Back to Journals » International Journal of Nanomedicine » Volume 13
Reverse micelle-lipid nanocapsules: a novel strategy for drug delivery of the plectasin derivate AP138 antimicrobial peptide
Authors Groo AC ![]() , Matougui N, Umerska A
, Matougui N, Umerska A ![]() , Saulnier P
, Saulnier P
Received 12 July 2018
Accepted for publication 11 September 2018
Published 15 November 2018 Volume 2018:13 Pages 7565—7574
DOI https://doi.org/10.2147/IJN.S180040
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Thomas Webster
Anne-Claire Groo,1 Nada Matougui,2 Anita Umerska,2,3 Patrick Saulnier2,4
1Normandie Univ, UNICAEN, CERMN - EA 4258, FR CNRS 3038 INC3M, SF 4206 ICORE, Caen, France; 2Micro & Nanomédecines Translationelles-MINT, UNIV Angers, INSERM U1066, CNRS UMR 6021, UBL Universite Bretagne Loire, Angers, France; 3Université de Lorraine, CITHEFOR, Nancy, France; 4Angers University Hospital, Angers, France
Introduction: Resistance to traditional antibiotics is an increasingly serious problem. Antimicrobial peptides (AMPs) have emerged as a new therapeutic class with great potential against infectious diseases, as they are less prone to induce resistance. Nanotechnology-based delivery strategies can improve the efficiency and stability of AMPs, particularly against proteolytic degradation. Lipid nanocapsules (LNCs) are a new generation of biomimetic nanocarriers and were used in this study to deliver peptides.
Methods: AMP-loaded reverse micelles (RM) were developed and incorporated into LNCs by the phase inversion process and the antimicrobial activity of the AMPs-loaded LNC was evaluated by the minimum inhibitory concentration method. We studied the activity of AMP solutions and AMP-loaded LNCs against Gram-positive and Gram-negative bacterial strains and then evaluated the encapsulation of a new cationic AMP called AP138. Finally, we analyzed the effect of enzymatic attack on AP138 and AP138-RM-LNCs after incubation with trypsin.
Results: AP138 was efficiently encapsulated in the LNCs (encapsulation efficiency = 97.8% at a drug loading of 0.151%), resulting in protection against degradation by proteases and the preservation of antimicrobial activity against Staphylococcus aureus, including methicillin-resistant Staphylococcus aureus.
Conclusion: This study shows that RM-LNCs are an excellent candidate system to deliver AMPs.
Keywords: nanoparticles, nanomedicine, antibacterial, AMP, methicillin-resistant Staphylococcus aureus, infection
Introduction
Bacterial infections constitute one of the most serious global public health threats in this century due to the rising incidence of nosocomial infections and the spread of multidrug-resistant bacteria. The first WHO global report on the surveillance of antimicrobial resistance, published in April 2014, collected data from national and international surveillance networks and shows the extent of this phenomenon in many parts of the world. A joint initiative of the WHO, Drugs for Neglected Diseases initiative, and Global Antibiotic Research and Development Partnership encourages research and development through public–private partnerships. By 2023, the partnership aims to develop and deliver up to four new treatments through improvement of existing antibiotics and acceleration of the entry of new antibiotic drugs.1
Antimicrobial peptides (AMPs) are present in all organisms as a component of the innate immune system. AMPs inhibit a wide array of microbes by perturbation of the mitochondrial membrane ionic balance and membrane pore formation. Thus, AMPs have pleiotropic effects and immense potential in diverse medical fields.2 These peptides represent a promising naturally derived new class of antibiotics.3 In various studies, they have shown activity against a wide variety of pathogens: Gram-positive and Gram-negative bacteria, including drug-resistant bacteria, fungi, viruses, and eukaryotic parasites. AMPs are generally composed of fewer than 50 amino acids, have a positive net charge, and are often amphiphilic.4 However, due to the inherent limitations of peptides as drugs, efforts have focused on increasing their stability and bioavailability.5 One of the strategies to overcome these drawbacks is the incorporation of AMPs into novel formulations, including nanocarriers, which are a promising peptide delivery system.6 For example, peptide-loaded nanoparticles increase the characteristics of tissue accumulation (increased tissue uptake and enhanced tissue permeation).7 Another advantage of the encapsulation of AMPs into nanocarriers is protection of the peptide against in vivo degradation, such as that by proteolytic enzymes.8 Lipid-based nanocarriers have generated increasing interest among nanoformulations because of their high degree of biocompatibility and versatility.9 Lange et al demonstrated the stability and efficacy of liposomes containing the AMP CM3.10 Moreover, lipid-based drug delivery systems offer the potential to be AMP carriers due to the affinity of the lipid components for the membrane.11
In this context, the original purpose of this study concerned the formulation of lipid nanoscale systems which combine all previously mentioned attributes. Lipid nanocapsules (LNCs) are a biomimetic drug delivery system composed of an oily core of medium triglycerides surrounded by a surfactant shell. These nanocapsules can be prepared without the use of toxic organic solvents by a low-energy process.12 They contain generally recognized as safe excipients and are compatible with scalable processes.13 The encapsulation of hydrophilic or hydrosoluble drugs into the lipid core was achieved by applying a simple idea that consists of incorporating a reverse micellar system to the LNC core. The micelle-loaded oil is incorporated into the oily continuous phase of the system, described above, at a temperature above the phase-inversion temperature (PIT). This process has been successfully applied to encapsulate dyes, such as fluorescein sodium salt,14 and the hydrophilic anticancer drugs doxorubicin hydrochloride15 and erlotinib hydrochloride, belonging to the tyrosine kinase inhibitor family.16 It could also be successfully applied to peptides. Moreover, the use of oleic Plurol® instead of Lipoid® in the formulation of the lipid core allows the entrapment of thermolabile molecules, such as plasmid DNA molecules, by decreasing the PIT.17
Plectasin is a cationic AMP that belongs to the class of defensins. It is produced by the saprophytic ascomycete Pseudoplectania nigrella.18,19 In contrast to other AMPs, which disrupt bacterial membranes, plectasin derivatives inhibit membrane biosynthesis by targeting the cellular precursor lipid II.18 The peptide AP138 is the product of an extensive lead optimization program based on a set of in silico generated plectasin variants.20 AP138 is active against Gram-positive bacteria, including Staphylococcus aureus (SA) and methicillin-resistant SA (MRSA), making it useful for the treatment of skin and soft-tissue infections.21 MRSA strains are highly resistant to many classes of antibiotics, including most β-lactams. Vancomycin is the drug of choice for the treatment of infections caused by MRSA; however, SA strains with elevated minimum inhibitory concentrations (MICs) against vancomycin are also emerging now.22 Thus, considerable effort has gone into developing alternatives to vancomycin for the treatment of MRSA.
In this context, our aim was to develop a new lipid-based carrier for AMP delivery, evaluate its physicochemical properties, and predict its biopharmaceutical behavior. The antimicrobial activity of AMPs against SA and MRSA was tested in vitro using MIC and time-kill assays. Then, we analyzed the effect of enzymatic attack on AP138 and AP138-LNCs by trypsin. This work allowed us to explore the potential of LNCs for the delivery of AMPs to treat bacterial infections.
Materials and methods
Materials
The lipophilic Labrafac® WL 1349 (“caprylic–capric acid triglycerides”) and oleic Plurol® CC 497 (polyglyceryl-3 dioleate; hydrophilic-lipophilic balance=3) were kindly provided by Gattefossé S.A. (Saint-Priest, France). Kolliphor® HS-15 (Sol), a mixture of free polyethylene glycol 660 (~30%) and 12-hydroxystearate of polyethylene glycol 660 (~70%), was provided by BASF (Ludwigshafen, Germany). Dioctyl sulfosuccinate sodium salt and trypsin were supplied by Sigma-Aldrich (St Louis, MO, USA). NaCl was purchased from Prolabo VWR International (Fontenay-sous-Bois, France). The AP138 AMP was synthesized and provided by PolyPeptide Laboratories (Limhamn, Sweden). HPLC-grade acetonitrile, methanol, and trifluoroacetic acid (TFA) were purchased from Fisher Bioblock (Illkirch, France). Deionized water was obtained from a Milli-Q plus system (Millipore, Paris, France).
Preparation of reverse micelles
Blank reverse micelles (RMs) were prepared by incubating dioctyl sulfosuccinate sodium salt for 3 hours at 45°C with Labrafac WL 1349 (1:10 w/w). AP138-RM was prepared by incubating AP138 (8 mg/g Labrafac WL 1349) with blank RMs in the presence of water (<4% w/w) for 2 hours at 45°C, after which excess AP138 (ie, non-solubilized remnants) was removed from the suspension by centrifugation at 4,000 rpm for 30 minutes at 20°C. We considered RMs to be formed when the mix was transparent, homogeneous, and stable, according to the method of Vrignaud et al.16
Preparation of RM-LNCs
RM-LNCs were prepared according to the method developed by Anton et al,14 corresponding to the PIT method described above, including several changes in the formulation. RM-LNCs were formed by introducing the RMs during the formulation process, just before the final dilution with water, when the system is known to be close to the micro-emulsion state. RM-LNCs were composed of 3.7% Oleic Plurol (w/w), 5.6% Kolliphor HS-15 (w/w), 14.1% lipophilic Labrafac WL 1349 (w/w), 74.3% water (w/w), 1.9% NaCl and 0.5% dioctyl sodium sulfosuccinate (w/w).
Briefly, Oleic Plurol, Kolliphor HS-15, lipophilic Labrafac WL 1349, water, and NaCl were mixed by magnetic stirring. Three cycles of heating and cooling between 30°C and 50°C were then carried out. During the last cycle, 1 mL RMs was added at 43°C and the mixture stirred for 40 seconds, followed by an irreversible shock induced by dilution with 2°C purified water. Magnetic stirring (at 300 rpm) was then applied to the LNC suspension for 5 minutes at room temperature. The structures of the RMs and RM-LNCs are represented in Figure 1.
 | Figure 1 Structure of RM and RM-LNC. |
Characterization of RM-LNCs
The average hydrodynamic diameter associated with the polydispersity index (PDI) was measured by dynamic light scattering using a ZetasizerNano series DTS 1060 (Malvern Instruments S.A., Worcestershire, UK). The zeta potential was calculated from the electrophoretic mobility using the Smoluchowski equation. All measurements were performed in triplicate at 25°C after dilution with deionized water. To determine AP138 drug loading (DL), three samples of RM-LNCs were prepared by dissolution of an exact quantity of RM-LNC suspension in a methanol/brine solution (final ratio: 10/73/17), vortexed, subjected to ultrasound treatment, and centrifuged at 15,000 rpm for 5 minutes to eliminate the residual components of the RM-LNCs and excess NaCl. A 20 μL aliquot of each filtrate was injected onto an HPLC column. Chromatography was performed using a Waters® 717 plus autosampler, Waters® 600 controller, and Waters® 2487 Dual Absorbance Spectrometer (Waters S.A., Saint-Quentin-en-Yvelynes, France) with a Symmetry shield® 250×4.60 mm column (Waters, Milford, Ireland) and an ultraviolet detector set at 200 nm. The flow rate was set to 1.2 mL/min. The gradient was obtained by mixing phase A (water/0.1% TFA) and phase B (water/acetonitrile/TFA: 200/800/0.85). Initially, the mobile phase composition was 90% A. A linear gradient was then applied to reach a composition of 65% A after 5 minutes, 40% A after 14 minutes, and 100% after 14.1 minutes, which was then maintained for 2 minutes before returning to the initial conditions. Quantification was achieved by comparing the peak area ratios of AP138 in the samples to a calibration curve obtained under the same experimental conditions. The AP138 peak had a retention time of 11.8 minutes. Data collection and integration were performed using Empower® 3 software. Linearity was observed between 5 and 100 μg/mL, with a correlation coefficient above 0.999. The detection and quantification limits were 1.2 and 3.5 μg/mL, respectively. The mean drug concentration (μg of AP138/g of RM-LNC dispersion) of each batch of RM-LNC dispersion and the SD were calculated for three samples. The ratio “experimental/theoretical AP138 concentration” (%) is an indicator of AP138 loss during AP138-loaded RM preparation. The experimental AP138 concentration corresponds to the total AP138 (free and encapsulated) in RM-LNC formulations. The theoretical AP138 concentration depends on the exact mass of the AP138 weighed to prepare the AP138-RMs. Encapsulation efficiency (EE) was obtained by separating the free from encapsulated fractions of AP138 by dialysis (100 kDa tubing, 24 hours in deionized water). The EE and DL were calculated using the following equations:
|
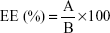
where A is the mass of encapsulated AP138 and B the total amount (mass) of AP138.
|
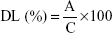
where C is the total weight of all components of the RM-LNCs (the encapsulated AP138 and the mass of surfactants and oil used for the preparation of the RM-LNCs).
Determination of antibacterial activity
The antibacterial effect of AP138-RM-LNCs and AP138 in solution was evaluated using two parameters: the MICs and time-kill assay. The strains used in this study were SA strain ATCC25923 and MRSA clinical strain 702E0196. For the determination of the MICs, bacterial suspensions with a turbidity equivalent to a McFarland 1.1 standard were prepared in a 0.85% NaCl solution. The bacterial suspensions were then further diluted 100 times with brain heart infusion medium (bioMerieux, Marcy l’Etoile, France).
The MIC was determined via a broth microdilution method. Briefly, serial dilutions of AP138 solution or RM-LNCs were prepared in the wells of a 96-well plate, followed by the addition of bacterial suspensions. Bacterial suspensions without peptides were used as a control. After 24 hours of incubation at 37°C, the MIC values were determined as the lowest concentration of the antimicrobial that inhibited visible growth of the microorganism. A difference of more than one dilution is necessary to claim a significant difference between two samples.
The time-kill assay experiments were performed at a concentration of 2× MIC in brain heart infusion. The bacterial counts were measured 0, 3, 6, and 24 hours after incubation at 37°C. At each timepoint, 100 μL bacterial suspension was plated on Columbia agar supplemented with sheep’s blood (Oxoïd, Dardilly, France). After incubating the agar plates overnight at 37°C, the colonies were counted. Each bacterial count was performed in triplicate.
Protease degradation assay
One obvious method for bacteria to inactivate AMPs is to produce peptidases and proteases that could affect peptide activity. Thus, preliminary tests of the stability of the peptides toward proteolysis were performed to establish whether the nanoformulations preserve peptide integrity. Trypsin was chosen as the model enzyme. Various ratios of trypsin:peptide:PBS were tested to reach the proteolytic concentration of trypsin for AP138. Free or encapsulated AP138 was incubated with a 1:2 (mass/mass) trypsin/peptide ratio at 37°C for up to 4 hours with gentle horizontal shaking. Negative controls were performed by incubation of the peptides in PBS without peptidases at 37°C for up to 4 hours. Aliquots were taken after 10, 20, 30, and 45 minutes and 1, 2, and 4 hours, diluted 10 times in methanol or 20 times in methanol:brine, and analyzed by reversed-phase HPLC to quantify the percentage of intact peptide.
Release study
The release of AP138 was monitored by dialysis. One milliliter of the sample was placed in a Float-A-Lyzer® G2 dialysis device with a 100 kDa molecular weight cut-off (Spectrum lab, Rancho Dominguez, CA, USA), prepared according to the manufacturer’s instructions, and incubated in PBS pH 7.4 (European Pharmacopeia, 9th ed.) with gentle magnetic shaking at 100 rpm at 37°C ± 1°C. At appropriate intervals, 100 μL samples were withdrawn, assayed, and replaced by fresh buffer. The amount of AP138 in the release medium was determined by HPLC as described previously. All measurements were performed in triplicate.
Results and discussion
Physicochemical properties of LNC suspensions
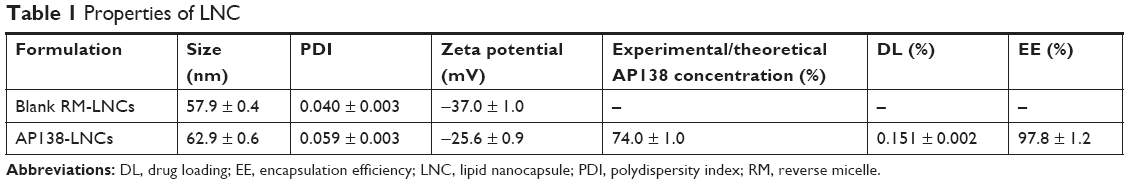
RM-LNCs were successfully prepared and AP138 encapsulated in the formulation. AP138-RM-LNCs had an average hydrodynamic diameter of 63 ± 1 nm and very narrow polydispersity (PDI=0.06), showing very low heterogeneity of the suspension (Table 1). AP138 encapsulation led to a slight size increase of the RM-LNCs and a lower negative zeta potential than that of blank RM-LNCs. This may be related to the positive charge of AP138 (Table 2). The EE reached 97.8% ± 1.2% at a DL of 0.151% ± 0.002%. This DL corresponds to a concentration of 285 μg of AP138/mL of LNC. The properties of AP138-RM were not tested, and the antimicrobial effect could be variable due to the instability of RMs in water. The advantage of new lipid-based carriers for AMP delivery is the obtention of a well-controlled system with a repeatable antimicrobial effect.
 | Table 1 Properties of LNC |
 | Table 2 Structure and properties of AP138 |
Antibacterial activity of formulations
The MIC values of the AP138 solution and AP138-RM-LNCs against SA and MRSA are presented in Table 3. Both the AP138 solution and AP138-RM-LNCs had MICs of 4 μg/mL against SA and 1–2 μg/mL against MRSA. There was no difference between the MICs of AP138 in solution and nano-encapsulated AP138. Non-loaded RM-LNCs showed no antibacterial activity at similar concentrations (data not shown). AP138-loaded RM-LNCs and the AP138 solution were evaluated by time-kill assay (Figure 2). The time-kill assays showed comparable results for the AP138 solution and AP138-RM-LNCs, as they were bactericidal from 18 to 24 hours following the beginning of the experiments. Peptides may display sensitivity to manufacturing processing conditions and excipients (ie, some surfactants, which can compromise peptide stability).23 Standard methods of nanovector preparation present several drawbacks, such as exposure of peptide to organic solvents, shear stress, and high or very low temperatures, which can modify the peptide morphology or denature it.24 Such chemical and physical modifications can lead to differences in bioperformance. However, the RM-LNC formulation process preserved peptide activity.
 | Table 3 MIC of AP138-LNC (in μg of AP138/mL) |
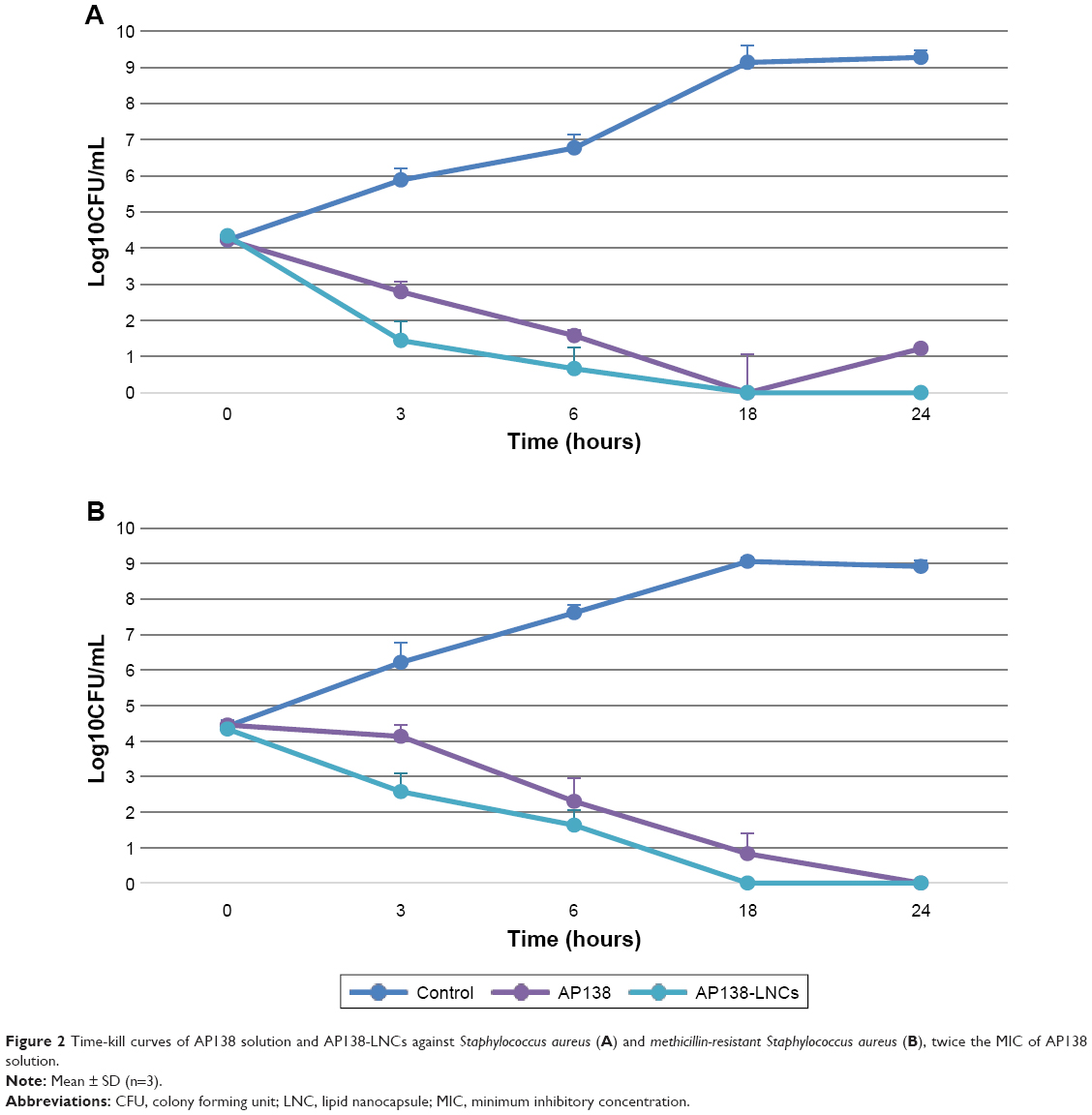 | Figure 2 Time-kill curves of AP138 solution and AP138-LNCs against Staphylococcus aureus (A) and methicillin-resistant Staphylococcus aureus (B), twice the MIC of AP138 solution. |
The existence of SA mutants that are hypersensitive to host defense peptides, such as plectasin, has been described previously.25 Recently, the MICs of plectasin derivatives against 14 studied strains was determined by Umerska et al and the MICs of AP138 varied 16-fold (range 0.125–4 μg/mL).26 This is in good agreement with previously reported data showing that the antibacterial spectrum of AP138 includes staphylococci such as MRSA.20 Garbacz et al reported that the MICs of AMPs for 215 isolates of SA from cystic fibrosis patients varied from 4 to 256 μg/mL.27 Wu et al showed that the MICs against SA ATCC 25923 of novel and control AMPs varied from 8 to 64 μg/mL.28 Compared to these results, the MIC of 1–4 μg/mL for AP138-RM-LNCs shows this formulation to be highly active against staphylococci. This study suggests that AP138-RM-LNCs may be a highly promising drug delivery system and acts a novel antibacterial agent against MRSA and SA. Moreover, lipid nanosystems could enhance bacterial uptake of AMPs by efficient fusion with infectious microbes. For example, drug-loaded liposomes can readily fuse with bacterial membranes and release the drug directly into the bacteria.29 It is now important to determine whether LNCs improve bacterial penetration of AMPs. This could be accomplished by studying antimicrobial penetration in biofilms.30 Indeed, biofilms are aggregations of infectious bacteria which protect the infected area against antibiotics and other therapies – they are common in many types of infection and are difficult to penetrate.
Release study
Approximately half of the dose of AP138 loaded into the RM-LNCs was released into PBS in the first 2 hours and up to 100% was released after 24 hours (Figure 3). AP138-RM-LNCs displayed much better and more rapid release than other delivery systems for which protein release can take days.31 Indeed, the encapsulated peptide may interact with the shell of the nanovector and its release could be affected, resulting in incomplete release.32 Drug release is an important parameter that affects the antibacterial activity of AMP-loaded LNCs,33 and release experiments can provide further insight into the in vitro antibacterial effect of the formulations. Boge et al demonstrated that the high affinity of AMPs to nanoparticles may retard their release, resulting in decreased amounts of free peptide available to exert their antibacterial activity.34 Indeed, if the drug is presented to bacteria in small quantities over a prolonged period, it may be more effectively neutralized by the microorganisms than if the same quantity is delivered all at once. Thus, the high affinity of the AMPs for nanovectors and prolonged release could result in the loss of antibacterial activity. However, this was not true for our delivery system. AMP release may occur rapidly after contact with the bacteria. However, if the amount of drug released results in a high drug concentration (higher than the MIC), prolonged release of the drug could be beneficial by allowing prolonged antibacterial action.
 | Figure 3 AP138 release. |
Protease degradation assay
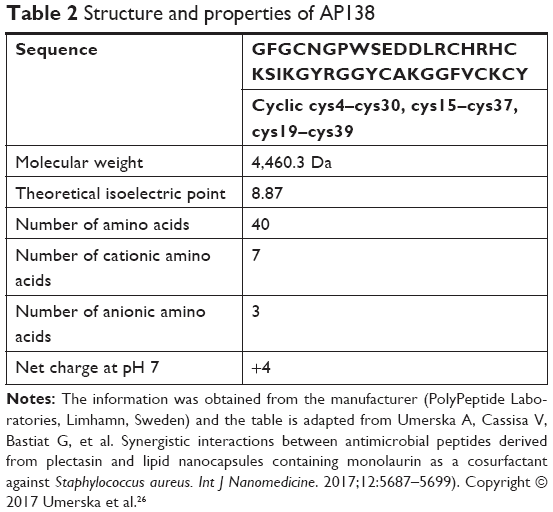
We first investigated the proteolytic stability of pure AP138 using the four clinically relevant enzymes Pseudomonas aeruginosa elastase, human neutrophil elastase, SA V8, and SA aurolysine. After incubation for 6 hours at 37°C with the elastases, the samples were separated by size using Tris-Tricine gel electrophoresis to assess peptide degradation by the enzymes. AP138 was not degraded by any of the four tested enzymes (data not shown). However, because AP138 contains basic residues (Arg, His, and Lys), as shown in Table 2, it can be cleaved by trypsin. Thus, we tested trypsin to evaluate the stability of AP138 against enzymatic degradation. As shown in Table 4, the properties of RM-LNCs were not modified by the incubation conditions, with or without trypsin. Their size, PDI, and zeta potential were unchanged after 2 hours of incubation.
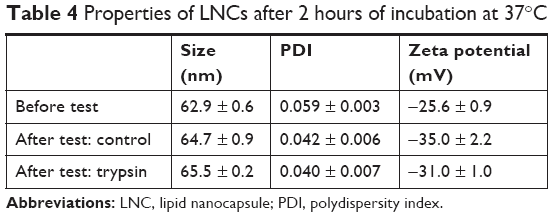 | Table 4 Properties of LNCs after 2 hours of incubation at 37°C |
AP138 in solution was not degraded in the absence of trypsin. Its concentration and that of AP138-RM-LNCs were not affected during incubation without trypsin (control), whereas they decreased during incubation with trypsin (Figure 4). In the presence of trypsin, the remaining AP138 concentration, as a function of time, was higher for the AP138 encapsulated in RM-LNCs. Indeed, the stability of AP138 in the RM-LNCs was significantly higher, for up to 120 minutes of incubation, than that of AP138 in solution, with the remaining ratio significantly higher for the AP138-RM-LNCs (P<0.05, Student’s t-test). Thus, the encapsulation of AP138 into RM-LNCs appears to limit its degradation. Antimicrobial activity may be impaired by susceptibility of the AMP to proteolytic degradation and undesirable interactions in the biological environment.35 Moreover, AMP proteolysis is a fairly common mechanism of bacterial resistance to AMPs36 induced by the bacterial production of proteases.37 A few studies have demonstrated a direct correlation between the in vivo contribution of a protease to virulence and resistance to AMPs.38,39 As a consequence, it is essential to evaluate AP138 degradation by a model protease. Encapsulation is particularly useful for protecting sensitive peptides against clinically relevant enzymes. For example, the peptide LL37 is degraded by infection-related enzymes from either bacteria (P. aeruginosa elastase [PE]) or human defense cells (leukocyte elastase [HNE]).40
 | Figure 4 AP138 degradation by trypsin. |
Protease stability is a pivotal consideration in the development of peptide-based drugs. Although AP138 appears to be stable in the presence of the four clinically relevant enzymes tested and in contact with SA (strain ATCC 25923) and MRSA (clinical strain 702E0196), such information on protease degradation is essential. Indeed, both SA and P. aeruginosa are known to develop resistance to AMP via mechanisms of proteolytic degradation.37 Many clinical strains of SA have been isolated that express various proteases which can contribute to varying sensitivity and resistance. Garbacz et al determined that the MICs of AMPs for 215 isolates of SA from cystic fibrosis patients can vary from 4 to 256 μg/mL.27 Encapsulating AP138 into LNCs appears to be of benefit concerning protease degradation from further strains other than tested SA. It reduced proteolytic susceptibility, resulting in a higher antibacterial effect after exposure to enzyme than that of AMP in solution. Encapsulation of peptides in RM-LNCs may modify their bioavailability and protect them against an unfavorable environment.
The effective and potent action of peptides makes them the drugs of choice for the treatment of numerous diseases.41 Peptides must be administrated through various routes to manifest their therapeutic effects. LNCs could be used as carriers for intravenous administration of peptides due to their small size and very good tolerance.42 Moreover, LNCs are a potential peptide drug carrier for oral delivery as they have been shown to be stable in gastrointestinal-simulated media,43 have demonstrated their ability to cross intestinal mucus,44 and show improved bioavailability.45 Moreover, the free surface of AMP-encapsulating nanoparticulate systems could allow their decoration or modification to improve their delivery to intended sites.46 Peptide delivery still presents many challenges, but peptide-loaded RM-LNCs show great promise for various applications.
Conclusion
Here, we have demonstrated that AMPs can be successfully loaded into LNCs using RMs with high loading efficiency. The AP138-RM-LNCs showed preserved antimicrobial activity and protease resistance. Indeed, the activity of AP138-RM-LNCs did not appear to be markedly different for resistant strains, for example, between the reference strains of SA and MRSA.
Acknowledgments
The research leading to these results received funding from the European Union’s Seventh Framework Programme (FP7/2007–2013) under grant agreement No. 604182 (http://ec.europa.eu.research). It was carried out within the Innovative Nanoformulation of Antimicrobial Peptides to Treat Bacterial Infectious Diseases (FORMAMP) project.
Disclosure
The authors report no conflicts of interest in this work.
References
World Health Organization. Antimicrobial Resistance: Global Report on Surveillance 2014. Geneva: World Health Organization; 2014. Available from: http://www.who.int/drugresistance/documents/surveillancereport/en/. Accessed October 25, 2018. | ||
Patel S, Akhtar N. Antimicrobial peptides (AMPs): the quintessential ‘offense and defense’ molecules are more than antimicrobials. Biomed Pharmacother. 2017;95:1276–1283. | ||
Reddy KV, Yedery RD, Aranha C. Antimicrobial peptides: premises and promises. Int J Antimicrob Agents. 2004;24(6):536–547. | ||
Pasupuleti M, Schmidtchen A, Malmsten M. Antimicrobial peptides: key components of the innate immune system. Crit Rev Biotechnol. 2012;32(2):143–171. | ||
Eckert R. Road to clinical efficacy: challenges and novel strategies for antimicrobial peptide development. Future Microbiol. 2011;6(6):635–651. | ||
Bi L, Yang L, Narsimhan G, Bhunia AK, Yao Y. Designing carbohydrate nanoparticles for prolonged efficacy of antimicrobial peptide. J Control Release. 2011;150(2):150–156. | ||
Ham AS, Cost MR, Sassi AB, Dezzutti CS, Rohan LC. Targeted delivery of PSC-RANTES for HIV-1 prevention using biodegradable nanoparticles. Pharm Res. 2009;26(3):502–511. | ||
Mahlapuu M, Håkansson J, Ringstad L, Björn C. Antimicrobial peptides: an emerging category of therapeutic agents. Front Cell Infect Microbiol. 2016;6:194. | ||
Matougui N, Boge L, Groo AC, et al. Lipid-based nanoformulations for peptide delivery. Int J Pharm. 2016;502(1–2):80–97. | ||
Lange CF, Hancock RE, Samuel J, Finlay WH. In vitro aerosol delivery and regional airway surface liquid concentration of a liposomal cationic peptide. J Pharm Sci. 2001;90(10):1647–1657. | ||
Piotrowska U, Sobczak M, Oledzka E. Current state of a dual behaviour of antimicrobial peptides-therapeutic agents and promising delivery vectors. Chem Biol Drug Des. 2017;90(6):1079–1093. | ||
Heurtault B, Saulnier P, Pech B, Proust JE, Benoit JP. A novel phase inversion-based process for the preparation of lipid nanocarriers. Pharm Res. 2002;19(6):875–880. | ||
Hureaux J, Lagarce F, Gagnadoux F, Clavreul A, Benoit JP, Urban T. The adaptation of lipid nanocapsule formulations for blood administration in animals. Int J Pharm. 2009;379(2):266–269. | ||
Anton N, Mojzisova H, Porcher E, Benoit JP, Saulnier P. Reverse micelle-loaded lipid nano-emulsions: new technology for nano-encapsulation of hydrophilic materials. Int J Pharm. 2010;398(1–2):204–209. | ||
Vrignaud S, Anton N, Gayet P, Benoit JP, Saulnier P. Reverse micelle-loaded lipid nanocarriers: a novel drug delivery system for the sustained release of doxorubicin hydrochloride. Eur J Pharm Biopharm. 2011;79(1):197–204. | ||
Vrignaud S, Hureaux J, Wack S, Benoit JP, Saulnier P. Design, optimization and in vitro evaluation of reverse micelle-loaded lipid nanocarriers containing erlotinib hydrochloride. Int J Pharm. 2012;436(1–2):194–200. | ||
Vonarbourg A, Passirani C, Desigaux L, et al. The encapsulation of DNA molecules within biomimetic lipid nanocapsules. Biomaterials. 2009;30(18):3197–3204. | ||
Schneider T, Kruse T, Wimmer R, et al. Plectasin, a fungal defensin, targets the bacterial cell wall precursor Lipid II. Science. 2010;328(5982):1168–1172. | ||
Mygind PH, Fischer RL, Schnorr KM, et al. Plectasin is a peptide antibiotic with therapeutic potential from a saprophytic fungus. Nature. 2005;437(7061):975–980. | ||
Lociuro S, Neve S, Kjærulf S, Nordkild P. AP138, a second generation plectasin, shows good bactericidal properties and long post-antibiotic effect. In: Final Programme; European Congress of Clinical Microbiology and Infectious Diseases; April 25-28, 2015; Copenhagen, Denmark. | ||
Stryjewski ME, Chambers HF. Skin and soft-tissue infections caused by community-acquired methicillin-resistant Staphylococcus aureus. Clin Infect Dis. 2008;46(Suppl 5):S368–S377. | ||
Venter H, Henningsen ML, Begg SL. Antimicrobial resistance in healthcare, agriculture and the environment: the biochemistry behind the headlines. Essays Biochem. 2017;61(1):1–10. | ||
Bak A, Leung D, Barrett SE, et al. Physicochemical and formulation developability assessment for therapeutic peptide delivery – a primer. AAPS J. 2015;17(1):144–155. | ||
Biswaro LS, da Costa Sousa MG, Rezende TMB, Dias SC, Franco OL. Antimicrobial peptides and nanotechnology, recent advances and challenges. Front Microbiol. 2018;9:855. | ||
Gottlieb CT, Thomsen LE, Ingmer H, Mygind PH, Kristensen HH, Gram L. Antimicrobial peptides effectively kill a broad spectrum of Listeria monocytogenes and Staphylococcus aureus strains independently of origin, sub-type, or virulence factor expression. BMC Microbiol. 2008;8(1):205. | ||
Umerska A, Cassisa V, Bastiat G, et al. Synergistic interactions between antimicrobial peptides derived from plectasin and lipid nanocapsules containing monolaurin as a cosurfactant against Staphylococcus aureus. Int J Nanomedicine. 2017;12:5687–5699. | ||
Garbacz K, Kamysz W, Piechowicz L. Activity of antimicrobial peptides, alone or combined with conventional antibiotics, against Staphylococcus aureus isolated from the airways of cystic fibrosis patients. Virulence. 2017;8(1):94–100. | ||
Wu X, Wang Z, Li X, et al. In vitro and in vivo activities of antimicrobial peptides developed using an amino acid-based activity prediction method. Antimicrob Agents Chemother. 2014;58(9):5342–5349. | ||
Theerthagiri R, Achuthanandan JM, Krishnamurthy S. Antimicrobial magnetosomes for topical antimicrobial therapy. In: Nanobiomaterials in Antimicrobial Therapy: Applications of Nanobiomaterials. Norwich (NY): William Andrew Publishing; 2016:67–101. | ||
Sharma P, Rozenbaum RT, Woudstra W, et al. A constant depth film fermenter to grow microbial biofilms. Protoc Exch. Epub 2017 Mar 8. | ||
Corrigan OI, Li X. Quantifying drug release from PLGA nanoparticulates. Eur J Pharm Sci. 2009;37(3–4):477–485. | ||
Mohammadi-Samani S, Taghipour B. PLGA micro and nanoparticles in delivery of peptides and proteins; problems and approaches. Pharm Dev Technol. 2015;20(4):385–393. | ||
Nehme H, Saulnier P, Ramadan AA, et al. Antibacterial activity of antipsychotic agents, their association with lipid nanocapsules and its impact on the properties of the nanocarriers and on antibacterial activity. Becker K, ed. PLoS One. 2018;13(1):e0189950. | ||
Boge L, Umerska A, Matougui N, et al. Cubosomes post-loaded with antimicrobial peptides: characterization, bactericidal effect and proteolytic stability. Int J Pharm. 2017;526(1–2):400–412. | ||
Brandelli A. Nanostructures as promising tools for delivery of antimicrobial peptides. Mini Rev Med Chem. 2012;12(8):731–741. | ||
Andersson DI, Hughes D, Kubicek-Sutherland JZ. Mechanisms and consequences of bacterial resistance to antimicrobial peptides. Drug Resist Updat. 2016;26:43–57. | ||
Nizet V. Antimicrobial peptide resistance mechanisms of human bacterial pathogens. Curr Issues Mol Biol. 2006;8(1):11–26. | ||
Sieprawska-Lupa M, Mydel P, Krawczyk K, et al. Degradation of human antimicrobial peptide LL-37 by Staphylococcus aureus-derived proteinases. Antimicrob Agents Chemother. 2004;48(12):4673–4679. | ||
Belas R, Manos J, Suvanasuthi R. Proteus mirabilis ZapA metalloprotease degrades a broad spectrum of substrates, including antimicrobial peptides. Infect Immun. 2004;72(9):5159–5167. | ||
Braun K, Pochert A, Lindén M, et al. Membrane interactions of mesoporous silica nanoparticles as carriers of antimicrobial peptides. J Colloid Interface Sci. 2016;475:161–170. | ||
Jain A, Jain A, Gulbake A, Shilpi S, Hurkat P, Jain SK. Peptide and protein delivery using new drug delivery systems. Crit Rev Ther Drug Carrier Syst. 2013;30(4):293–329. | ||
Hureaux J, Lagarce F, Gagnadoux F, et al. Toxicological study and efficacy of blank and paclitaxel-loaded lipid nanocapsules after i.v. administration in mice. Pharm Res. 2010;27(3):421–430. | ||
Roger E, Lagarce F, Benoit JP. The gastrointestinal stability of lipid nanocapsules. Int J Pharm. 2009;379(2):260–265. | ||
Groo AC, Saulnier P, Gimel JC, et al. Fate of paclitaxel lipid nanocapsules in intestinal mucus in view of their oral delivery. Int J Nanomedicine. 2013;8(1):4291. | ||
Groo AC, Bossiere M, Trichard L, Legras P, Benoit JP, Lagarce F. In vivo evaluation of paclitaxel-loaded lipid nanocapsules after intravenous and oral administration on resistant tumor. Nanomedicine. 2015;10(4):589–601. | ||
Liu C, Kou Y, Zhang X, Cheng H, Chen X, Mao S. Strategies and industrial perspectives to improve oral absorption of biological macromolecules. Expert Opin Drug Deliv. 2018;15(3):223–233. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.