Back to Journals » International Medical Case Reports Journal » Volume 19
Retroperitoneal Malignant Triton Tumor in a 6-Year-Old Child Mimicking Wilms Tumor: A Case Report
Authors Ilumbulumbu MK, Nganza SK, Elomba DM ![]() , Posite CM
, Posite CM ![]() , Sylvie VV, Bahati MM, Bahati FL, Ndanga E, Vahaviraki FM, Bomongo EK, Sokoni RM, Kirongozi L, Moreels R
, Sylvie VV, Bahati MM, Bahati FL, Ndanga E, Vahaviraki FM, Bomongo EK, Sokoni RM, Kirongozi L, Moreels R
Received 21 December 2025
Accepted for publication 26 May 2026
Published 3 June 2026 Volume 2026:19 587145
DOI https://doi.org/10.2147/IMCRJ.S587145
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tanvi Dhere
Michel Kalongo Ilumbulumbu,1 Sifa Katungu Nganza,1,2 Daniel Maboso Elomba,1,3,4 Charles Malisaba Posite,5,6 Vissa Visavingi Sylvie,1 Moise Muhindo Bahati,1 Felix Lukoo Bahati,1 Emile Ndanga,7 Francisca Masika Vahaviraki,7 Emmanuel Kalongo Bomongo,1,8 Rachel Masika Sokoni,1,9 Lalia Kirongozi,1 Réginald Moreels1
1Surgery Department, UNICHIR, Beni, Democratic Republic of the Congo; 2Surgery Department, Université Catholique du Graben, Butembo, Democratic Republic of the Congo; 3Anaesthesia and Critical Care Department, Muhimbili University, Dar-Es-Salam, Republic of Tanzania; 4Anaesthesia and Critical Care Department, Université Catholique du Graben, Butembo, Democratic Republic of the Congo; 5Pathology Department, Kampala International University Western Campus, Ishaka, Uganda; 6Pathology Department, Université Catholique du Graben, Butembo, Democratic Republic of the Congo; 7Radiology Department, UNICHIR, Beni, Democratic Republic of the Congo; 8Orthopedics and Traumatology Department, Muhimbili University of Health and Allied Sciences, Dar-Es-Salam, Republic of Tanzania; 9Obstetrics and Gynecology department, Kampala International University Western Campus, Ishaka, Uganda
Correspondence: Daniel Maboso Elomba, Anaesthesia and Critical Care Department, Muhimbili University of Health and Allied Sciences, Dar-Es-Salam, Republic of Tanzania, Email [email protected]
Background: Malignant Triton tumor (MTT) is a rare, aggressive variant of malignant peripheral nerve sheath tumor (MPNST) featuring divergent rhabdomyoblastic differentiation. Comprising less than 5% of MPNST cases, MTT is associated with a poor prognosis, exhibiting a five-year survival rate of only 27.5%. Its occurrence in the pediatric retroperitoneum is exceedingly rare and frequently mimics more common malignancies, such as Wilms tumor.
Case Presentation: A 6-year-old female presented with a four-month history of a painful right flank mass and lower limb functional impairment, without clinical stigmata of neurofibromatosis type 1 (NF1). Ultrasonography revealed a 349 mL vascularized retroperitoneal mass, leading to a provisional diagnosis of Wilms tumor. Exploratory laparotomy identified a tumor adherent to the vertebral column, necessitating intracapsular excision. Histopathological and immunohistochemical analysis confirmed MTT with rhabdomyoblastic differentiation and an immunoprofile suggestive of NF1 association. Despite palliative chemotherapy (cyclophosphamide and vincristine), the patient experienced rapid tumor recurrence and progressive clinical deterioration, culminating in death three weeks post-intervention.
Conclusion: This case highlights the diagnostic complexity of pediatric retroperitoneal MTT, which can be clinically and radiologically indistinguishable from nephroblastoma. In resource-limited settings, where advanced molecular diagnostics are scarce, maintaining a high index of clinical suspicion and ensuring multidisciplinary management are paramount. Early histopathological confirmation is critical to addressing the rapid progression and therapeutic resistance characteristic of this malignancy.
Keywords: malignant triton tumor, wilms tumor, retroperitoneal tumor, pediatric oncology, malignant peripheral nerve sheath tumor, neurofibromatosis type 1
Introduction
Malignant Triton tumor (MTT) is a rare and highly aggressive neoplasm classified as a subtype of malignant peripheral nerve sheath tumor (MPNST) exhibiting rhabdomyoblastic differentiation, a histological hallmark that distinguishes it from conventional MPNST and confers a significantly worse prognosis.1,2 First described by Masson in 1932 and subsequently named after the mythological Triton salamander by Woodruff et al in 1973, MTT accounts for fewer than 5% of all MPNST cases and remains among the rarest soft tissue sarcomas encountered in clinical practice.1 The global incidence of MPNST is estimated at approximately one per 100,000 persons per year, making MTT exceedingly uncommon in absolute terms. Reported series are largely limited to isolated case reports and small institutional series, reflecting the paucity of systematically collected data.3,4
In the pediatric population, MTT is particularly uncommon, with most reported cases occurring in adolescents aged 12–16 years; however, cases in younger children have been documented.4,5 The overall five-year survival rate for MTT is approximately 27.5%, with a median survival of 23.9 months, substantially worse than that of conventional MPNST, which carries a five-year survival rate of approximately 34–52% depending on grade, extent of disease, and NF1 status.4,5 Among pediatric patients, retroperitoneal localization represents an especially uncommon and surgically challenging presentation, further complicating both diagnosis and definitive management.3
A well-established pathophysiological relationship exists between MTT and neurofibromatosis type 1 (NF1), an autosomal dominant neurocutaneous syndrome caused by loss-of-function mutations in the NF1 gene on chromosome 17q11.2, encoding neurofibromin, a negative regulator of the RAS/MAPK signaling pathway.1,2 NF1-associated tumors arise through biallelic inactivation of NF1, resulting in constitutive RAS activation and uncontrolled cellular proliferation. Approximately 50–65% of MTT cases occur in the context of NF1, and NF1-associated MTT carries a worse prognosis relative to sporadic cases, with higher rates of local recurrence and systemic metastasis.4,5 However, as illustrated by the present case, NF1 stigmata may be clinically absent at the time of tumor presentation, particularly in young children, rendering the clinical diagnosis of NF1 unreliable without molecular genetic confirmation.3
The accurate preoperative differentiation of MTT from more common pediatric retroperitoneal malignancies, in particular Wilms tumor (nephroblastoma), which accounts for approximately 6–7% of all childhood cancers and is the most common pediatric renal malignancy-represents a significant diagnostic challenge.6 Both entities may present with an abdominal mass, flank pain, and a large heterogeneous retroperitoneal lesion on ultrasonography. Computed tomography (CT) and magnetic resonance imaging (MRI) can provide important morphological information, including lesion origin, vascular involvement, and the presence of intralesional hemorrhage or necrosis; however, the radiological features of MTT and Wilms tumor demonstrate considerable overlap, and imaging alone is insufficient to establish a definitive diagnosis.7 Positron emission tomography (PET) may add functional metabolic data but remains inaccessible in many low-resource settings. Cases of retroperitoneal MTT have also been misdiagnosed as gastrointestinal stromal tumors (GISTs), neuroblastoma, and rhabdomyosarcoma prior to histopathological evaluation, further highlighting the imperative of tissue-based diagnosis.8 In resource-constrained environments, where advanced cross-sectional imaging may be unavailable or delayed, the risk of diagnostic error is compounded, and the clinical presentation may be the primary basis upon which surgical decision-making proceeds.
Herein, we report the case of a 6-year-old female with a retroperitoneal MTT that was clinically and sonographically misdiagnosed as a Wilms tumor; we further delineate the diagnostic and surgical trajectory while situating the clinico-pathological findings within the existing literature. This report contributes to the sparse body of evidence regarding pediatric retroperitoneal MTT and underscores the necessity of systematic immunohistochemical evaluation and a high index of clinical suspicion in the differential diagnosis of pediatric abdominal masses, particularly within resource-limited clinical environments.
Case Presentation
On 13 August 2024, a 6-year-old girl was referred to the Surgical Unit of UNICHIR Hospital in Beni, Democratic Republic of the Congo, with a four-month history of a progressive right flank swelling and functional impairment of the right lower limb. According to the patient’s parents, the mass was initially painless and was first noted approximately four months prior to presentation; it had subsequently enlarged progressively, and localized flank pain and right lower extremity weakness had developed over the preceding weeks. Initial treatment at a peripheral health facility had been ineffective, prompting referral for advanced evaluation and management. The patient’s bowel and urinary functions were reported as normal, and no history of fever was elicited. Her past medical history was notable for treated rheumatic fever, a complete vaccination history, and recurrent malaria episodes. There was no prior surgical history. She was the third of five children; no family history of hereditary disease, neurofibromatosis, or pediatric malignancy was reported, and her siblings were reportedly in good health.
On physical examination, the patient appeared in moderate distress and was ambulating with the assistance of a cane. Vital signs were within normal limits. Notably, there were no café-au-lait spots, axillary freckling, Lisch nodules, or other clinical stigmata of neurofibromatosis type 1. Abdominal examination revealed a large, irregular, mildly tender mass extending from the right hypochondrium to the pelvis. Regional lymph nodes were non-palpable. On the basis of the clinical presentation, a provisional diagnosis of Wilms tumor was established.
Abdominal ultrasonography demonstrated a large, heterogeneous, solid-cystic, Doppler-vascularized retroperitoneal mass in the right flank, with an estimated volume of approximately 349 mL. The bilateral kidneys, liver, and spleen appeared sonographically normal. Laboratory investigations were unremarkable: white blood cell count 9,230/mm3 (neutrophils 45%), hemoglobin 10.4 g/dL, and hematocrit 32.6%. Advanced cross-sectional imaging (CT or MRI) was not available at the treating institution at the time of presentation.
Given the sonographic findings, the clinical urgency of the presentation, and the unavailability of preoperative biopsy facilities, a decision was made to proceed with exploratory laparotomy, consistent with the standard surgical approach for presumed Wilms tumor in resource-limited settings, where nephrectomy following upfront surgery is an accepted management pathway in the absence of contraindications.
Intraoperative findings revealed a large retroperitoneal tumor with intimate adherence to the vertebral column (Figure 1). The liver, spleen, bilateral kidneys, and intestines were macroscopically normal and free of tumor involvement. An intracapsular excision was performed, necessitating ligation of a major nerve that was encased by tumor tissue. The resected specimen was forwarded to a reference pathology laboratory at the Saint-Jean Campus in Belgium for histopathological and immunohistochemical analysis (Figure 2).
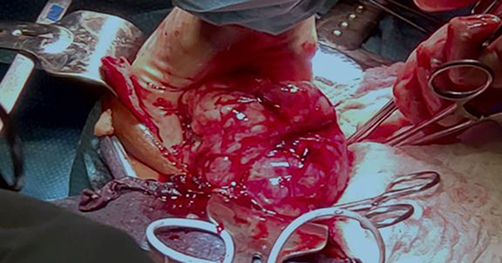 |
Figure 1 Intraoperative photograph demonstrating the large retroperitoneal tumor with intimate adherence to the vertebral column. The liver, bilateral kidneys, spleen, and intestines were macroscopically normal. Intracapsular excision was performed with ligation of an encased major nerve. |
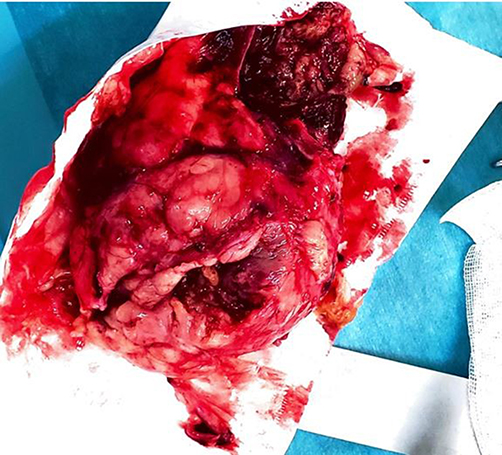 |
Figure 2 Gross pathological specimen of the resected retroperitoneal tumor following intracapsular excision, demonstrating the heterogeneous cut surface with areas of solid tumor and central softening. |
Microscopic examination revealed a variably cellular spindle cell neoplasm composed of epithelioid cells arranged within a loose stroma, with prominent mitotic activity. Immunohistochemical profiling demonstrated positivity for BRG1 and INI1 (SMARCB1), with intermediate expression of SOX10, S100 protein, and neuron-specific enolase (NSE); and negativity for H3K27me3, MUC4, epithelial membrane antigen (EMA), glial fibrillary acidic protein (GFAP), and pan-cytokeratin (PanCK). These findings were consistent with a malignant Triton tumor exhibiting rhabdomyoblastic differentiation and an immunoprofile suggestive of NF1 association. No molecular genetic testing for NF1 mutation was performed, as this facility was not available at the treating institution.
The immediate postoperative course was complicated by urinary retention on postoperative day two, which was managed conservatively. The patient demonstrated progressive recovery of right lower limb function with physiotherapy. However, three weeks postoperatively, follow-up abdominal ultrasonography revealed two recurrent intraperitoneal masses with central liquefaction and demonstrable Doppler flow, associated with peripheral lymphadenopathy, mild bowel dilation, and a moderate intraperitoneal fluid collection (Figure 3). Hepatic and renal function parameters remained within normal limits.
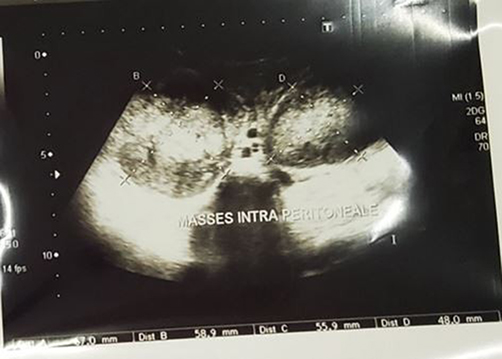 |
Figure 3 Postoperative abdominal ultrasonography performed three weeks following surgical resection, demonstrating two recurrent intraperitoneal masses with central liquefaction, demonstrable Doppler vascular flow, peripheral lymphadenopathy, mild bowel dilation, and a moderate intraperitoneal fluid collection, consistent with rapid tumor recurrence. |
Following documentation of tumor recurrence and reassessment of hepatic and renal function, palliative systemic chemotherapy was initiated, comprising cyclophosphamide and vincristine, with morphine-based analgesia for pain control. Azathioprine was included as part of the regimen. Despite these therapeutic measures, the patient’s clinical condition deteriorated progressively, characterized by escalating hypertension and rapid tumor growth. A fatal outcome occurred on the tenth day of follow-up after recurrence. This rapid and fatal clinical course is consistent with the well-documented biological aggressiveness of MTT, in which early local recurrence rates of up to 50% have been reported and long-term survival following incomplete resection is exceedingly rare.
Discussion
The present case illustrates the profound diagnostic complexity that may arise when a rare and aggressive retroperitoneal malignancy presents in the pediatric age group with clinical and radiological features mimicking those of a far more common condition. Wilms tumor remains the most frequent pediatric renal malignancy, accounting for approximately 6–7% of all childhood cancers and representing the dominant diagnostic consideration in any child presenting with a unilateral abdominal mass.6 The overlapping features, a large retroperitoneal mass in a young child, in the absence of distinctive clinical markers, led to the provisional diagnosis of Wilms tumor in this case, a diagnostic error that is well-recognized in the MTT literature.3,6,8
Definitive diagnosis in this case was established through comprehensive histopathological and immunohistochemical analysis, performed at an international reference laboratory, which identified the defining biphasic morphology of MTT, a malignant peripheral nerve sheath tumor component (evidenced by SOX10, S100, and NSE positivity) in combination with rhabdomyoblastic differentiation.6,9 The immunohistochemical profile, particularly the intermediate expression of SOX10 and S100, together with BRG1 and INI1 positivity and negativity for epithelial and glial markers, effectively excluded alternative diagnoses, including rhabdomyosarcoma, neuroblastoma, clear-cell sarcoma of the kidney, and synovial sarcoma. The retention of SMARCB1/INI1 expression further differentiated this tumor from epithelioid sarcoma and other INI1-deficient neoplasms.6,9 Loss of H3K27me3 expression, which is characteristic of a subset of MPNST associated with NF1 in adults, was observed in this case; however, its prognostic significance in pediatric MTT requires further investigation.
Although the immunoprofile was interpreted as suggestive of NF1 association, the patient exhibited no clinical stigmata of neurofibromatosis at the time of presentation, and no molecular genetic confirmation of an NF1 germline mutation was performed. This constitutes an important limitation of the current report. The absence of clinical NF1 features does not exclude the diagnosis, as cutaneous manifestations may be subtle or absent in young children, and internal NF1-associated tumors may precede the development of externally visible signs. Genetic counseling and germline testing of the NF1 gene are strongly recommended for all pediatric patients presenting with MPNST or MTT, irrespective of the clinical phenotype.1,4,5
The role of imaging in the preoperative diagnosis of retroperitoneal MTT warrants specific discussion. In the present case, abdominal ultrasonography demonstrated a large heterogeneous mass, but could not characterize the lesion further or differentiate it from a Wilms tumor. CT and MRI, which offer superior soft-tissue characterization, delineation of vascular anatomy, and assessment of surgical resectability, were unavailable at the treating institution. Prior series have demonstrated that MTT may exhibit heterogeneous enhancement, intralesional hemorrhage, central necrosis, and ill-defined margins on CT and MRI, features that may overlap with Wilms tumor but can suggest a sarcomatous diagnosis in the appropriate clinical context.7 PET-CT may additionally identify metabolically active disease foci and locoregional nodal involvement; however, this modality remains inaccessible in most sub-Saharan African health facilities. This case highlights the diagnostic vulnerability created by limited imaging resources in low-resource settings, where the clinical presentation alone may be insufficient to raise suspicion for a rare diagnosis and where preoperative biopsy may not be feasible.
With respect to treatment, the standard of care for MTT, as for MPNST generally, is complete surgical resection with widely negative margins, which represents the most important prognostic determinant.4–6 The role of adjuvant chemotherapy in MTT remains incompletely defined due to the rarity of the condition and the absence of prospective randomized data. Regimens commonly employed for high-grade soft tissue sarcomas, including ifosfamide and doxorubicin, have demonstrated limited but measurable response rates in MPNST and have been applied to MTT in selected cases.6,9,10 Radiotherapy may reduce the risk of local recurrence following margin-positive resection, particularly in anatomical locations that do not permit re-excision, though its benefit in retroperitoneal MTT has not been formally established.6 In the present case, only palliative systemic chemotherapy could be offered following tumor recurrence, as the patient’s performance status and the extent of disease precluded repeat surgical intervention. The chemotherapy regimen employed (cyclophosphamide and vincristine) is broadly consistent with salvage approaches used for pediatric sarcomas in resource-limited settings, though its evidence base for MTT specifically is limited.
Limitations
Several important limitations of this report merit explicit acknowledgment. This is a single-case report, and no generalizable conclusions regarding treatment outcomes or diagnostic strategies can be drawn. The absence of preoperative CT or MRI represents a significant constraint on the completeness of radiological characterization. Surgical margin status, a critical prognostic variable in sarcoma management, was not formally reported in the pathological assessment and therefore could not be analyzed. Molecular genetic confirmation of NF1 association was not obtained. These limitations reflect the realities of practicing in a resource-limited environment and are acknowledged as inherent constraints of this report.
Conclusion
This case of pediatric retroperitoneal MTT highlights the diagnostic complexities inherent in rare sarcomas that mimic common malignancies like Wilms tumor. Characterized by aggressive rhabdomyoblastic differentiation and a high propensity for rapid local recurrence, MTT’s clinical course in this patient underscores its poor prognosis. Definitive diagnosis necessitates rigorous histopathological and immunohistochemical evaluation to distinguish it from other pediatric abdominal masses, particularly in resource-limited settings where advanced imaging and biopsy may be inaccessible. Consequently, clinicians must maintain a high index of suspicion for MTT, even absent NF1 stigmata, and prioritize timely, multidisciplinary management and international collaborative research to optimize diagnostic accuracy and therapeutic outcomes for this rare entity.
Ethics Approval
Institutional review board approval was not required for a single anonymized case report according to local institutional policy.
Consent for Publication
Written informed consent was obtained from the patient’s mother for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Acknowledgment
The authors would like to thank Dr Réginald Moreels –Humanitarian Surgeon- and the Fondation Unichir Dr Réginald Moreels for their generous support. We are grateful for the sponsorship provided to three of the authors during their Master’s studies and assistance in transporting biopsy samples to Belgium for specialized histopathological analysis, which was crucial for this case report.
Author Contributions
All authors made a significant contribution to the work reported, took part in drafting and reviewing the article, gave final approval of the version to be published, have agreed on the journal to which the article has been submitted, and agree to be accountable for all aspects of the work.
Disclosure
The authors declare that they have no conflicts of interest in this work.
References
1. Mocellin S. Malignant Triton Tumor. In: Soft Tissue Tumors. Cham: Springer; 2021. doi:10.1007/978-3-030-58710-9_168
2. Heredia‑Montaño M, Navarro-Tovar F, Díaz-Barrientos CZ, et al. Malignant peripheral nerve sheath tumor (Triton tumor) located in mesentery. Gaceta Mexicana de Oncología. 2018;17:69–7. doi:10.24875/j.gamo.M18000110
3. Hou Z, Wang C, Li L, et al. Retroperitoneal malignant triton tumor in an infant: case report and literature review. Transl Pediatr. 2020;9:567–572. doi:10.21037/TP.2020.03.12
4. Zhao A, Ding D, Li X, et al. Malignant triton tumor in a child: case report and literature review. Cancer Manag Res. 2019;11:10759–10766. doi:10.2147/CMAR.S221110
5. Bins RB, Pinzon CE, da Silva Pereira LD, et al. Malignant triton tumor of the kidney in a child: case report. Int J Surg Case Rep. 2021;85(C):106252. doi:10.1016/j.ijscr.2021.106252
6. Rebegea L, Niculet E, Craescu M, et al. Clinical, histological, immunohistochemical aspects in a rare malignant peripheral nerve sheath (Triton) tumor. Modern Med. 2021;28.
7. Amirah Idalis I, et al. Iconographic review and radio‑histological concordance of retroperitoneal tumors in children. 2022.
8. Baía C, Martins P, Serralva M, et al. An atypical location for Malignant Triton Tumor – a case report. Braz. J. Case Report. 2023;4:72–77
9. Agarwal K, et al. Teratoid Wilms Tumor: case report and literature review. Pediatr Nephrol Nephrol. 2020;8:1. doi:10.22037/jpn.v8i4.32079
10. Martín Palacios‑Acosta J, Marisol SM, Angélica LH, et al. Tumor “tritón” del retroperitoneo. Acta Pediatr Mex. 2013;34:263–267.
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.