Fail. 2015;8(2):252–260. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.