Back to Journals » Drug Design, Development and Therapy » Volume 20
Research Progress on the Mechanism and Targeted Intervention of G Protein-Coupled Bile Acid Receptor 1 (GPBAR1)-Mediated “Inflammation-Apoptosis-Metabolism-Microcirculation” Regulatory Network in Hepatitis B-Associated Liver Failure
Authors Cui C ![]() , Sun Y, Zhang K, Shi J, Wang K, Gao S
, Sun Y, Zhang K, Shi J, Wang K, Gao S
Received 11 October 2025
Accepted for publication 12 March 2026
Published 25 March 2026 Volume 2026:20 573597
DOI https://doi.org/10.2147/DDDT.S573597
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Tin Wui Wong
Chao Cui,1 Yu Sun,2 Kaiyue Zhang,2 Jingfei Shi,3 Kai Wang,2,4 Shuai Gao4
1Scientific Research Department, Qilu Hospital of Shandong University Dezhou Hospital, Dezhou, People’s Republic of China; 2Department of Infectious Disease, Qilu Hospital of Shandong University Dezhou Hospital, Dezhou, People’s Republic of China; 3Department of Clinical and Basic Medicine, Shandong First Medical University, Jinan, People’s Republic of China; 4Department of Hepatology, Qilu Hospital of Shandong University, Jinan, People’s Republic of China
Correspondence: Kai Wang, Email [email protected]; Shuai Gao, Email [email protected]
Abstract: Hepatitis B-associated liver failure (HBV-LF) is a severe hepatic disease induced by hepatitis B virus infection, characterized by massive hepatocyte necrosis, rapid liver dysfunction, uncontrolled inflammation, bile acid metabolism disorders and immune microenvironment imbalance. It has a high short-term mortality of 40%– 70% worldwide. Current mainstream treatments, including antiviral and organ support therapies, only control viral replication and maintain organ function, but cannot effectively reverse the excessive inflammatory cascade and metabolic disorders that drive HBV-LF progression. This review systematically summarizes the molecular mechanisms by which GPBAR1 regulates the four core pathological processes of HBV-LF: uncontrolled inflammation, massive hepatocyte apoptosis, bile acid metabolism disorders and hepatic microcirculation disturbance, and clarifies its value as a potential therapeutic target. G protein-coupled bile acid receptor 1 (GPBAR1/TGR5) is a key membrane receptor for bile acids and critically regulates hepatic physiology and pathology. Given the limited direct evidence in HBV-LF, we cautiously extrapolate relevant mechanisms from chronic hepatitis B, acute liver injury and metabolic liver diseases based on shared pathological features, while noting that these mechanisms require further validation in HBV-LF models and clinical samples. We review the structure, distribution, activation and signaling pathways of GPBAR1, focusing on its protective role in HBV-LF: inhibiting excessive inflammation via NF-κB and Keap1-Nrf2 pathways, reducing hepatocyte apoptosis through mitochondrial and PI3K/Akt pathways, maintaining bile acid balance by regulating CYP7A1/CYP8A1, and improving hepatic immunity and microcirculation via NKT, CCL2/CCR2 and ET-1 axes. Abnormal GPBAR1 expression correlates with HBV-LF severity and poor prognosis. We also summarize preclinical progress of GPBAR1 agonists (e.g. INT-767, BAR501) and compare GPBAR1-targeted strategies with other GPCR therapies. This review provides a theoretical basis for mechanistic research and target identification of HBV-LF.
Keywords: hepatitis b-associated liver failure, g protein-coupled bile acid receptor 1, uncontrolled inflammation, bile acid metabolism disorder, immune microenvironment imbalance
Introduction
Hepatitis B is one of the infectious diseases with the most severe global disease burden and also a major public health challenge.1 Although positive progress has been made in global hepatitis B prevention and control, with the prevalence of HBsAg in the general population continuing to decline, the total number of HBV-infected individuals remains relatively large.2 Without scientific and systematic treatment, patients with hepatitis B are prone to progress to severe diseases such as liver cirrhosis, HCC, and liver failure. Among these, HBV-LF is characterized by a critical condition, extremely high mortality (the short-term mortality rate is up to 40%-70% globally), and lacks specific and effective targeted therapeutic strategies. It poses a serious threat to patients’ life and health, and has become a key clinical challenge in the treatment of HBV-related severe liver diseases.3
The pathogenesis of liver failure is extremely complex, involving multiple processes such as viral infection, immune response, and metabolic disturbance.4 In recent years, with the deepening of research, the role of GPBAR1 in liver diseases has gradually attracted attention. While GPBAR1 has been widely studied in CHB, HBV-related HCC, NAFLD, and other liver injuries, its regulatory role in the acute and fulminant process of HBV-LF remains less systematically summarized. Existing studies have suggested that GPBAR1 is associated with multiple abnormal pathological phenomena related to HBV-LF, such as abnormal platelet activation, immune cell dysfunction, and intrahepatic vascular endothelial damage,5 but the systematic regulatory mechanism and its potential as a therapeutic target have not been fully elucidated.
This review focuses exclusively on HBV-LF and aims to present an unbiased summary of the current understanding of GPBAR1 in this disease. Where direct experimental or clinical evidence for HBV-LF is limited, we cautiously extrapolate and integrate mechanisms validated in CHB, HBV-related acute liver injury, and non-HBV ALF models with explicit cautionary statements for all extrapolations; this extrapolation is based on shared core pathological cascades (uncontrolled inflammation, massive hepatocyte apoptosis, bile acid metabolic disorders, and hepatic microcirculation disturbance) and does not represent definitive evidence for HBV-LF. As a member of the GPCR superfamily, GPBAR1 is also an important membrane receptor for bile acids. It is widely distributed in various tissue cells, and is particularly highly expressed in liver non-parenchymal cells.6 By recognizing ligands such as bile acids and activating downstream signaling pathways, GPBAR1 participates in the regulation of liver physiological functions, including the maintenance of glucose homeostasis, regulation of energy metabolism, and modulation of lipid metabolism.7
Bile acids not only participate in the digestion and absorption of dietary lipids but also act as signaling molecules. By activating membrane receptors such as GPBAR1 and nuclear receptors such as FXR, they play a crucial role in the occurrence and development of liver diseases.8 For HBV-LF specifically: (1) direct evidence for GPBAR1-FXR crosstalk in HBV-LF bile acid metabolism is absent; (2) most GPBAR1-FXR bile acid regulatory mechanisms are validated in CHB, NAFLD, and cholestatic liver disease (non-LF models); (3) in HBV-LF, acute bile acid metabolic disorder is driven by massive hepatocyte necrosis and cholestasis, whereas chronic non-LF models involve progressive bile acid dysregulation—these distinct pathological triggers may lead to different GPBAR1-FXR signaling outcomes. During the progression of HBV-LF, bile acid metabolism is disrupted, and the expression and function of GPBAR1 also undergo significant changes. These changes may further affect hepatic inflammatory responses, hepatocyte apoptosis, and the balance of bile acid metabolism, thereby exacerbating the disease. In-depth research on the mechanism by which GPBAR1 participates in HBV-LF is of great significance for revealing the pathogenesis of HBV-LF and developing new therapeutic strategies.
At present, the treatment for HBV-LF mainly includes antiviral therapy, supportive therapy, and liver transplantation, but these therapeutic approaches still have certain limitations.9 Therefore, exploring new therapeutic targets and strategies is extremely urgent. As a potential therapeutic target, GPBAR1 provides a new direction for the treatment of HBV-LF (Figure 1). Meanwhile, comparing GPBAR1 with other GPCR-based therapeutic strategies (such as those targeting the bile acid receptor FXR, AT1 receptor, and cannabinoid receptors) helps to identify the most promising targets for alleviating severe inflammation and liver failure in HBV-LF, and provides a reference for the optimization of clinical treatment regimens.
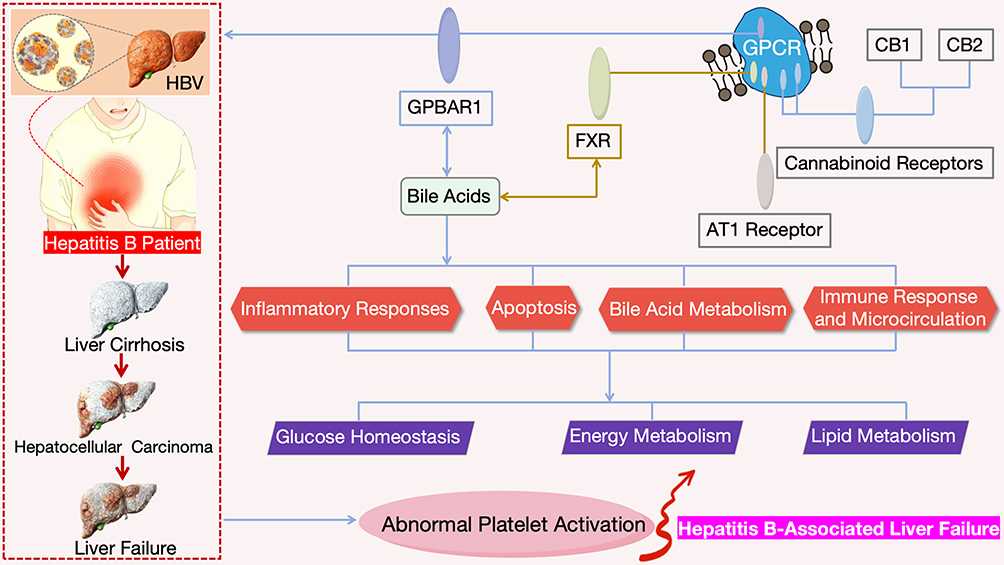 |
Figure 1 Schematic Diagram of the Research Framework for Therapeutic Targets of HBV-LF Based on GPBAR1. |
Review Methodology
Literature Search Strategy
This review was conducted in accordance with the PRISMA guidelines for narrative systematic reviews. The electronic databases including PubMed, Web of Science, Embase, Scopus, and CNKI were systematically searched for relevant studies. The key search terms were a combination of: G Protein-Coupled Bile Acid Receptor 1, GPBAR1, TGR5, Hepatitis B-Associated Liver Failure, HBV-LF, Acute-on-Chronic Liver Failure, Inflammation, Apoptosis, Metabolism, Microcirculation, Bile Acid Metabolism, Immune Microenvironment, and Targeted Intervention. The search was limited to English and Chinese peer-reviewed original research articles, review articles, and meta-analyses; conference abstracts and unpublished data were excluded to ensure the reliability of the included literature.
Time Frame
The literature search was performed from January 2000 to September 2025, covering the latest research progress of GPBAR1 and HBV-LF over the past 25 years. This time frame was selected to include the initial characterization of GPBAR1, the in-depth research on its signaling pathways, and the latest evidence of its role in HBV-related liver diseases, ensuring the timeliness and comprehensiveness of the review.
Inclusion and Exclusion Criteria
Inclusion Criteria
- Studies focused on the biological function of GPBAR1 in liver diseases, especially HBV-LF, CHB, and acute liver failure;
- Studies exploring the molecular mechanisms of GPBAR1 regulating inflammation, apoptosis, bile acid metabolism, and hepatic microcirculation;
- Studies investigating GPBAR1 agonists/antagonists and their preclinical application in HBV-related liver injury models;
- Studies reporting the expression, methylation, and prognostic value of GPBAR1 in HBV-LF patients;
- Systematic reviews and meta-analyses summarizing the role of GPCR family in liver failure.
Exclusion Criteria
- Studies focused on GPBAR1 in non-liver diseases (eg., cardiovascular diseases, neurological diseases) without correlation to liver injury;
- Studies on HBV infection without involving liver failure or severe hepatic pathological changes;
- Duplicate publications, low-quality studies with incomplete data, and studies without clear experimental design;
- Studies on other bile acid receptors (eg., FXR) without mentioning the crosstalk with GPBAR1.
Review Type
This review is a narrative systematic review. Unlike a quantitative meta-analysis, it systematically collates, analyzes, and summarizes the qualitative evidence of GPBAR1 in HBV-LF, including molecular mechanisms, preclinical research, and therapeutic potential. Given the limited direct clinical evidence of GPBAR1 in HBV-LF, this review reasonably extrapolates the mechanistic evidence from CHB, acute liver injury, and metabolic liver diseases to HBV-LF based on shared core pathological features, and further organizes and interprets the evidence to form a comprehensive regulatory network of GPBAR1 in HBV-LF.
Overview of GPBAR1
As a key membrane receptor in the bile acid signaling pathway, GPBAR1 is closely associated with liver physiological metabolism and pathological processes. With the deepening of research on the pathogenesis of HBV-LF, the role of GPBAR1 in regulating hepatic inflammatory responses, maintaining bile acid metabolic balance, and promoting cell damage repair has gradually attracted attention, making it a crucial entry point for analyzing the pathological mechanism of this disease and developing targeted therapeutic strategies. This section will elaborate on three aspects of GPBAR1: its molecular structural characteristics, tissue distribution patterns, and its activation mechanism along with downstream signal transduction pathways, laying a theoretical foundation for subsequent discussions on its specific role and mechanism in HBV-LF.
Structure and Distribution of GPBAR1: Molecular Architecture and Hepatic Expression Profile
GPBAR1, also known as TGR5, is a key member of the GPCR superfamily. Its structure resembles an elaborately constructed molecular framework, composed of 335 amino acid residues arranged in an orderly manner. The iconic 7-transmembrane topological structure serves as the core architecture for its biological functions.10 Each transmembrane domain contains 20–25 hydrophobic amino acid residues, which fold skillfully into an α-helical secondary structure. These α-helices are parallel to each other, alternately traversing the phospholipid bilayer of the cell membrane, and eventually converge to form a unique “helix bundle” three-dimensional conformation.11 This conformation acts like a precision lock cylinder, providing a solid structural basis for GPBAR1 to specifically recognize ligands such as bile acids and initiate signal transduction.12
A more in-depth analysis reveals that the transmembrane α-helices are closely connected by 3 extracellular loops and 3 intracellular loops.13 The extracellular region, including the extracellular loops and the N-terminus, acts like a sensitive “scout” that first participates in the ligand recognition and initial binding stages. It accurately captures bile acid molecules through specific amino acid sequences and spatial conformations. In contrast, the intracellular region, composed of the intracellular loops and the C-terminus, functions as a key “relay runner” in a signal relay race. After GPBAR1 is activated by ligands, this region rapidly interacts with downstream signaling molecules such as G proteins, initiating complex intracellular signal transduction cascades.14 This typical structural feature is not only a unique identifier of GPBAR1 but also a shared molecular hallmark of the GPCR superfamily, endowing GPBAR1 with a crucial role as a signal transduction “hub” on the cell membrane (Figure 2). Upon stimulation by ligands such as bile acids, GPBAR1 can act like a well-trained dancer, activating downstream pathways through delicate conformational changes and deeply participating in the regulation of physiological functions in tissues such as the liver. Its active involvement ranges from the maintenance of bile acid metabolic balance to the precise regulation of lipid metabolism.15
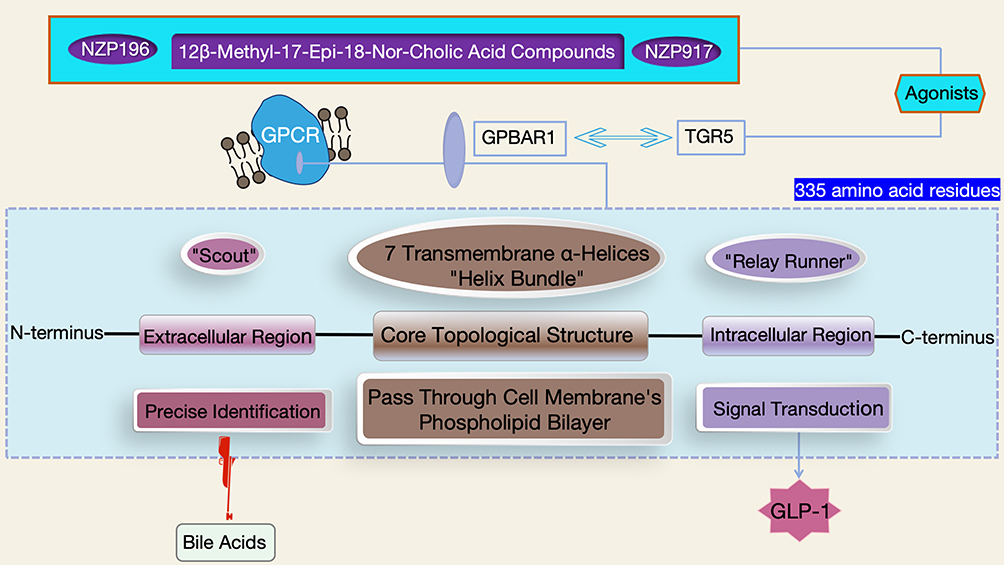 |
Figure 2 Schematic Diagram of the Molecular Structure of GPBAR1 (Including the 7-Transmembrane Topological Structure and Intracellular/Extracellular Functional Domains). |
At the forefront of molecular targeting research, researchers have ingeniously designed and identified non-bile acid dual agonists of FXR/GPBAR1 (such as isonicotinamide derivatives) in vitro.16 To evaluate the efficacy of these novel compounds, the research team first fed C57BL/6J mice a high-fat diet—defined as a choline-deficient L-amino acid diet containing 60% fat and 0.1% methionine—for 1 week, followed by administration of compound 20p. The experimental results were encouraging: compound 20p at doses of 10 mg/kg and 30 mg/kg acted like a “magic key”—it successfully activated GPBAR1, significantly increased plasma GLP-1 levels, and thereby effectively reduced blood glucose. Meanwhile, it also precisely activated FXR, induced the expression of SHP mRNA, and decreased the level of hepatic CYP7A1. These series of experimental results conclusively confirm that compound 20p is a non-bile acid dual agonist of FXR/GPBAR1. This finding provides highly valuable reference for in-depth exploration of GPBAR1’s structure and function, as well as for addressing therapeutic challenges in related diseases, and also points out the direction for the subsequent development of novel metabolic regulatory drugs.
Furthermore, researchers successfully screened two 12β-methyl-17-epi-18-nor-cholic acid compounds, namely NZP196 and 917, from a diverse bile acid library consisting of synthetic 12β-methyl-18-nor-cholic acid and its C17-epimers.17 These two compounds, regarded as “promising rising stars”, are glycine conjugates of 6α-ethyl-substituted 12β-methyl-18-nor-cholic acid with side chains located on the α-face of the steroid skeleton, and they act as potent GPBAR1 agonists. Strong evidence from SEAP reporter gene assays confirmed that they exhibit no activity on FXR, demonstrating high GPBAR1 selectivity. This discovery not only provides a brand-new tool for in-depth research on the structure and function of GPBAR1 but also offers a new compound basis for the treatment of complex diseases such as HBV-LF, which is expected to play a significant role in future clinical research.
From the macroscopic perspective of tissue distribution, GPBAR1 acts like an omnipresent guardian, widely present in various tissue cells.18 Within the liver—a vital metabolic organ—GPBAR1 is mainly expressed in non-parenchymal cells such as LSECs, Kupffer cells, and biliary epithelial cells, while its presence is barely detectable in hepatocytes.19 This unique tissue distribution feature, like a mysterious code, strongly suggests that GPBAR1 plays a specific and irreplaceable role in the regulation of liver physiological functions. It may exert key effects in processes such as maintaining liver internal environment homeostasis, regulating immune responses, and participating in the enterohepatic circulation of bile acids.20
In the critical field of drug development, based on the Rule of Five theory, researchers have taken a novel approach by adding polyethylene glycol of appropriate length to the GPBAR1 pharmacophore. This ingenious structural modification is like equipping the drug with a precise navigation system, successfully enabling the identification of GPBAR1 agonists that either exert systemic effects or act only within the intestinal lumen.21 This breakthrough achievement has injected new vitality into subsequent drug development targeting GPBAR1, providing crucial ideas and directions for structural modification. It helps researchers design innovative drugs with better efficacy and stronger targeting, bringing new hope for overcoming diseases related to GPBAR1.
Activation of GPBAR1: Ligand Selectivity and Cross-Organ Crosstalk
In the complex physiological regulatory network of the human body, bile acids, as the endogenous ligands of GPBAR1, exhibit highly sophisticated ligand specificity in regulating GPBAR1 (Figure 3). This specificity is concentrated in the modulation of NLRP3 inflammasome activation, thereby achieving precise control over the inflammatory response of macrophages.22,23
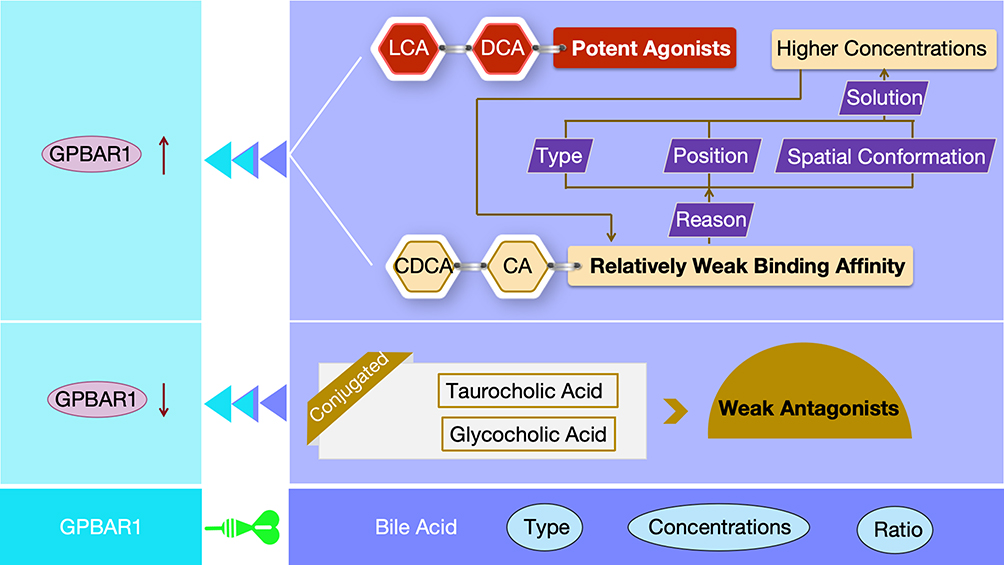 |
Figure 3 Schematic Diagram of the Differences in Activation of GPBAR1 by Different Types of Bile Acids. |
At the molecular structural level, different types of bile acids exhibit significant differences in their binding affinity to GPBAR1 and their ability to activate GPBAR1, due to marked variations in their chemical structures. LCA and DCA, by virtue of their unique chemical structures, are classified as potent agonists.24 At physiological concentrations, they can specifically bind to GPBAR1, induce conformational changes in GPBAR1, initiate the receptor activation process, and rapidly trigger a series of complex downstream signal cascades, thereby playing a key role in physiological processes such as maintaining the body’s physiological balance.25 For bile acids such as CDCA and CA, factors including the type, position, or spatial conformation of certain groups in their chemical structures result in relatively weak binding affinity to GPBAR1.26 Typically, these bile acids require higher concentrations to bind sufficiently to GPBAR1 and induce receptor activation, thereby exerting their physiological effects and participating in the regulation of physiological processes such as bile acid metabolism and lipid metabolism.27
Conjugated bile acids, such as taurocholic acid and glycocholic acid, form new structural forms through binding to amino acids.28 This modification greatly alters their interaction mode with GPBAR1, resulting in significantly weaker ability to activate GPBAR1. Some conjugated bile acids can even act only as weak antagonists, exerting a negative regulatory effect on GPBAR1 activity in the cellular microenvironment to maintain intracellular signal balance and prevent physiological dysfunction caused by excessive activation of GPBAR1.29 This ligand selectivity derived from differences in the chemical structure of bile acids provides a crucial molecular basis for researchers to further explore and precisely regulate the GPBAR1 signaling pathway. It also indicates that under different physiological and pathological conditions, targeted intervention in GPBAR1-related physiological processes is expected to be achieved by regulating the type, concentration, and ratio of bile acids—thereby offering new strategies for the treatment of related diseases. For instance, in diseases associated with abnormal bile acid metabolism, attempts can be made to regulate GPBAR1 activity by modifying bile acid composition to alleviate disease symptoms.
Notably, the bile acid profile is significantly altered in patients with HBV-LF, characterized by markedly elevated levels of conjugated bile acids (eg., TCDCA, GCDCA) and reduced proportions of potent GPBAR1 agonists including LCA and DCA. Such a shift in bile acid composition tends to attenuate GPBAR1 activation in the HBV-LF liver microenvironment, which may further weaken the anti-inflammatory and hepatoprotective effects mediated by GPBAR1 signaling. However, direct clinical evidence linking the HBV-LF-specific bile acid signature to GPBAR1 activity remains limited and requires further verification.
In-depth studies have revealed that conjugated bile acids, such as TCDCA and GCDCA, play an indispensable role in macrophage-mediated inflammatory disease models.30 Taking the severe acute pancreatitis model as an example, TCDCA and GCDCA can specifically activate the GPBAR1 pathway in macrophages.31 They first bind to GPBAR1, inducing conformational changes in GPBAR1, which then recruits and activates downstream G proteins. The activated G proteins further trigger a series of effector molecules; through complex and precise molecular events—including protein-protein interactions, phosphorylation modifications, and the activation/inhibition of signaling molecules—they effectively suppress the activation of the NLRP3 inflammasome. Inhibition of NLRP3 inflammasome activation reduces the production and release of downstream inflammatory factors, thereby significantly alleviating the inflammatory response, mitigating the degree of pancreatic tissue damage, and improving disease progression. This finding not only uncovers the critical regulatory role of conjugated bile acids in the pathogenesis of severe acute pancreatitis but also opens up a novel direction for the therapeutic strategies of this disease. It provides a theoretical basis for the development of new GPBAR1-targeted therapeutic drugs, such as drugs that can promote the production of TCDCA and GCDCA or enhance their binding ability to GPBAR1.32
In the research process of HBV-LF, it is particularly critical to fully draw on the mechanism of bile acids in other disease models when comprehensively and in-depth analyzing the activation and signal transduction mechanisms of GPBAR1.33 The common inflammatory regulatory mechanism of the bile acid-GPBAR1-NLRP3 axis revealed in severe acute pancreatitis research strongly suggests that GPBAR1 is highly likely to participate in the occurrence and development of hepatic inflammation through similar signaling pathways under the complex and dynamically changing inflammatory microenvironment of HBV-LF.
During the process of hepatocyte injury caused by persistent HBV infection, multiple factors—including viral infection, immune response, and hepatocellular metabolic disturbance—lead to severe disorders in bile acid metabolism, which in turn cause significant changes in the levels of specific bile acids. These altered bile acid levels may directly affect the activation state of GPBAR1: by regulating the activity of the NLRP3 inflammasome, they influence the inflammatory response of liver immune cells such as macrophages and Kupffer cells. When macrophages are overactivated, they release a large number of proinflammatory cytokines, further exacerbating hepatic inflammatory damage; meanwhile, abnormal function of Kupffer cells may impair their ability to effectively clear pathogens and damaged cells, resulting in persistent inflammation and aggravated liver tissue injury.34,35 Ultimately, these complex interactions exert a profound impact on the disease progression and outcome of HBV-LF. This finding opens up a new research direction for exploring the role and mechanism of GPBAR1 in HBV-LF. From the perspective of common inflammatory regulation, it is expected to bring breakthrough progress in the clinical treatment and new drug development of HBV-LF—for example, developing drugs that can regulate bile acid levels or GPBAR1 activity to interfere with the inflammatory response and improve liver function. One potential approach could be the development of a drug that modulates bile acid levels to maintain GPBAR1 in a moderately activated state, thereby inhibiting the excessive activation of the NLRP3 inflammasome and alleviating hepatic inflammation.
Furthermore, existing studies have shown that chronic liver disease-induced muscle atrophy in mice is accompanied by excessive activation of the ubiquitin-proteasome system. This is specifically manifested by a significant upregulation in the protein levels of atrophy-related ubiquitin ligases such as atrogin-1/MAFbx and MuRF-1, along with a marked increase in oxidative stress markers. The occurrence and development of this phenomenon depend on the expression of the bile acid membrane receptor GPBAR1.36 In the context of chronic liver disease, impaired liver function leads to bile acid metabolic imbalance. Abnormal bile acid signals are transmitted to muscle tissue via GPBAR1, which may disrupt normal intracellular signal transduction in muscle cells, disturb the balance between protein synthesis and degradation, and result in excessive muscle protein degradation. Meanwhile, enhanced oxidative stress further damages muscle cells, ultimately triggering muscle atrophy. This finding suggests that in HBV-LF research, in addition to focusing on local liver lesions, great attention must also be paid to the systemic complications it induces. The GPBAR1 signaling pathway may serve as a key bridge connecting liver lesions to complications such as muscle atrophy, providing new clues for in-depth understanding of the systemic pathophysiological processes of HBV-LF. This is conducive to formulating more comprehensive and effective therapeutic strategies—strategies that not only focus on liver function recovery but also take into account the prevention and treatment of systemic complications, thereby improving patients’ quality of life and prognosis. For example, in the treatment of HBV-LF, in addition to liver-protective and antiviral therapies targeting the liver, consideration could be given to regulating the GPBAR1 signaling pathway to prevent or alleviate complications such as muscle atrophy.
Signal Transduction of GPBAR1: CAMP-PKA Axis vs. β-Arrestin Axis
GPBAR1 is a GPCR for secondary bile acids. The signal transduction network of activated GPBAR1 is characterized by multi-pathway synergy, and its biological effects are mainly mediated through the following two types of pathways (Figure 4).
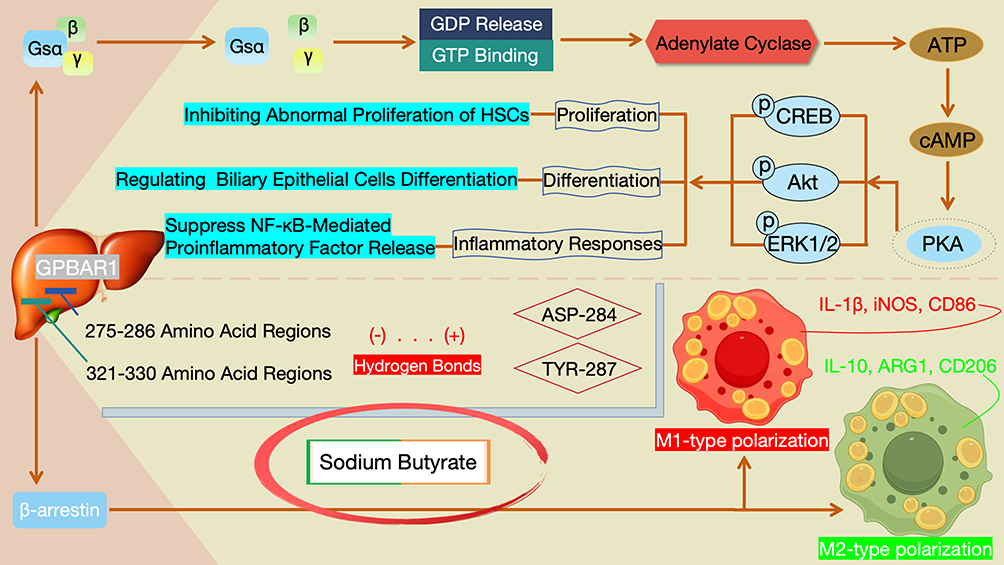 |
Figure 4 Schematic Diagram of the GPBAR1 Signaling Transduction Network: Molecular Mechanisms and Cross-Talk Between Canonical and Non-Canonical Pathways. |
Gs protein-coupled classical pathway: In the liver, activated GPBAR1 first undergoes conformational coupling with Gs proteins, which promotes the dissociation of the α subunit (Gsα) from the βγ subunits of Gs proteins. This dissociation is accompanied by the release of GDP and binding of GTP, thereby activating Gsα.37 Activated Gsα can directly activate adenylate cyclase on the cell membrane, catalyzing the conversion of intracellular ATP to cAMP and leading to a rapid increase in intracellular cAMP levels.38 As a key second messenger, cAMP further activates PKA: after activation, PKA phosphorylates downstream target proteins (such as transcription factors or kinases including CREB, Akt, and ERK1/2), and participates in regulating biological processes such as cell proliferation (eg., inhibiting abnormal proliferation of HSCs), differentiation (eg., regulating the differentiation of biliary epithelial cells), and inflammatory responses (eg., suppressing the release of proinflammatory factors mediated by the NF-κB pathway).5,39
In addition to the classical Gs-PKA pathway, the activation of GPBAR1 can also recruit β-arrestin proteins to initiate non-classical signal transduction.40 Relevant studies have further confirmed the function of this non-classical pathway, with the mechanism of action of sodium butyrate as a typical example: through surface plasmon resonance, limited proteolysis mass spectrometry, and molecular docking verification, sodium butyrate identifies GPBAR1 as its target. It can bind to the 275–286 and 321–330 amino acid regions of GPBAR1 and form hydrogen bonds with ASP-284 and TYR-287. In the in vitro lipopolysaccharide-induced macrophage polarization model, sodium butyrate can reverse the lipopolysaccharide-induced upregulation of M1-type polarization markers (IL-1β, iNOS, CD86) and simultaneously restore the expression of M2-type polarization markers (IL-10, ARG1, CD206) by activating the GPBAR1/β-arrestin2 pathway. Thus, it inhibits the polarization of macrophages toward the pro-inflammatory M1 type and promotes their polarization toward the anti-inflammatory M2 type.41 This mechanism not only clarifies the role of the GPBAR1/β-arrestin2 non-classical pathway in regulating cell functions but also provides a reference for exploring the inflammatory regulatory effect of this pathway in HBV-LF.
In the field of NSCLC cell research, GPBAR1 plays a crucial role in the disease progression.42 Studies have shown that two specific agonists, R399 and INT-777, exhibit distinct regulatory effects on the growth and apoptosis of NSCLC cells.43 After R399 induces GPBAR1 activation, it promotes GRK2 to phosphorylate GPBAR1 at the S310, S321, S323, and S324 sites. This phosphorylation further facilitates the binding of GPBAR1 to β-arrestin 1, thereby redirecting the signal transduction toward the β-arrestin 1 signaling pathway. The activated β-arrestin 1 signaling pathway ultimately stimulates the proliferation of NSCLC cells by activating the YAP signal. In contrast, INT-777 preferentially activates the GPBAR1-Gs signaling pathway, which inhibits YAP to suppress cell proliferation and induce cell apoptosis. These findings indicate that under the action of different agonists, GPBAR1 differentially regulates tumor cell behaviors through classical and non-classical signaling pathways. This not only provides new insights for understanding tumor growth mechanisms and the development of related drugs but also reflects the complexity and diversity of the GPBAR1 signaling pathway in different disease contexts. It also offers certain references for exploring the potential similar regulatory mechanisms of GPBAR1 in HBV-LF.
In the complex pathological process of HBV-LF, the signaling pathway mediated by GPBAR1 does not operate in isolation. Instead, it forms an elaborate regulatory network through highly coordinated “cross-talk”.44 On one hand, the classical cAMP-PKA pathway can dynamically regulate the affinity and binding mode between β-arrestin and GPBAR1 via the phosphorylation modification of β-arrestin, thereby affecting the activation efficiency and duration of β-arrestin-dependent non-classical signals. On the other hand, the GPBAR1 internalization process mediated by β-arrestin can inversely affect the coupling efficiency of Gs proteins, altering the activation kinetics of signaling molecules (such as cAMP and PKA) in the classical pathway. This bidirectional and synergistic regulatory mode endows GPBAR1 with the ability to flexibly adjust signal output according to the cellular microenvironment (eg., fluctuations in bile acid concentration, changes in inflammatory stress levels). It ensures that GPBAR1 accurately participates in the maintenance of liver physiological homeostasis (eg., balance of bile acid synthesis, transport, and enterohepatic circulation) and the repair of pathological damage (eg., regulation of extracellular matrix metabolism during liver fibrosis, modulation of immune cell recruitment and inflammatory factor release during inflammatory injury). This complex yet orderly signal regulatory network provides a key signaling pathway perspective for in-depth analysis of the role of GPBAR1 in the pathogenesis of HBV-LF.
Association Between GPBAR1 and HBV-LF
As a key receptor in the bile acid signaling pathway, GPBAR1 plays a crucial regulatory role in the occurrence and development, disease assessment, and prognosis prediction of HBV-LF. Its abnormal expression, changes in methylation status, and signaling pathway regulatory mechanisms provide core directions for investigating the pathological mechanisms of HBV-LF and exploring therapeutic targets.
Expression Changes of GPBAR1 in Patients with HBV-LF
GPBAR1 is closely associated with liver failure in the context of this disease. A study involving 33 critically ill patients with liver failure showed that serum bile acids from 67% (22 cases) of the patients could significantly activate GPBAR1. These bile acids severely impaired monocyte function, notably reducing the monocytes’ ability to release pro-inflammatory cytokines such as TNF-α and IL-6 upon bacterial stimulation. In contrast, no such changes were observed in serum bile acids from healthy individuals or in monocytes that do not express GPBAR1.45 Additionally, serum bile acids capable of activating GPBAR1 are a risk factor for death in patients with liver failure, independent of disease severity. This suggests that GPBAR1 and bile acid metabolism may serve as potential targets for the treatment of immune dysfunction in liver failure.
The aberrant promoter methylation and dysregulated expression of GPBAR1 have been reported in HBV-related liver diseases including CHB and HBV-HCC (non-LF models).46 This evidence, obtained exclusively from CHB and HBV-HCC (non-LF conditions), is cautiously extrapolated to HBV-LF with the following caveats: (1) the extrapolation is based solely on shared core pathological processes (excessive inflammation, bile acid metabolic disturbance, and progressive hepatocyte injury); (2) no direct evidence of GPBAR1 methylation in HBV-LF patients has been reported to date; (3) the epigenetic regulation of GPBAR1 may differ between chronic non-LF HBV liver disease and acute/fulminant HBV-LF due to distinct pathological kinetics (acute massive hepatocyte necrosis vs. chronic progressive injury). In HBV-LF, hypermethylation of the GPBAR1 promoter is a prominent, disease-specific epigenetic alteration that leads to a significant downregulation of GPBAR1 mRNA and protein expression levels, and this epigenetic change is closely correlated with the clinical severity, disease progression, and poor prognosis of HBV-LF. Importantly, this specific GPBAR1 promoter hypermethylation event holds great potential as a novel, non-invasive epigenetic biomarker for HBV-LF, which can be used for disease stratification, prognosis prediction, and even the identification of patients who may benefit from GPBAR1-targeted therapeutic interventions.
The Impact of Abnormal GPBAR1 Expression on the Pathogenesis and Prognosis of HBV-LF
In terms of the association between GPBAR1 and HBV-LF, low GPBAR1 expression is closely correlated with the severity of the disease and poor prognosis in patients with HBV-LF. Studies have revealed that GPBAR1 can influence the progression of liver fibrosis by regulating mitochondrial fission in HSCs (CHB/liver fibrosis model, non-LF).47 Specifically, activation of GPBAR1 using INT-777 inhibits the activation of the human HSCs line LX-2 induced by the HBx protein (in vitro non-LF model). This effect is associated with reduced mitochondrial fission and can alleviate liver fibrosis; this anti-fibrotic mechanism is extrapolated to HBV-LF with caution, as liver fibrosis is a chronic pathological process and HBV-LF is characterized by acute massive hepatocyte necrosis rather than progressive fibrosis (fibrosis may be a pre-existing condition in ACLF but not the core pathological driver of acute liver failure). In contrast, liver fibrosis is exacerbated in GPBAR1 knockout mice, while the mitochondrial fission inhibitor Mdivi-1 can mitigate fibrosis. These findings indicate that GPBAR1 mediates liver fibrosis by regulating mitochondrial fission in HSCs, which may be associated with HBV-LF. This suggests that GPBAR1 is a potential biomarker for evaluating the prognosis of patients with HBV-LF.
HBV-related HCC often progresses to liver failure in advanced stages; however, the anti-inflammatory and anti-tumor mechanisms of GPBAR1 observed in HCC (non-LF, chronic proliferative model) are extrapolated to the prevention of HBV-LF with critical caution: (1) HCC is driven by uncontrolled cell proliferation and malignant transformation, while HBV-LF is driven by acute massive cell death; (2) the GPBAR1-mediated signaling pathways regulating proliferation may not overlap with those regulating apoptosis/necrosis in HBV-LF; (3) these mechanisms are only applicable to the subset of HBV-LF secondary to advanced HCC, not de novo HBV-LF induced by acute viral exacerbation. GPBAR1 acts as a negative regulator of the STAT3 signaling pathway, and its deficiency promotes chemically induced hepatocarcinogenesis and acute liver injury.48 These findings support the protective role of GPBAR1 in both chronic liver tumor progression and acute liver failure episodes.
Abnormal methylation of the GPBAR1 promoter can serve as a potential biological biomarker for evaluating the prognosis of patients with ACHBLF.49 Compared with patients with CHB and healthy controls, patients with ACHBLF showed significant differences in the methylation status of the GPBAR1 promoter and mRNA expression levels in peripheral blood mononuclear cells: the methylation frequency of the GPBAR1 promoter was significantly increased in ACHBLF patients, while the corresponding mRNA expression level was significantly decreased. Further analysis revealed that the methylation status of the GPBAR1 promoter not only distinguishes ACHBLF patients from CHB patients with high specificity but is also closely associated with poor survival outcomes in ACHBLF patients. Moreover, in predicting the 3-month mortality of ACHBLF patients, the efficacy of this methylation indicator is significantly superior to that of the Model for End-Stage Liver Disease score.
The Association of GPBAR1 with Other Liver Diseases
To better understand the role of GPBAR1 in HBV-LF, we briefly summarize its function in other liver injuries (predominantly alcoholic liver injury and NASH, as well as drug-induced liver injury and MASH). All mechanistic insights derived from these non-viral liver disease models are presented solely for supplementary contextual reference, and critical distinctions are drawn between these alcohol/metabolic-associated mechanisms and the yet-to-be-validated mechanisms of GPBAR1 in viral hepatitis-induced HBV-LF. These contents are only for supplementary mechanistic reference, and all discussions are explicitly transitioned back to and integrated with the pathological mechanisms and therapeutic target value of HBV-LF; no direct extrapolation of alcohol/NASH-validated GPBAR1 mechanisms to HBV-LF is made, and this review does not expand on the therapeutic applicability of these mechanisms in other liver diseases. These diseases share similar core pathological processes (inflammation, hepatocyte apoptosis, and metabolic dysregulation) with HBV-LF, providing a tentative theoretical basis for exploratory mechanistic inference to HBV-LF (rather than definitive mechanistic confirmation). The specific mechanisms and supporting evidence are provided below, as shown in Table 1.
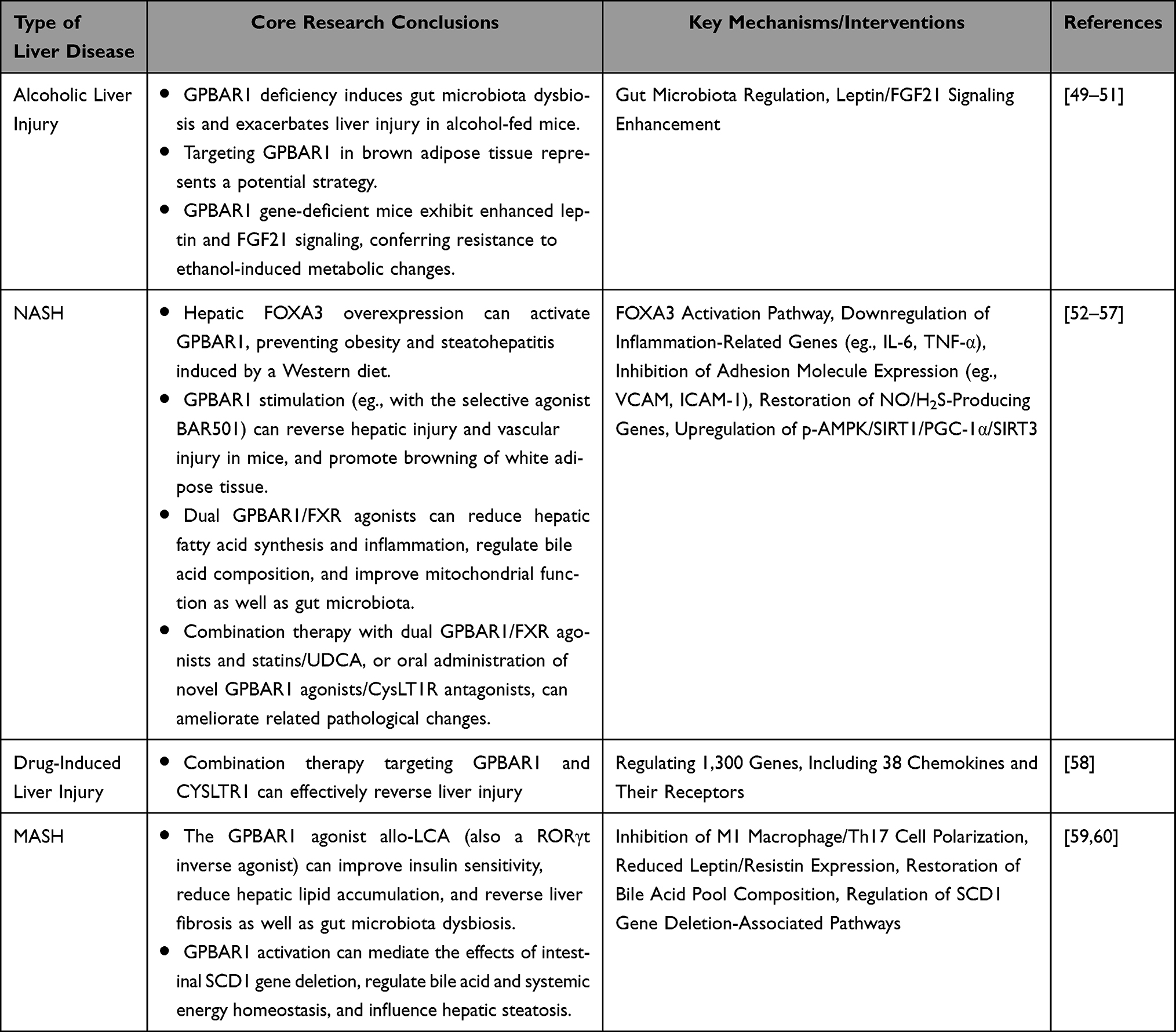 |
Table 1 Mechanisms of Action and Research Evidence of GPBAR1 in Alcoholic Liver Injury, NASH, Drug-Induced Liver Injury, and MASH |
The Mechanism of GPBAR1 in HBV-LF
HBV-LF is triggered by HBV infection, characterized by massive hepatocyte necrosis and acute liver failure as the core features, accompanied by uncontrolled inflammation and bile acid metabolism disorders. HBV-LF is characterized by interconnected core pathological processes (uncontrolled inflammation, massive hepatocyte apoptosis, bile acid metabolism disorder, and hepatic immune-microcirculation imbalance), which form a vicious cycle driving disease progression. As the core regulatory molecule, GPBAR1 does not regulate these processes independently; instead, it mediates an integrated intracellular and intercellular regulatory network (synthesized from Figures 5–9) that links the four pathological processes, and the regulatory effects of GPBAR1 on a single process can further reverse or alleviate the other three pathological abnormalities. This integrated network is the core molecular basis for GPBAR1’s protective role in HBV-LF, and its dysfunction will break the balance of the entire network, exacerbating the vicious cycle of HBV-LF pathogenesis. As a key bile acid receptor, GPBAR1 can regulate metabolism, inhibit inflammation, protect against cell apoptosis, and modulate immunity. It forms a “protective network” through multiple mechanisms in HBV-LF, while its dysfunction exacerbates liver injury. Elucidating this mechanism can provide a basis for targeted therapy, which is specifically elaborated from four core mechanisms. The four mechanisms are discussed in a progressive manner: from the initiating pathological process (uncontrolled inflammation, Regulation of Hepatic Inflammatory Response) to the core effector process (hepatocyte apoptosis, Effects on Hepatocyte Apoptosis), then to the metabolic link (bile acid metabolism disorder, Regulation of Bile Acid Metabolism), and finally to the terminal protective process (immune-microcirculation regulation, Regulation of Immune Response and Microcirculation in HBV-LF by GPBAR1). Each process is not independent, and the cross-regulation between them is highlighted to form an integrated regulatory network, avoiding isolated and repetitive discussion.
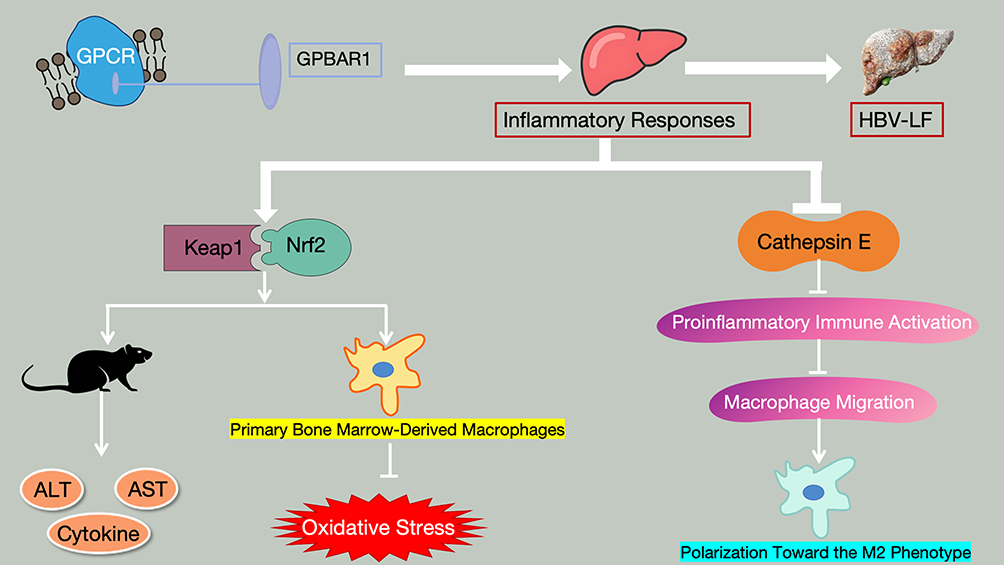 |
Figure 5 Schematic Diagram of the Core Mechanisms by Which GPBAR1 Regulates Hepatic Inflammatory Responses in HBV-LF (Keap1-Nrf2 Pathway and Cathepsin E Inhibition Pathway). |
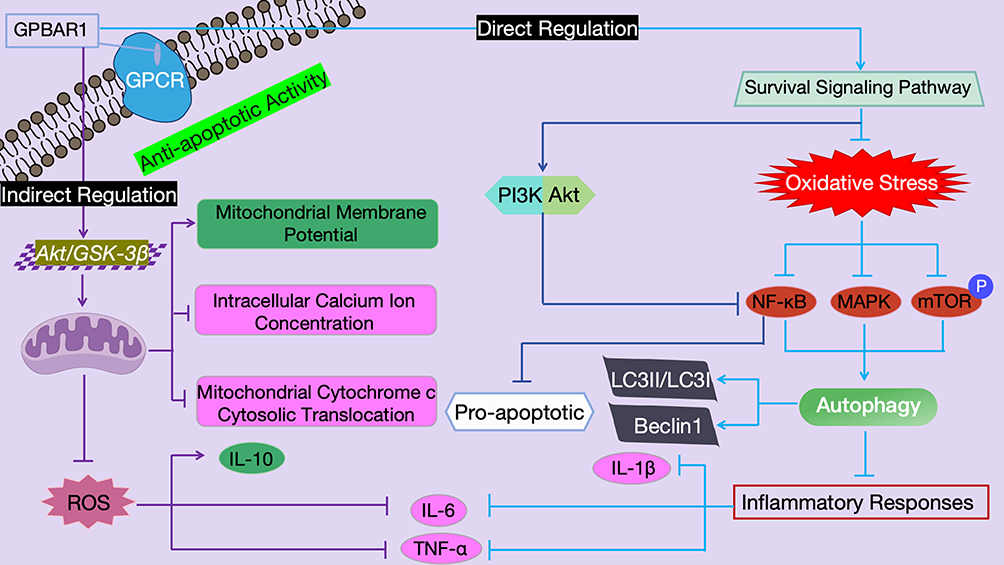 |
Figure 6 Schematic Diagram of the Mechanism by Which GPBAR1 Regulates Hepatocyte Apoptosis Through the Mitochondrial Apoptotic Pathway and Survival Signaling Pathway. |
 |
Figure 7 Schematic Diagram of the Mechanism by Which GPBAR1 Regulates Bile Acid Metabolism and Related Pathological Cycles in HBV-LF. |
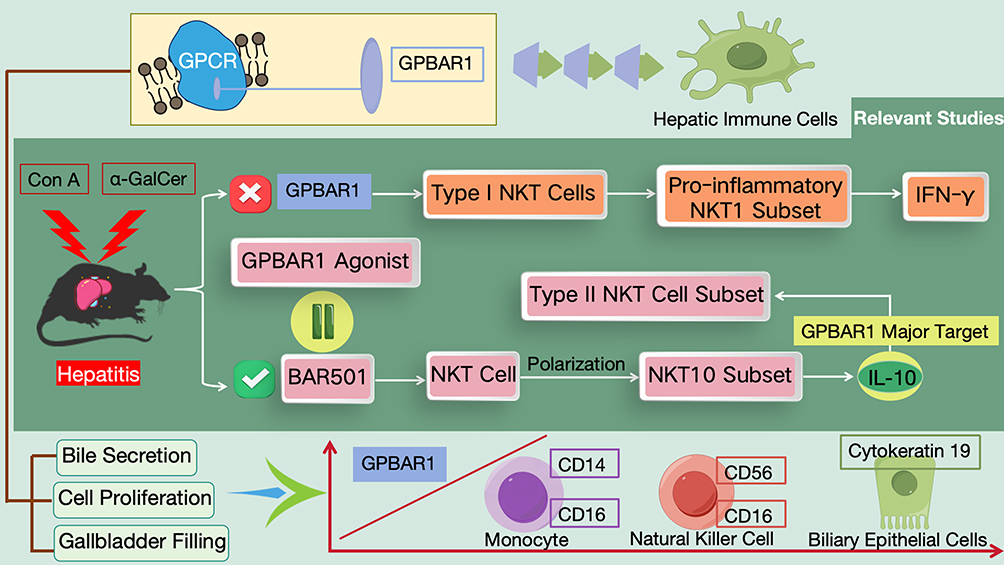 |
Figure 8 Schematic Diagram of the Molecular Mechanism by Which GPBAR1 Participates in Hepatic Immune Defense by Regulating NKT Cell Populations (Pro-Inflammatory/Regulatory Differentiation). |
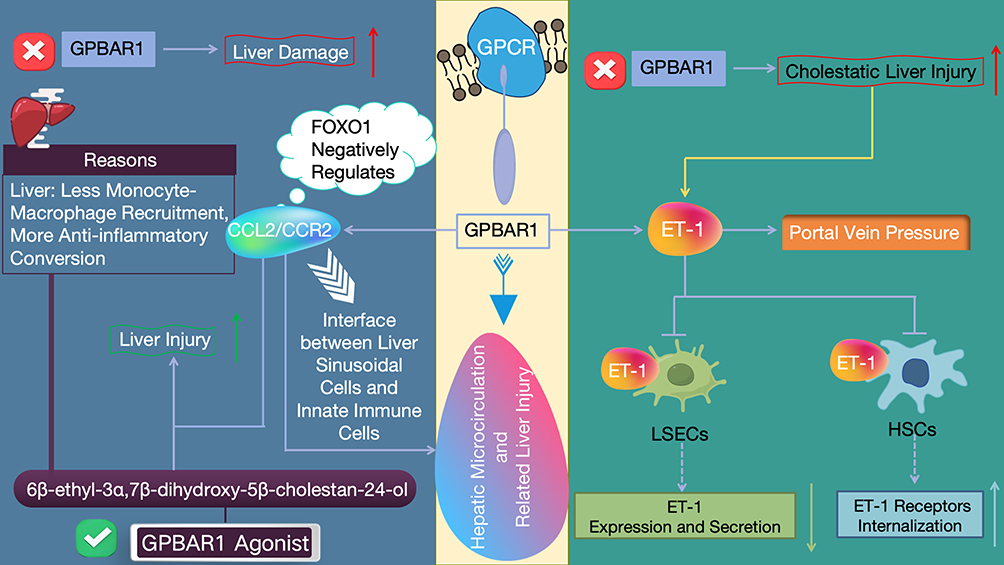 |
Figure 9 Schematic Diagram of the Core Mechanisms by Which GPBAR1 Regulates Hepatic Microcirculation and Liver Injury (Negative Regulation of the CCL2/CCR2 Axis + Inhibition of ET-1 Signaling). Symbol Definitions: • Red cross (✗): Denotes genetic knockout or functional ablation of the GPBAR1 gene, indicating complete loss of GPBAR1 expression and signaling activity. • Green checkmark (✓): Identifies the small-molecule compound 6β-ethyl-3α,7β-dihydroxy-5β-cholestan-24-ol as a selective GPBAR1 agonist that activates GPBAR1 signaling. • Red upward arrow (↑): Indicates a statistically significant increase in the severity of the corresponding pathological endpoint (liver damage, cholestatic liver injury, or portal vein pressure) in GPBAR1-deficient conditions. • Green upward arrow (↑): Represents the protective effect of GPBAR1 activation (via its agonist) that leads to a reduction in the severity of liver injury—specifically, the enhanced attenuation of liver damage through inhibited monocyte-macrophage recruitment and promotion of anti-inflammatory macrophage polarization. • Blue downward arrow (↓): Represents the inhibitory/downregulatory effect of GPBAR1 activation on pathological downstream processes, including hepatic microcirculation dysfunction and liver injury. • White downward arrow with a chevron tail (↧): Signifies the FOXO1-mediated negative regulatory effect of GPBAR1 activation on the expression and activity of the CCL2/CCR2 signaling axis. |
Regulation of Hepatic Inflammatory Response
In the development and progression of HBV-LF, excessive and uncontrolled inflammatory response is one of the core pathological mechanisms driving massive hepatocyte necrosis and rapid deterioration of liver function.61 As a key organ for immune response, after HBV infection, viral components (eg., HBV DNA, HBsAg, HBx) can be recognized by innate immune cells in the liver, such as Kupffer cells and dendritic cells, via pattern recognition receptors (eg., TLR4, RIG-I). This recognition activates downstream signaling pathways and triggers the release of a large number of proinflammatory cytokines (eg., TNF-α, IL-6, IL-1β), which further recruit neutrophils, lymphocytes, and other cells to infiltrate the liver, forming an “inflammatory cascade amplification effect” and ultimately leading to extensive hepatocyte injury.62,63 As an important inflammatory regulatory molecule, GPBAR1 plays a crucial role in maintaining hepatic inflammatory homeostasis by precisely targeting the NF-κB signaling pathway to inhibit excessive hepatic inflammatory response.64 Studies have confirmed that the bile acid receptor GPBAR1 can antagonize NF-κB activation by enhancing the interaction between IκBα and β-arrestin 2, thereby inhibiting hepatic inflammation in mice. Activation of GPBAR1 reduces LPS-induced hepatic inflammation in wild-type mice (this effect is absent in GPBAR1 knockout mice)65 (LPS-induced acute liver injury model, non-HBV-LF). This anti-inflammatory mechanism is extrapolated to HBV-LF and suggests GPBAR1 is a potential therapeutic target for immune and inflammatory liver diseases; caution is warranted as LPS-induced sterile inflammation differs from HBV-induced viral immune inflammation, the core inflammatory driver of HBV-LF.
In the regulation of hepatic inflammatory responses, GPBAR1 can be activated by LCA and DCA. Activated GPBAR1 promotes the production of cAMP, and the increased cAMP further reduces the generation of proinflammatory cytokines, thereby exerting an anti-inflammatory effect.66 During the pathological progression of liver failure, the expression of GPBAR1 shows dynamic changes: its expression level increases in the early stage to exert a protective effect on the brain; however, as the disease progresses, its expression decreases when the level of ammonia in the body rises.67 It should be noted that hepatic encephalopathy, a common complication of acute liver failure, may be associated with the changes in GPBAR1 expression and the aforementioned inflammatory regulation process in its occurrence and development.68
GPBAR1 participates in the HBV-LF-associated pathological processes by regulating hepatic inflammatory responses, which is specifically achieved through two key mechanisms (Figure 5): First, GPBAR1 can activate the Keap1-Nrf2 signaling pathway to enhance the liver’s resistance to ischemia-reperfusion injury, while reducing the release of inflammatory molecules and regulating innate immunity (ischemia-reperfusion liver injury model, non-HBV-LF). This mechanism, verified in non-HBV-LF in vivo and in vitro experiments, is extrapolated to HBV-LF with caution: ischemia-reperfusion injury is a mechanical/hypoxic stress, whereas HBV-LF involves viral infection and immune-mediated injury; the Keap1-Nrf2 pathway may play a conserved role in oxidative stress regulation, but its specific activation mode and downstream targets in HBV-LF remain unconfirmed. In in vivo studies, the injection of si-GPBAR1 into wild-type mice significantly aggravated liver tissue injury, whereas the injection of the GPBAR1 agonist (INT-777) reversed this injury. This result was confirmed by the detection of serum ALT and AST, as well as observations of liver tissue injury, detection of cytokine expression, liver immunohistochemical analysis, and TUNEL staining. In the in vitro hypoxia/reoxygenation model of primary bone marrow-derived macrophages, overexpression of GPBAR1 activated by INT-777 reduced hypoxia/reoxygenation-induced oxidative stress and inflammatory responses by regulating the Keap1-Nrf2 pathway; si-GPBAR1 completely reversed this effect, which was consistent with the results of in vivo experiments.69 Second, GPBAR1 can inhibit cathepsin E to suppress proinflammatory immune activation, thereby inhibiting macrophage migration and promoting their polarization toward the M2 phenotype. Ultimately, this protects the liver from ischemia-reperfusion injury caused by insulin resistance.70
Notably, the interaction between GPBAR1 signaling and NLRP3 inflammasome activation, a key regulatory axis in hepatic inflammation, has no direct evidence from HBV-LF human tissue samples to date; all current mechanistic evidence for this pathway is primarily derived from non-HBV-LF animal models (eg., LPS-induced acute liver injury, severe acute pancreatitis, and ischemia-reperfusion liver injury models) and in vitro cellular models. No clinical validation using human HBV-LF liver tissue, peripheral blood mononuclear cells, or serum samples has been reported for the GPBAR1-NLRP3 inflammasome regulatory pathway in the context of viral hepatitis-induced liver failure.
Notably, the anti-inflammatory effect of GPBAR1 is the initiating link of its integrated regulatory network in HBV-LF (Figure 5). Inhibiting the NF-κB-mediated inflammatory cascade not only directly reduces pro-inflammatory cytokine-induced hepatocyte apoptosis (Effects on Hepatocyte Apoptosis) but also downregulates the expression of bile acid synthetic enzymes (CYP7A1/CYP8A1) induced by inflammatory stress (Regulation of Bile Acid Metabolism), and alleviates inflammatory factor-mediated LSEC damage (a prerequisite for microcirculation disorder, Regulation of Immune Response and Microcirculation in HBV-LF by GPBAR1). This section focuses only on the direct anti-inflammatory mechanism of GPBAR1 and its upstream signaling initiation; the downstream effects on microcirculation and immune cells are elaborated in their respective dedicated sections to avoid repetitive discussion.
Effects on Hepatocyte Apoptosis
In the pathological process of HBV-LF, the abnormal increase in hepatocyte apoptosis is one of the core links leading to the rapid deterioration of liver function.71 HBV infection can induce hepatocyte death and functional abnormalities through multiple pathways: HBx protein (a key viral protein in HBV-LF) can induce ferroptosis via EZH2-mediated inhibition of SLC7A11 (in vitro HBV hepatocyte injury model, direct relevance to HBV-LF),72 block the G1/S transition of the hepatocyte cycle and cause liver function loss and cell death in the regenerated liver of HBx transgenic mice73 (transgenic chronic CHB model, non-LF; extrapolated to HBV-LF with caution, as cell cycle arrest in chronic regeneration differs from acute apoptotic necrosis in HBV-LF), and disrupt the protective effect of Bcl-2 against Fas-mediated hepatic apoptosis. This process is associated with HBx-induced changes in mitochondrial membrane (cytochrome c release) and activation of caspase 3.74 Meanwhile, pro-inflammatory factors such as TNF-α and IL-1β produced by excessive intrahepatic inflammatory responses can further exacerbate hepatocyte apoptosis by activating caspase family proteases (eg., caspase-1).75 As an important regulatory molecule for cell survival, activated GPBAR1 can exert anti-apoptotic effects through multi-dimensional mechanisms, effectively maintaining hepatocyte survival and liver function stability.76
The cytoprotective, anti-apoptotic activity of GPBAR1 is orchestrated through two functionally integrated signaling axes (Figure 6). Direct regulation of the mitochondrial apoptotic pathway: Mitochondria serve as the “core hub” for the regulation of cell apoptosis, and the stability of their membrane potential directly determines the cell survival status.77 By activating the Akt/GSK-3β pathway, GPBAR1 improves mitochondrial function (eg., stabilizing mitochondrial membrane potential, reducing intracellular calcium ion concentration, and decreasing cytosolic translocation of mitochondrial cytochrome c), regulates cell proliferation and apoptosis, inhibits inflammatory responses by reducing the production of ROS, increasing the expression of the anti-inflammatory factor IL-10, and decreasing the expression of the pro-inflammatory factors IL-6 and TNF-α78 (non-HBV, non-LF liver injury model; extrapolated to HBV-LF with the following caveats: (1) this mechanism is validated in chemical-induced liver injury, not HBV-induced injury; (2) the Akt/GSK-3β pathway may be differentially regulated by HBV viral proteins (eg., HBx) in HBV-LF; (3) mitochondrial dysfunction in HBV-LF involves both viral direct damage and immune-mediated oxidative stress, unlike the single stress in non-LF models). Indirect regulation of the survival signaling pathway: In addition to directly regulating mitochondrial function, the activation of GPBAR1 can also downregulate the NF-κB and MAPK pathways by antagonizing oxidative stress, while inhibiting mTOR phosphorylation. This process thereby induces cell apoptosis, enhances autophagy (autophagic activity is reflected by an increased LC3II/LC3I ratio and upregulated Beclin1 expression), and suppresses inflammatory responses (manifested by decreased expression of the pro-inflammatory factors IL-1β, IL-6, and TNF-α).79,80 Furthermore, GPBAR1 can reduce NF-κB-mediated pro-apoptotic signal transduction by activating the PI3K/Akt pathway (another important survival signaling pathway).81
In liver and white adipose tissue, GPBAR1—sensing secondary bile acids—functionally antagonizes the primary bile acid receptor FXR in the control of autophagy (metabolic liver disease model, non-LF; no direct evidence for this GPBAR1-FXR crosstalk in HBV-LF).82 This regulatory mechanism is extrapolated to HBV-LF with extreme caution: (1) it is validated in metabolic disorders (eg., NAFLD) rather than viral liver failure; (2) the bile acid pool composition in HBV-LF (acute cholestasis) is very different from that in metabolic liver disease (chronic dysmetabolism); (3) the role of autophagy in HBV-LF (protective vs. detrimental) remains controversial and unlinked to GPBAR1-FXR crosstalk to date. Through a cAMP-CREB axis, GPBAR1 actively licenses autophagy during fasting; genetic deletion of the receptor shifts the bile-acid pool toward FXR agonism by eliminating murine deoxycholic acid, an endogenous FXR antagonist generated via the CYP2C70 pathway. The resulting FXR dominance precipitates a selective autophagy defect that is fully reversible by FXR inhibition. Conversely, FXR activation in the fed state terminates autophagy by counteracting CREB-mediated transcription of Atgs; conversely, loss of FXR erases the post-prandial suppression of Atgs and partially relieves nutrient-dependent inhibition of the autophagic program.
Hepatocyte apoptosis is the core effector link of GPBAR1’s integrated regulatory network in HBV-LF (Figure 6), and its regulation by GPBAR1 forms a bidirectional cross-talk with other pathological processes: on the one hand, GPBAR1-mediated inhibition of mitochondrial apoptosis and activation of PI3K/Akt pathway reduce the release of DAMPs from apoptotic hepatocytes, which further suppresses Kupffer cell activation and inflammatory factor secretion (Regulation of Hepatic Inflammatory Response), and alleviates DAMP-induced bile acid metabolic disorder in hepatocytes and cholangiocytes (Regulation of Bile Acid Metabolism); on the other hand, the reduction of massive hepatocyte apoptosis preserves the structural integrity of the liver, which maintains the normal function of LSECs and hepatic microcirculation (Regulation of Immune Response and Microcirculation in HBV-LF by GPBAR1) and restores the enterohepatic circulation of bile acids. This section focuses on the direct anti-apoptotic mechanisms of GPBAR1 and its role as a downstream effector of inflammatory signaling; detailed discussions of bile acid metabolism, immune cell regulation, and hepatic microcirculation are presented in Regulation of Bile Acid Metabolism and 5.4, respectively, to avoid content overlap and ensure conceptual progression.
Regulation of Bile Acid Metabolism
Bile acid metabolism disorder is one of the highly characteristic pathological features in the progression of HBV-LF, and it is closely associated with the abnormal regulation of GPBAR1 (Figure 7). Under physiological conditions, as a key receptor for bile acids, GPBAR1 is deeply involved in the regulation of the enterohepatic circulation of bile acids and the maintenance of metabolic homeostasis: upon activation by bile acids, GPBAR1 can promote the reabsorption efficiency of bile acids in epithelial cells at the terminal ileum through signal transduction. This process effectively reduces the abnormal accumulation of bile acids in the liver.83 Normally, the liver continuously synthesizes bile acids and secretes them into the biliary tract; if bile acids cannot be timely recycled via the enterohepatic circulation and properly disposed of, excessive accumulation will directly exert chemical toxicity on hepatocytes, damage the hepatocyte membrane structure, and cause hepatocyte edema or even necrosis. The reabsorption regulation mediated by GPBAR1 can significantly reduce the risk of such toxic damage. Meanwhile, GPBAR1 can also maintain metabolic homeostasis by regulating the expression of key enzymes in the bile acid synthesis pathway. For instance, it can finely regulate the activity of cholesterol 7α-hydroxylase (CYP7A1). As the rate-limiting enzyme in bile acid synthesis, the expression level and activity of CYP7A1 directly determine the synthesis rate of bile acids. GPBAR1 inhibits or activates the expression of this enzyme through signal transduction, preventing excessive or insufficient bile acid synthesis, thereby maintaining the stability of the intrahepatic bile acid pool.84,85
In addition to CYP7A1, CYP8A1, which is involved in the CDCA branch-specific reaction in the bile acid synthesis pathway, functions to catalyze the conversion of 7α-hydroxy-4-cholesten-3-one into 7α,12α-dihydroxy-4-cholesten-3-one. It serves as a key enzyme in the synthesis of 12α-hydroxylated bile acids (such as cholic acid). GPBAR1 can also influence the ratio of 12α-hydroxylated to non-12α-hydroxylated bile acids by regulating the expression of CYP8A1, thereby further optimizing the compositional structure of the bile acid pool.86 This precise regulation of key enzymes involved in bile acid synthesis can prevent liver toxicity caused by the excessive accumulation of a single type of bile acid. Particularly in the context of HBV-LF, dysregulation of GPBAR1 can lead to abnormal expression of CYP8A1, disrupting the balance of bile acid components and exacerbating hepatocyte injury. Restoring the normal regulation of CYP8A1 by GPBAR1 can assist in ameliorating bile acid metabolic disorders, providing a potential avenue for alleviating liver damage in HBV-LF.
Research has confirmed that in chronic liver diseases, GPBAR1 serves as a crucial target for bile acid-induced circadian rhythm sleep disorders. The underlying mechanism is as follows: upon activation, GPBAR1 upregulates the expression of Per2 and promotes its phosphorylation via the ERK and CK1ε pathways. Phosphorylated Per2 can inhibit its own nuclear translocation and relieve transcriptional repression, thereby accelerating the circadian rhythm cycle. Meanwhile, GPBAR1 also weakens the light-entrained response mediated by gastrin-releasing peptide-positive neurons and the light-induced phase shift of the suprachiasmatic nucleus through the GPBAR1-Per2 axis.87
During the process of GPBAR1 regulating bile acid metabolism in HBV-LF, the mechanisms related to colorectal liver metastasis can be referred to: intestinal microbiota-derived LCA can activate GPBAR1 in cancer-associated fibroblasts, prompting them to upregulate the secretion of CCL3, which in turn recruits MDSCs to the metastatic microenvironment. Meanwhile, CCL2 produced by MDSCs can exert a compensatory pro-inflammatory effect that suppresses immune suppression via CCR2 signaling. This provides a new direction for immunotherapy in related diseases.88 In addition, the process of GPBAR1 regulating bile acid metabolism is also associated with MASH, and its mechanism holds reference value for understanding HBV-LF. As a secondary bile acid, conjugated LCA can activate GPBAR1 in liver hepatocytes. The activated GPBAR1 binds to CD36 and further recruits the E3 ubiquitin ligase TRIM21, leading to the degradation of BBOX1, a key enzyme in de novo carnitine biosynthesis. This process promotes lipotoxicity-induced hepatocyte death and inflammatory responses, driving the transition from MASLD to MASH. Notably, the absence of GPBAR1 in hepatocytes can significantly block this progression. Targeted therapy against GPBAR1 holds the potential to restore carnitine biosynthesis, providing a potential strategy for intervening in this pathological transition and related liver diseases, including HBV-LF.89
GPBAR1 governs the composition of bile acids and the function of the gallbladder to protect the liver from damage caused by bile acid overload.90 More importantly, bile acid metabolic disorders can, in turn, exacerbate metabolic imbalances through aberrant activation of GPBAR1. Under abnormal GPBAR1 conditions, the activating effect of bile acids on it deviates from the normal regulatory pathway, leading to an abnormal elevation of cAMP levels within the liver.91 As a crucial intracellular second messenger, abnormal cAMP levels further disrupt the intracellular signaling network in hepatocytes, impairing their normal metabolic functions and survival status. This creates a vicious cycle of “bile acid metabolic disorders - GPBAR1 abnormalities - abnormal cAMP levels - aggravated hepatocyte injury”, ultimately driving the progression of HBV-LF. In exploring the mechanism by which GPBAR1 participates in HBV-LF, relevant studies have demonstrated that endotoxemia and septic shock induced by sepsis result in a significantly higher total circulating bile acid level compared to healthy controls, although the magnitude of increase is often mild in most cases. However, in patients with septic shock complicated by severe liver failure, the circulating bile acid levels are markedly and substantially higher than those in healthy controls. Moreover, the ability of circulating bile acids to activate GPBAR1 in these patients is also significantly enhanced compared to healthy controls, which can trigger immunosuppression.92 This suggests that regulating bile acid metabolism may hold clinical significance in improving the disease course and prognosis of septic shock in such patients, providing evidence for understanding the role of GPBAR1 in HBV-LF.
Bile acid metabolism disorder is the metabolic link connecting all pathological processes in GPBAR1’s regulatory network (Figure 7), and GPBAR1-mediated balance of bile acid metabolism exerts a “multi-target regulatory effect” on HBV-LF: first, correcting abnormal bile acid composition inhibits bile acid-induced GPBAR1 over-activation and NLRP3 inflammasome-mediated inflammation (Regulation of Hepatic Inflammatory Response); second, reducing bile acid cytotoxicity alleviates mitochondrial damage and hepatocyte apoptosis (Effects on Hepatocyte Apoptosis); third, restoring the enterohepatic circulation of bile acids modulates the secretion of CCL2/ET-1 by LSECs (the molecular basis of microcirculation regulation, Regulation of Immune Response and Microcirculation in HBV-LF by GPBAR1). This section focuses on the metabolic regulatory mechanism of GPBAR1 and its cross-linking role; the specific effects on inflammation, apoptosis and microcirculation are only briefly linked to their respective sections without repetitive elaboration.
Regulation of Immune Response and Microcirculation in HBV-LF by GPBAR1
In addition to the aforementioned mechanisms, GPBAR1 is hypothesized to participate in the pathogenesis of HBV-LF through two pathways: modulating the function of hepatic immune cells and influencing hepatic microcirculation. This section represents the terminal protective link of GPBAR1’s integrated regulatory network in HBV-LF (Figures 8–9), and discusses the regulatory mechanism from the cellular level (immune cell polarization) to the tissue level (hepatic microcirculation stability), realizing the conceptual advancement from single molecular pathway to tissue microenvironment regulation; all content is independent of the previous sections and avoids repetitive discussion of inflammation, apoptosis and metabolism. Notably, there is no direct experimental or clinical evidence validating these two pathways in HBV-LF models or HBV-LF patients; all mechanistic evidence is derived from non-HBV liver injury mouse models (eg., Con A, acetaminophen-induced liver injury) and healthy human cell studies, and the following mechanisms are extrapolated to HBV-LF based on shared core pathological processes (immune cell dysfunction, hepatic microcirculation disturbance) with explicit caution: no clinical relevance for HBV-LF has been demonstrated to date. As a GPCR that can be activated by both primary and secondary bile acids, GPBAR1 plays a prominent role in this process. Its expression exhibits specific cellular localization characteristics, primarily being expressed in macrophages, natural killer cells, and sinusoidal cells (such as LSECs). These cells precisely undertake crucial physiological functions: macrophages constitute a vital part of the liver’s innate immune system and are involved in immune defense and inflammatory regulation; sinusoidal cells (such as LSECs) serve as the core force in maintaining hepatic microcirculatory stability.93 The expression of GPBAR1 in these functionally critical cells provides a potential structural and functional basis for its involvement in the pathogenesis of HBV-LF by regulating hepatic immune cell function and influencing hepatic microcirculation.
In terms of regulating the function of hepatic immune cells, GPBAR1 exerts regulatory effects on the polarization and function of NKT cells (Figure 8): on one hand, it promotes the generation of regulatory NKT cells (such as NKT10 cells), and on the other hand, it inhibits the activation of inflammatory NKT cells (such as NKT1 cells), thereby modulating the hepatic immune microenvironment. Relevant studies have experimentally proven the following in non-HBV, acute liver injury mouse models (Con A or α-GalCer-induced hepatitis): GPBAR1 gene knockout exacerbates liver injury and also biases type I NKT cells towards the pro-inflammatory NKT1 subset (capable of producing IFN-γ). Conversely, treatment with the selective GPBAR1 agonist BAR501 can facilitate the recovery of wild-type mice from acute liver injury, shift NKT cell polarization towards the regulatory NKT10 subset (capable of secreting the anti-inflammatory cytokine IL-10), and significantly increase the IL-10-secreting type II NKT cell subset. RNA sequencing has revealed that IL-10 is a major target of GPBAR1, and in the Con A model, IL-10 gene knockout abolishes the protective effect of GPBAR1 agonists. This indicates that GPBAR1 can regulate hepatic immunity by modulating the hepatic NKT cell population and the GPBAR1-IL-10 axis.94 Hypothetically, this GPBAR1-IL-10-NKT cell regulatory axis may modulate the hepatic immune microenvironment in HBV-LF; however, this hypothesis has not been tested in any HBV-LF in vitro/in vivo model or clinical sample, and no clinical relevance for HBV-LF patients has been implied or confirmed. Additionally, GPBAR1 can regulate bile secretion, cell proliferation, and gallbladder filling.95 In the liver, its expression level is positively correlated with monocyte markers (CD14, CD16), natural killer cell markers (CD56, CD16), and the commonly used marker for biliary epithelial cells, cytokeratin 19. This correlation only provides a preclinical potential clue for the association between GPBAR1 expression and monocyte/natural killer cell infiltration/activation in the normal and chronic liver injury liver; it has not been validated in HBV-LF, and no inference of clinical relevance to HBV-LF can be made from this finding.96
In the regulation of hepatic microcirculation and related liver injury, GPBAR1 plays a pivotal role (Figure 9). It is expressed in various non-parenchymal cells in the liver, including biliary epithelial cells, liver-resident macrophages, LSECs, activated HSCs, and so on. Moreover, it can act at the interface between liver sinusoidal cells and innate immune cells, maintaining hepatic microenvironmental stability and regulating liver injury through different mechanisms. On one hand, GPBAR1 can influence hepatic microcirculation and liver injury by regulating the CCL2/CCR2 signaling pathway. Experimentally proven in non-HBV mouse models: (1) In acetaminophen-induced acute liver injury mice, GPBAR1 gene knockout exacerbates liver damage, while the GPBAR1 agonist 6β-ethyl-3α,7β-dihydroxy-5β-cholestan-24-ol protects against liver injury by reducing monocyte-derived macrophage recruitment and promoting their anti-inflammatory polarization; mechanistically, GPBAR1 activation negatively regulates the CCL2/CCR2 axis via FOXO1 at the liver sinusoid/macrophage interface.97 (2) In cholestatic liver injury mice (non-HBV), GPBAR1 knockout increases disease susceptibility, and GPBAR1 activation modulates hepatic ET-1 signaling through two mechanisms: reducing ET-1 secretion in LSECs and promoting ET-1 receptor internalization in HSCs, thereby regulating portal vein pressure.98 Notably, no direct evidence has demonstrated the effects of GPBAR1 agonists on portal pressure in viral hepatitis or HBV-related liver injury models to date; the regulatory effect of GPBAR1 on portal vein pressure is exclusively validated in non-HBV cholestatic liver injury mouse models, and no extrapolation to HBV-LF or HBV-related chronic liver disease has been confirmed by preclinical or clinical studies. Hypothetically, the GPBAR1-CCL2/CCR2 and GPBAR1-ET-1 axes may regulate hepatic microcirculation in HBV-LF; however, this hypothesis is unvalidated in HBV-LF models/patients, and no clinical relevance for HBV-LF has been established.
As a hypothetical terminal protective link and functional closed loop of GPBAR1’s integrated regulatory network in HBV-LF (Figures 8 and 9), the regulation of hepatic immune response and microcirculation is proposed to form a positive feedback with upstream inflammation, apoptosis and metabolism based on extrapolated non-HBV evidence: if validated in HBV-LF, GPBAR1-mediated NKT cell polarization and CCL2/CCR2 inhibition would reduce inflammatory cell infiltration (Regulation of Hepatic Inflammatory Response), decrease hepatocyte damage (Effects on Hepatocyte Apoptosis), restore bile acid metabolism (Regulation of Bile Acid Metabolism), and ultimately reverse the vicious cycle of HBV-LF pathogenesis. This section completes the conceptual advancement of GPBAR1’s regulatory mechanism from single pathway to integrated network, and no repetitive content with the previous sections is included.
Critical Comparative Analysis of GPBAR1-Based and Other GPCR-Targeted Therapeutic Strategies in HBV-LF: Mechanistic Advantages and Rationale for Prioritization
GPCRs are key signaling molecules in the regulation of hepatic physiological functions and the pathological progression of liver diseases. Functional abnormalities in multiple members of the GPCR family, such as GPBAR1, FXR, AT1 receptor, and cannabinoid receptors (CB1/CB2), are either directly associated or extrapolated to be associated with the onset and development of severe liver inflammation and HBV-LF. This section conducts a critical, HBV-LF-centric comparative analysis of GPCR-targeted therapeutic strategies, with the core goal of elucidating why GPBAR1 is the mechanistically and therapeutically superior target for HBV-LF over other GPCRs. We strictly focus on the four core pathological processes of HBV-LF (uncontrolled inflammation, massive hepatocyte apoptosis, bile acid metabolic disorders, and hepatic microcirculation disturbance) to evaluate each GPCR’s therapeutic potential, explicitly distinguish findings directly relevant to HBV-LF from those extrapolated from non-LF models, and eliminate non-essential descriptive content that does not inform GPBAR1-based intervention for HBV-LF. A concise, analytical comparative summary is presented in Table 2.
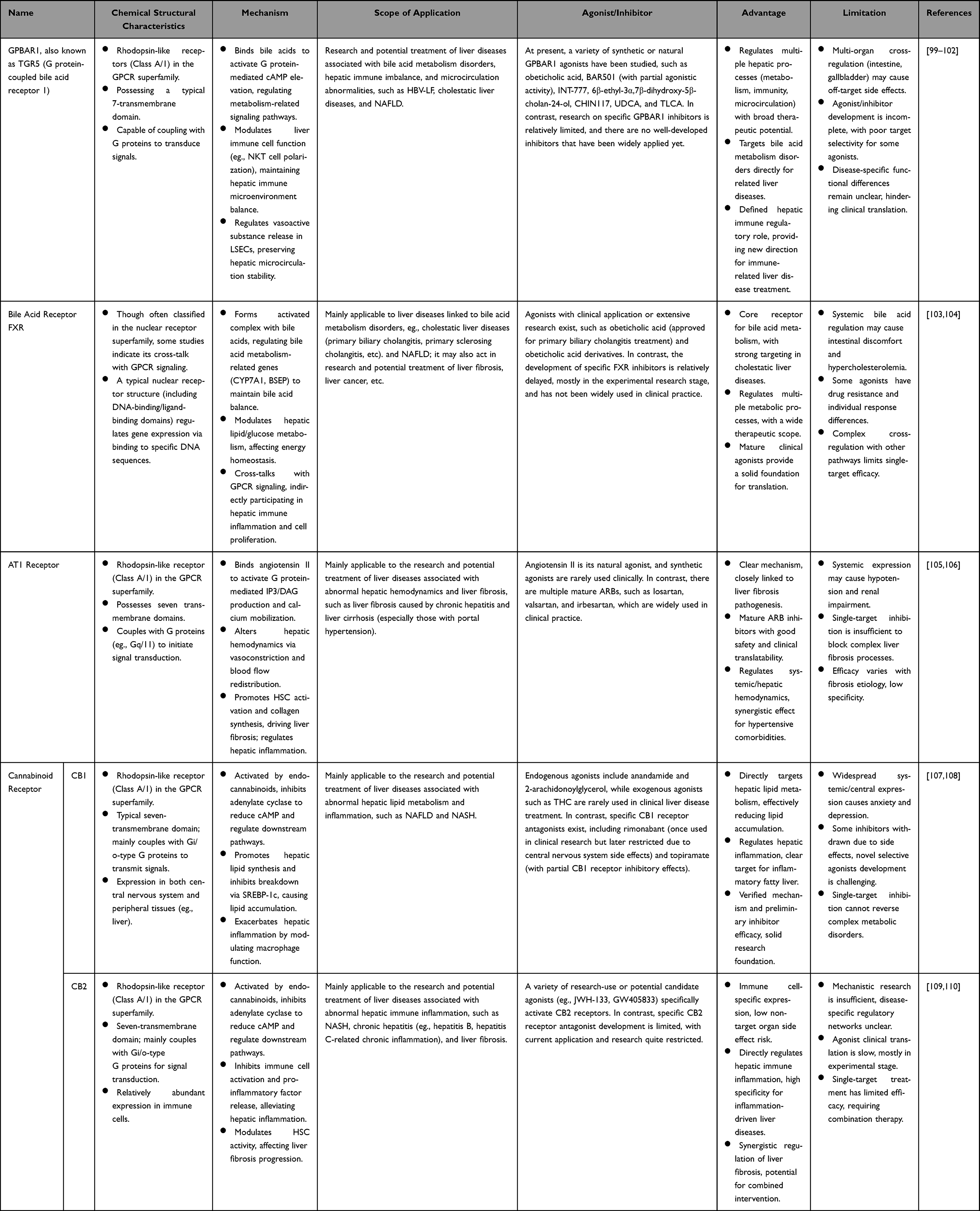 |
Table 2 Therapeutic Strategies Targeting GPCRs Associated with Liver Diseases |
Therapeutic Strategies Based on GPBAR1
Given the unique and core regulatory role of GPBAR1 in mediating the “Inflammation-Apoptosis-Metabolism-Microcirculation” integrated regulatory network specific to HBV-LF (a regulatory feature not shared by other GPCRs), developing therapeutic drugs targeting GPBAR1 holds unmatched broad prospects for the prevention and treatment of HBV-LF, and is the preferred GPCR-targeted strategy for this disease. Currently, a variety of GPBAR1 agonists have been developed and their protective effects have been directly verified in preclinical models of ACHBLF (ACLF, the main clinical subtype of HBV-LF) and related pathological stimuli (eg., HBV infection combined with acute inflammatory stress); in these animal experiments, GPBAR1 agonists can alleviate hepatic inflammation, reduce hepatocyte damage, and exert a robust protective effect against HBV-ACLF (direct evidence for this subtype of HBV-LF). However, their protective effects on acute liver failure induced by de novo severe HBV infection (another subtype of HBV-LF) have not been validated in preclinical models and are thus extrapolated from HBV-ACLF models with caution. Despite these promising preclinical findings demonstrating high experimental translational promise, GPBAR1 agonists have not yet entered any clinical trials for HBV-LF patients, and their clinical readiness is severely limited by the lack of HBV-LF–specific clinical trials and clinical evidence. However, their translation into clinical application for HBV-LF patients still faces many challenges, such as the poor tissue specificity of agonists, potential off-target side effects, and insufficient efficacy in advanced HBV-LF, which require further in-depth research and structural optimization for clinical adaptability.
Bile acids, the endogenous ligands of GPBAR1, are synthesized through the primary pathway for cholesterol catabolism and play a vital role in maintaining hepatic lipid homeostasis, a key pathological link that aggravates hepatocyte injury and promotes the progression from CHB to HBV-LF.111 As physiological amphipathic substances, bile acids mediate the absorption, distribution, metabolism, and excretion of nutrients and exogenous substances in the liver; more importantly, they act as key signaling molecules that activate nuclear receptor FXR and membrane receptor GPBAR1 synergistically. Upon activation, GPBAR1 modulates hepatic glucose, lipid, and energy metabolism, and inhibits excessive inflammatory response, thereby attenuating the cumulative liver damage induced by persistent HBV infection and blocking the progression to HBV-LF.112 In the exploration of GPBAR1-based therapeutic strategies for HBV-LF, INT-767—a dual agonist of FXR and GPBAR1—has demonstrated significant anti-HBV activity and hepatic protective effects, providing a crucial translational direction for the prevention and treatment of HBV-LF.113 In vitro and in vivo experiments confirmed that INT-767 can effectively inhibit multiple key steps of HBV infection, with a dual and synergistic mechanism: On one hand, it directly binds to HBV virions and potently inhibits viral entry into hepatocytes, with inhibitory activity significantly superior to natural bile acids; on the other hand, it activates the FXR/GPBAR1 dual signaling pathway to suppress HBV replication and transcription in host cells, and simultaneously alleviates HBV-induced hepatic inflammation and hepatocyte apoptosis. In humanized liver chimeric mouse models of HBV infection, INT-767 significantly delayed the elevation of HBsAg, HBeAg, and HBV DNA, reduced the level of covalently cccDNA, and markedly attenuated hepatic pathological damage and prevented the development of liver failure phenotypes (direct evidence for the prevention of HBV-LF from chronic HBV infection). Caution is warranted when extrapolating these findings to established HBV-LF: this model only validates the preventive effect of INT-767, not its therapeutic effect on overt HBV-LF with massive hepatocyte necrosis and established liver dysfunction, and no preclinical model has tested INT-767 in established HBV-LF to date. These preclinical results fully support the experimental promise of INT-767 for HBV-LF intervention, but its clinical readiness remains completely unproven due to the absence of HBV-LF–specific clinical trials and limited clinical safety and efficacy data in patients with liver failure. These findings confirm that dual regulation targeting GPBAR1 and FXR is a promising anti-HBV strategy, and the GPBAR1 component is the key to its efficacy in HBV-LF: GPBAR1 mediates the rapid inhibition of uncontrolled inflammation and hepatic microcirculation disturbance (the acute, life-threatening core pathological processes of HBV-LF), while FXR only modulates chronic bile acid metabolism disorder (a secondary pathological feature in established HBV-LF). This dual regulation can reduce the cumulative liver injury caused by chronic HBV infection, block the progression from CHB to HBV-LF, and even alleviate established HBV-LF—an efficacy profile that cannot be achieved by single-targeting of FXR, AT1, CB1, or CB2 receptors in HBV-LF.
Other GPCR Therapeutic Strategies
Bile Acid Receptor FXR
FXR, a bile acid-activated nuclear receptor with metabolic regulatory and anti-inflammatory properties, may participate in the regulation of HBV replication, hepatic homeostasis, and inflammatory injury in HBV-LF. FXRα can be inhibited by FGF11, which impairs FXRα-mediated transcriptional activation of HBV promoters, thereby suppressing HBV gene expression, replication and promoter activity (in vitro HBV replication model, direct relevance to HBV infection; extrapolated to HBV-LF with caution, as viral replication suppression may not reverse acute massive hepatocyte necrosis in established HBV-LF).114 In HBV-related hepatic injury, FXR expression is negatively regulated by ACSL4;115 modulation of ACSL4/FXR signaling may restore hepatic bile acid homeostasis, alleviate inflammatory damage, and thus inhibit the progression from chronic HBV injury to HBV-LF.
FXR agonists exhibit promising anti-HBV and hepatoprotective potential for HBV-LF intervention: GW4064 has a confirmed direct antiviral effect against HBV,116 and Vonafexor, another FXR agonist, showed good tolerability and safety in a phase Ib clinical trial for CHB, reducing HBsAg levels and demonstrating synergistic antiviral effects when combined with pegylated interferon-α2a (CHB clinical trial, non-LF).117 This anti-HBV effect in CHB is extrapolated to HBV-LF with critical caution: (1) the trial was conducted in stable CHB patients, not HBV-LF patients with acute liver failure; (2) FXR agonists target viral replication and chronic inflammation, which are not the core therapeutic targets for established HBV-LF (focused on organ support and anti-necrosis); (3) the safety and efficacy of FXR agonists in HBV-LF patients (with severe liver dysfunction) are completely unstudied. While FXR signaling is also involved in HBV-associated hepatocarcinogenesis via the regulation of HBV X protein variants118,119 and tumor microenvironment,120 this anti-tumorigenic mechanism may additionally contribute to the prevention of liver failure in advanced HBV-related HCC.
Compared with GPBAR1, FXR has inherent mechanistic limitations in HBV-LF treatment: FXR only regulates bile acid metabolism and chronic inflammation (secondary pathological processes in HBV-LF), and cannot mediate the integrated regulation of inflammation, apoptosis, and microcirculation (the acute core pathological processes of HBV-LF). In addition, FXR agonists lack direct efficacy in reversing massive hepatocyte apoptosis and hepatic microcirculation disturbance in established HBV-LF, which are the key therapeutic targets of GPBAR1 agonists. Thus, FXR is a supplementary target rather than a preferred target for HBV-LF.
AT1 Receptor
In patients with chronic HBV infection, chronic hepatic injury leads to hyperactivation of the renin-angiotensin-aldosterone system, which is directly associated with the progression of hepatic fibrosis, portal hypertension, and the development of acute-on-chronic HBV-LF (clinical observational direct evidence).121 Clinical and population-based studies have shown that ACEI/ARB inhibitors, which target the AT1 receptor pathway, can reduce the risk of liver cirrhosis in HBV-infected patients and exert hepatoprotective effects by alleviating hepatic fibrosis and portal hypertension (CHB/liver cirrhosis clinical studies, non-LF models);122 this protective effect is extrapolated to the prevention of HBV-LF in chronic HBV patients with caution, with the following limitations: (1) the studies were conducted in stable CHB patients at risk of cirrhosis, not patients with impending or established HBV-LF; (2) ACEI/ARB inhibitors target chronic fibrosis and portal hypertension, not the core pathological processes of HBV-LF (acute uncontrolled inflammation, massive hepatocyte apoptosis/necrosis); (3) the safety and efficacy of ACEI/ARB inhibitors in HBV-LF patients with severe liver dysfunction and potential hemodynamic instability have not been evaluated in clinical trials. The AT1 receptor gene 1166A/C polymorphism, however, is not associated with genetic susceptibility to HBV-induced liver cirrhosis (non-LF finding, no relevance to HBV-LF genetic susceptibility has been investigated).123
Compared with GPBAR1, AT1 receptor has obvious therapeutic limitations in HBV-LF: AT1 receptor only targets hepatic fibrosis and portal hypertension (chronic pathological processes that predispose to HBV-LF), and has no regulatory effect on the acute uncontrolled inflammation, massive hepatocyte apoptosis, and bile acid metabolic disorder that drive the progression of established HBV-LF. In contrast, GPBAR1 can simultaneously regulate these acute and chronic pathological processes, making it a more comprehensive and effective target for HBV-LF.
CB1
CB1 receptor signaling is implicated in HBV-mediated hepatic inflammatory injury and endoplasmic reticulum stress, which are key pathological drivers of HBV-LF. Exogenous HBV envelope proteins (LHBs, MHBs) can induce endoplasmic reticulum stress in hepatic cells expressing CB1, upregulating stress-related molecules and activating downstream pro-inflammatory signaling pathways, while CB1 deletion or pharmacological inhibition can abrogate this stress response and reduce HBV-induced hepatocellular damage.124 Clinically, elevated CB1 expression has also been observed in liver tissues from HBV-infected patients with severe chronic hepatic injury,125 suggesting a potential pro-inflammatory role of CB1 in HBV-associated liver damage. However, all evidence for CB1 in HBV-related liver disease is derived from non-LF models, including CHB in vitro cell models, HBV transgenic mouse models of chronic liver injury, and clinical samples from stable CHB patients; no direct experimental or clinical evidence links CB1 signaling to acute hepatocyte necrosis, massive liver parenchymal damage, or the development of overt HBV-LF. Furthermore, the therapeutic potential of CB1 inhibition has not been tested in any HBV-LF animal model or clinical cohort to date, and the functional relevance of CB1 to the acute, fulminant pathological processes of HBV-LF (eg., uncontrolled inflammatory cascade, rapid hepatocyte apoptosis/necrosis) remains entirely unelucidated.
Compared with GPBAR1, CB1 has no clinical application value in HBV-LF: CB1 mainly regulates hepatic lipid metabolism and chronic inflammation, and its inhibition can only alleviate mild hepatic steatosis in chronic HBV infection (non-LF). For established HBV-LF, CB1 inhibition has no effect on massive hepatocyte apoptosis and hepatic microcirculation disturbance, and even has potential side effects (eg., immune dysfunction), which is far inferior to the protective effect of GPBAR1 activation in HBV-LF.
CB2
CB2 receptor signaling modulates hepatic inflammation and fibrosis, two core pathological processes that contribute to the progression of chronic HBV infection to acute-on-chronic HBV-LF. The CB2 rs35761398 gene RR genotype has been identified as an independent predictor of extensive necrotizing inflammation in CHB patients, indicating that CB2 genetic variation is closely associated with the severity of hepatic inflammation in chronic HBV infection.126 Additionally, cannabidiol, a phytocannabinoid acting on the CB2 receptor, exerts anti-inflammatory and anti-fibrotic effects in liver injury models by inhibiting inflammatory cell proliferation, alleviating hepatic inflammatory responses, and inducing apoptosis of activated hepatic stellate cells.127 Cautionary notes for CB2 in HBV-LF: (1) All mechanistic and clinical evidence for CB2 in HBV-related liver disease is validated in CHB (chronic inflammation/fibrosis, non-LF) and non-HBV liver injury models, with no direct evidence obtained from HBV-LF models or clinical samples; (2) CB2 genetic variation is linked to inflammatory severity in stable CHB, but its association with HBV-LF susceptibility, disease severity, or clinical prognosis remains uninvestigated and unconfirmed; (3) cannabidiol has no direct anti-HBV viral activity, and its anti-inflammatory and anti-fibrotic effects in chronic CHB have not been translated or validated in acute or acute-on-chronic HBV-LF, where acute massive hepatocyte necrosis rather than chronic fibrosis is the core pathological driver. Thus, the therapeutic potential of CB2 targeting for HBV-LF is currently speculative and requires further preclinical and clinical research specifically in HBV-LF settings.
Compared with GPBAR1, CB2 has limited therapeutic potential in HBV-LF: CB2 only modulates chronic hepatic inflammation by regulating immune cell function, and cannot regulate bile acid metabolism disorder, massive hepatocyte apoptosis, or hepatic microcirculation disturbance in HBV-LF. In addition, CB2 agonists have no direct anti-HBV activity and no protective effect in acute-on-chronic HBV-LF models, which is significantly less effective than GPBAR1 agonists that can simultaneously inhibit HBV replication and alleviate acute liver injury.
Conclusions and Prospects
As a key membrane receptor for bile acids, GPBAR1 acts as an emerging and potential regulatory target rather than a fully validated central regulator in the pathological progression of HBV-LF. This review systematically summarizes the molecular mechanisms by which GPBAR1 participates in the “Inflammation-Apoptosis-Metabolism-Microcirculation” regulatory network, and clarifies its translational value as a potential therapeutic target for HBV-LF. However, the conclusion that GPBAR1 serves as a central regulator in HBV-LF is not fully supported by current human clinical data, and most related mechanistic evidence is cautiously extrapolated from chronic hepatitis B, acute liver injury, and metabolic liver disease models with shared pathological features rather than direct HBV-LF–specific evidence.
Critical knowledge gaps in this field must be emphasized and addressed in future studies:
First, the expression profile of human GPBAR1 in HBV-LF remains unclear. The exact expression level, cellular localization, and dynamic changes of GPBAR1 in liver tissues and clinical samples of HBV-LF patients have not been fully elucidated, and large‑scale clinical cohort studies are still lacking.
Second, the safety of GPBAR1 agonists in patients with liver failure has not been evaluated. The pharmacokinetic characteristics, adverse reactions, and clinical tolerance of GPBAR1 agonists in HBV-LF patients with severe liver dysfunction are unknown, and targeted safety verification is urgently needed.
Third, the disease-stage specificity of GPBAR1 signaling in HBV-LF is undefined. Whether GPBAR1 exerts stage‑dependent regulatory effects in the early, progressive, and terminal stages of HBV-LF, as well as its optimal intervention window, still requires systematic exploration.
Abnormal expression or functional deficiency of GPBAR1 destroys the integrity of its “Inflammation-Apoptosis-Metabolism-Microcirculation” integrated regulatory network in HBV-LF, and impairs the cross-regulation between the four core pathological processes: it not only directly exacerbates a single pathological abnormality (eg., excessive inflammation) but also breaks the positive feedback loop of mutual inhibition between pathways, leading to the simultaneous deterioration of inflammation, apoptosis, bile acid metabolism disorder and immune-microcirculation imbalance, and ultimately accelerating the progression of HBV-LF. In-depth analysis of the molecular mechanisms by which GPBAR1 participates in HBV-LF not only helps to clarify the pathogenic essence of this disease, but also provides theoretical support and potential targets for the development of novel targeted therapeutic strategies. Future studies need to further develop highly efficient and selective GPBAR1 agonists and other targeted intervention approaches, and verify their efficacy and safety through rigorous clinical research specifically in HBV-LF to provide new treatment options for patients with HBV-LF. Meanwhile, combining GPBAR1-targeted therapy with existing treatments such as antiviral therapy and organ function support therapy is expected to synergistically improve the overall therapeutic effect of HBV-LF and enhance the long-term prognosis of patients.
Abbreviations
α-GalCer, α-galactosylceramide; ACEI/ARB, renin-angiotensin system inhibitors; ACEIs, angiotensin-converting enzyme inhibitors; ACHBLF, acute-on-chronic hepatitis B liver failure; ACSL4, acyl-CoA synthetase long-chain family member 4; AFP, alpha-fetoprotein; Akt, Protein Kinase B; ALT, alanine transaminase; AMPK, AMP-Activated Protein Kinase; ARBs, AT1 receptor antagonists; ARG1, Arginase 1; ARNI, angiotensin receptor neprilysin inhibitor; AST, aspartate transaminase; AT1, Angiotensin II Type 1; Atgs, autophagy-related genes; ATP, Adenosine Triphosphate; atrogin-1, atrophy-related gene 1; BBOX1, γ-butyrobetaine hydroxylase 1; Bcl-2, B-cell Lymphoma 2; BiP, Binding Immunoglobulin Protein; BSEP, Bile Salt Export Pump; c-Rel, v-rel avian reticuloendotheliosis viral oncogene homolog; CA, cholic acid; cAMP, cyclic adenosine monophosphate; CB1 Receptor, Cannabinoid Receptor Type 1; CB2 Receptor, Cannabinoid Receptor Type 2; cccDNA, covalently closed circular DNA; CCL, C-C Motif Chemokine Ligand; CCR2, C-C Motif Chemokine Receptor 2; CD, Cluster of Differentiation; CDCA, chenodeoxycholic acid; cGAS, Cyclic GMP-AMP Synthase; CHB, chronic hepatitis B; CHOP, C/EBP Homologous Protein; CK1ε, casein kinase 1ε; CNKI, China National Knowledge Infrastructure; Con A, concanavalin A; CREB, cAMP-response element binding protein; CYP2C70, Cytochrome P450 Family 2 Subfamily C Member 70; CYP7A1, cytochrome P450 7A1; CYP8A1, Cytochrome P450, Family 8, Subfamily A, Polypeptide 1; CysLT1R, cysteinyl leukotriene receptor 1; DAG, diacylglycerol; DAMPs, damage-associated molecular patterns; DCA, deoxycholic acid; DEN, diethylnitrosamine; DNA, Deoxyribonucleic Acid; eGFR, estimated glomerular filtration rate; eIF2α, Eukaryotic Initiation Factor 2 Alpha; ERK, Extracellular Signal-Regulated Kinase; ET-1, Endothelin-1; EZH2, Enhancer of Zeste Homolog 2; FGF, Fibroblast Growth Factor; FOXA3, Forkhead Box Protein A3; FOXO1, Forkhead Box O1; FXR, Farnesoid X Receptor; GADD153, Growth Arrest and DNA Damage-Inducible Protein 153; GCDCA, glycochenodeoxycholic acid; GDP, Guanosine Diphosphate; GLP-1, glucagon-like peptide-1; GPBAR1, G protein-coupled bile acid receptor 1; GPCR, G protein-coupled receptor; GRK2, G Protein-Coupled Receptor Kinase 2; GRP78, Glucose-Regulated Protein 78; Gs, stimulatory G protein; Gsα, stimulatory G protein alpha subunit; GSK-3β, Glycogen Synthase Kinase-3 Beta; GTP, Guanosine Triphosphate; GZMB, granzyme B; HBeAg, hepatitis B e antigen; HBsAg, hepatitis B virus surface antigen; HBV, hepatitis B virus; HBV-LF, Hepatitis B-Associated Liver Failure; HBx, HBV X protein; HCC, Hepatocellular Carcinoma; HCV, hepatitis C virus; HSCs, hepatic stellate cells; HSPB8, Heat Shock Protein Beta 8; ICAM-1, Intercellular Adhesion Molecule-1; IFN-γ, Interferon-gamma; IL, Interleukin; iNOS, inducible Nitric Oxide Synthase; INT-767, 6alpha-Ethyl-3alpha,7alpha,23-trihydroxy-24-nor-5beta-cholan-23-sulfate; IP-10, Interferon-inducible Protein 10; IP3, inositol trisphosphate; Keap1, Kelch-like ECH-associated Protein 1; KNG1, kininogen 1; LC3, Microtubule-Associated Protein 1 Light Chain 3; LCA, lithocholic acid; LHBs, Large Hepatitis B Virus Surface Protein; LSECs, liver sinusoidal endothelial cells; MAFbx, Muscle Atrophy F-box protein; MAPK, Mitogen-Activated Protein Kinase; MASH, metabolic dysfunction-associated steatohepatitis; MASLD, metabolic dysfunction-associated steatotic liver disease; MCP-1, Monocyte Chemoattractant Protein-1; Mdivi-1, Mitochondrial division inhibitor 1; MDSCs, myeloid-derived suppressor cells; MHBs, Middle Hepatitis B Virus Surface Protein; MMP, Matrix Metalloproteinase-2; mTOR, Mechanistic Target of Rapamycin; MuRF-1, Muscle RING Finger-1; NAFLD, non-alcoholic fatty liver disease; NASH, non-alcoholic steatohepatitis; NF-κB, Nuclear Factor kappa-light-chain-enhancer of Activated B Cells; NKT, Natural Killer T cell; NLRP3, NOD-like receptor pyrin domain-containing protein 3; NO, Nitric Oxide; Nrf2, Nuclear Factor Erythroid 2-Related Factor 2; NSCLC, non-small cell lung cancer; Per2, period circadian protein 2; PERK, Protein Kinase R-like Endoplasmic Reticulum Kinase; PGC-1α, Peroxisome Proliferator-Activated Receptor Gamma Coactivator 1-Alpha; pgRNA, pregenomic RNA; PI3K, Phosphatidylinositol 3-Kinase; PKA, Protein Kinase A; PRISMA, Preferred Reporting Items for Systematic Reviews and Meta-Analyses; RIG-I, Retinoic Acid-Inducible Gene I; RORγt, Retinoid-Related Orphan Receptor Gamma T; ROS, reactive oxygen species; SCD1, Stearoyl-CoA Desaturase 1; si-GPBAR1, GPBAR1 small interfering RNA; SIRT, Sirtuin; SLC7A11, Solute Carrier Family 7 Member 11; SREBP-1c, Sterol Regulatory Element-Binding Protein-1c; STAT3, Signal Transducer and Activator of Transcription 3; STING, Stimulator of Interferon Genes; TCDCA, taurochenodeoxycholic acid; TGR5, Takeda G protein‑coupled receptor 5; THC, tetrahydrocannabinol; TLCA, taurolithocholic acid; TLR4, Toll-Like Receptor 4; TNF-α, Tumor Necrosis Factor-alpha; UDCA, ursodeoxycholic acid; VCAM, Vascular Cell Adhesion Molecule; XBP1, X-box Binding Protein 1; YAP, Yes-Associated Protein.
Consent for Publication
All authors have given their consent for the publication of this review article. The content of this article has not been published or submitted for publication elsewhere.
Acknowledgments
The authors extend their sincere gratitude to all researchers who have contributed to this study.
Funding
The Project Supported by National Natural Science Foundation of China (82272313) and Natural Science Foundation of Shandong Province (ZR2022MH006).
Disclosure
The authors declare that they have no competing interests. There are no financial or personal relationships that could inappropriately influence the work reported in this review article.
References
1. Szmaragd C, Balloux F. The population genomics of hepatitis B virus. Mol Ecol. 2007;16(22):4747–31. doi:10.1111/j.1365-294X.2007.03564.x
2. Wong GL. Updated guidelines for the prevention and management of chronic hepatitis b-world health organization 2024 compared with China 2022 HBV guidelines. J Viral Hepat. 2024;31(Suppl 2):13–22. doi:10.1111/jvh.14032
3. Hui Z, Yu W, Fuzhen W, et al. New progress in HBV control and the cascade of health care for people living with HBV in China: evidence from the fourth national serological survey, 2020. Lancet Reg Health West Pac. 2024;51:101193. doi:10.1016/j.lanwpc.2024.101193
4. Li G, He J, Shi J, et al. Bioprinting functional hepatocyte organoids derived from human chemically induced pluripotent stem cells to treat liver failure. Gut. 2025;74(7):1150–1164. doi:10.1136/gutjnl-2024-333885
5. Reusswig F, Reich M, Wienands L, Herebian D, Keitel-Anselmino V, Elvers M.The bile acid receptor TGR5 inhibits platelet activation and thrombus formation. Platelets. 2024;35;1.2322733.
6. Li M, Guo J, Meng J, et al. Synbiotic Intervention with Inulin and Lactiplantibacillus plantarum LPm77 attenuates type 2 diabetes via enhanced tudca metabolism and gut-liver axis modulation. J Agric Food Chem. 2025;73(31):19442–19459. doi:10.1021/acs.jafc.5c03515
7. Di Ciaula A, Khalil M, Baffy G, Portincasa P. Advances in the pathophysiology, diagnosis and management of chronic diarrhoea from bile acid malabsorption: a systematic review. Eur J Intern Med. 2024;128:10–19. doi:10.1016/j.ejim.2024.07.008
8. Zhou H, Mu X, Hu H, et al. DHLCA alleviates diabetic kidney disease via TGR5/FXR activation and gut microbiota remodeling. Drug Des Devel Ther. 2025;19:6469–6485. doi:10.2147/DDDT.S530823
9. Liu T, Wang H, Zhao Y, Wang YX, Xing X, Gao P. Drug development for chronic hepatitis B functional cure: recent progress. World J Hepatol. 2025;17(4):105797. doi:10.4254/wjh.v17.i4.105797
10. Yang F, Mao C, Guo L, et al. Structural basis of GPBAR activation and bile acid recognition. Nature. 2020;587(7834):499–504. doi:10.1038/s41586-020-2569-1
11. Fiorucci S, Distrutti E. Linking liver metabolic and vascular disease via bile acid signaling. Trends Mol Med. 2022;28(1):51–66. doi:10.1016/j.molmed.2021.10.005
12. Urbani G, Rondini E, Distrutti E, Marchianò S, Biagioli M, Fiorucci S. Phenotyping the chemical communications of the intestinal microbiota and the host: secondary bile acids as postbiotics. Cells. 2025;14(8):595. doi:10.3390/cells14080595
13. Xu L, Wu X, Long L, et al. TGR5 attenuates DOCA-salt hypertension through regulating histone H3K4 methylation of ENaC in the kidney. Metabolism. 2025;165:156133. doi:10.1016/j.metabol.2025.156133
14. Zhou T, Ismail A, Francis H. Bile acids in autoimmune liver disease: unveiling the nexus of inflammation, inflammatory cells, and treatment strategies. Cells. 2023;12(23):2725. doi:10.3390/cells12232725
15. Ma K, Tang D, Yu C, Zhao L. Progress in research on the roles of TGR5 receptor in liver diseases. Scand J Gastroenterol. 2021;56(6):717–726. doi:10.1080/00365521.2021.1903547
16. Miyata S, Kawashima Y, Sakai M, et al. Discovery, optimization, and evaluation of non-bile acid FXR/TGR5 dual agonists. Sci Rep. 2021;11(1):9196. doi:10.1038/s41598-021-88493-0
17. Luxenburger A, Harris LD, Ure EM, et al. The discovery of 12β-methyl-17-epi-18-nor-bile acids as potent and selective TGR5 agonists. Eur J Med Chem. 2023;250:115143. doi:10.1016/j.ejmech.2023.115143
18. Castel J, Botzanowski T, Brooks I, et al. Deciphering molecular determinants of GPBAR1-Gs protein interactions by HDX-MS and cryo-EM. Sci Rep. 2025;15(1). doi:10.1038/s41598-025-16529-w
19. Thibaut MM, Bindels LB. Crosstalk between bile acid-activated receptors and microbiome in entero-hepatic inflammation. Trends Mol Med. 2022;28(3):223–236. doi:10.1016/j.molmed.2021.12.006
20. Wu L, Feng J, Li J, et al. The gut microbiome-bile acid axis in hepatocarcinogenesis. Biomed Pharmacother. 2021;133:111036. doi:10.1016/j.biopha.2020.111036
21. Hoguet V, Lasalle M, Maingot M, et al. Beyond the rule of 5: impact of pegylation with various polymer sizes on pharmacokinetic properties, structure-properties relationships of mpegylated small agonists of tgr5 receptor. J Med Chem. 2021;64(3):1593–1610. doi:10.1021/acs.jmedchem.0c01774
22. Yan W, Zhang K, Guo J, Xu L. Bile acid-mediated gut-liver axis crosstalk: the role of nuclear receptor signaling in dynamic regulation of inflammatory networks. Front Immunol. 2025;16:1595486. doi:10.3389/fimmu.2025.1595486
23. Shi Y, Su W, Zhang L, et al. TGR5 regulates macrophage inflammation in nonalcoholic steatohepatitis by modulating nlrp3 inflammasome activation. Front Immunol. 2021;11:609060. doi:10.3389/fimmu.2020.609060
24. Sun B, Xie W, Li X, et al. Inulin enhanced rifaximin-inhibited colon cancer pulmonary metastasis by flora-regulated bile acid pathway. Int J Biol Macromol. 2024;275(Pt 1):133582. doi:10.1016/j.ijbiomac.2024.133582
25. Zheng BX, Yi Y, Wang XW, et al. Geniposide via enema alleviates colitis by modulating intestinal flora and bile acid metabolites, inhibiting S100A8/S100A9/NF-κB, and promoting TGR5 inhibition of NLRP3 inflammasome. Phytomedicine. 2025;142:156791. doi:10.1016/j.phymed.2025.156791
26. Kuhre RE, Wewer Albrechtsen NJ, Larsen O, et al. Bile acids are important direct and indirect regulators of the secretion of appetite- and metabolism-regulating hormones from the gut and pancreas. Mol Metab. 2018;11:84–95. doi:10.1016/j.molmet.2018.03.007
27. Li G, L. Guo G. Farnesoid X receptor, the bile acid sensing nuclear receptor, in liver regeneration. Acta Pharm Sin B. 2015;5(2):93–98. doi:10.1016/j.apsb.2015.01.005
28. Kowal JM, Haanes KA, Christensen NM, Novak I. Bile acid effects are mediated by ATP release and purinergic signalling in exocrine pancreatic cells. Cell Commun Signal. 2015;13:28. doi:10.1186/s12964-015-0107-9
29. Tyagi A, Kumar V. The gut microbiota-bile acid axis: a crucial regulator of immune function and metabolic health. World J Microbiol Biotechnol. 2025;41(7):215. doi:10.1007/s11274-025-04395-7
30. Wu L, Wang Y, Zhu S, et al. Changes in plasma bile acids are associated with gallbladder stones and polyps. BMC Gastroenterol. 2020;20(1):363. doi:10.1186/s12876-020-01512-8
31. Zhang ZY, Guo XL, Liu JT, et al. Conjugated bile acids alleviate acute pancreatitis through inhibition of TGR5 and NLRP3 mediated inflammation. J Transl Med. 2024;22(1):1124. doi:10.1186/s12967-024-05922-0
32. Feng S, Xie X, Li J, et al. Bile acids induce liver fibrosis through the NLRP3 inflammasome pathway and the mechanism of FXR inhibition of NLRP3 activation. Hepatol Int. 2024;18(3):1040–1052. doi:10.1007/s12072-023-10610-0
33. An P, Fan Y, Wang Q, et al. Cholic acid activation of GPBAR1 does not induce or exacerbate acute pancreatitis but promotes exocrine pancreatic secretion. Biochem Biophys Res Commun. 2024;735:150825. doi:10.1016/j.bbrc.2024.150825
34. Xu M, Liu Z, Pan Q, et al. Neuroprotective liver portal area macrophages attenuate hepatic inflammation. Nat Immunol. 2025;26(7):1048–1061. doi:10.1038/s41590-025-02190-y
35. Tang J, Song H, Li S, et al. TMEM16F expressed in kupffer cells regulates liver inflammation and metabolism to protect against listeria monocytogenes. Adv Sci. 2024;11(39):e2402693. doi:10.1002/advs.202402693
36. Abrigo J, Campos F, Gonzalez F, et al. Sarcopenia induced by chronic liver disease in mice requires the expression of the bile acids membrane receptor TGR5. Int J Mol Sci. 2020;21(21):7922. doi:10.3390/ijms21217922
37. Masyuk TV, Masyuk AI, Lorenzo Pisarello M, et al. TGR5 contributes to hepatic cystogenesis in rodents with polycystic liverdiseases through cyclic adenosine monophosphate/Galphas signaling. Hepatology. 2017;66(4):1197–1218. doi:10.1002/hep.29284
38. Ay Ü, Leníček M, Haider RS, et al. Microbially conjugated bile salts found in human bile activate the bile salt receptors TGR5 and FXR. Hepatol Commun. 2024;8(4):e0383. doi:10.1097/HC9.0000000000000383
39. Zhuang L, Jia N, Zhang L, et al. Gpbar-1/cAMP/PKA signaling mitigates macrophage-mediated acute cholestatic liver injury via antagonizing NLRP3-ASC inflammasome. Biochim Biophys Acta Mol Basis Dis. 2024;1870(6):167266. doi:10.1016/j.bbadis.2024.167266
40. Hu MM, He WR, Gao P, et al. Virus-induced accumulation of intracellular bile acids activates the TGR5-beta-arrestin-SRC axis to enable innate antiviral immunity. Cell Res. 2019;29(3):193–205. doi:10.1038/s41422-018-0136-1
41. Liu M, Xie WJ, Zhang X, Wu W, Li G, Wang L. Sodium butyrate regulates macrophage polarization by TGR5/beta-arrestin2 in vitro. Mol Med. 2025;31(1):31. doi:10.1186/s10020-025-01096-7
42. Liu X, Chen B, You W, Xue S, Qin H, Jiang H. The membrane bile acid receptor TGR5 drives cell growth and migration via activation of the JAK2/STAT3 signaling pathway in non-small cell lung cancer. Cancer Lett. 2018;412:194–207. doi:10.1016/j.canlet.2017.10.017
43. Ma L, Yang F, Wu X, et al. Structural basis and molecular mechanism of biased GPBAR signaling in regulating NSCLC cell growth via YAP activity. Proc Natl Acad Sci U S A. 2022;119(29):e2117054119. doi:10.1073/pnas.2117054119
44. Zangerolamo L, Carvalho M, Barbosa HCL. The critical role of the bile acid receptor tgr5 in energy homeostasis: insights into physiology and therapeutic potential. Int J Mol Sci. 2025;26(14):6547. doi:10.3390/ijms26146547
45. Leonhardt J, Haider RS, Sponholz C, et al. Circulating bile acids in liver failure activate tgr5 and induce monocyte dysfunction. Cell Mol Gastroenterol Hepatol. 2021;12(1):25–40. doi:10.1016/j.jcmgh.2021.01.011
46. Han LY, Fan YC, Mu NN, et al. Aberrant DNA methylation of G-protein-coupled bile acid receptor Gpbar1(TGR5) is a potential biomarker for hepatitis B Virus associated hepatocellular carcinoma. Int J Med Sci. 2014;11(2):164–171. doi:10.7150/ijms.6745
47. Sun L, Shao Y, Zhuang Z, et al. Targeting TGR5 to mitigate liver fibrosis: inhibition of hepatic stellate cell activation through modulation of mitochondrial fission. Int Immunopharmacol. 2024;140:112831. doi:10.1016/j.intimp.2024.112831
48. Chen WD, Yu D, Forman BM, Huang W, Wang YD. Deficiency of G-protein-coupled bile acid receptor Gpbar1 (TGR5) enhances chemically induced liver carcinogenesis. Hepatology. 2013;57(2):656–666. doi:10.1002/hep.26019
49. Pokhrel S, Dilts M, Stahl Z, et al. Tgr5-/- mice are protected from ethanol-induced metabolic alterations through enhanced leptin and Fgf21 signaling. Hepatol Commun. 2023;7(5):e0138. doi:10.1097/HC9.0000000000000138
50. Spatz M, Ciocan D, Merlen G, et al. Bile acid-receptor TGR5 deficiency worsens liver injury in alcohol-fed mice by inducing intestinal microbiota dysbiosis. JHEP Rep. 2021;3(2):100230. doi:10.1016/j.jhepr.2021.100230
51. Fan M, Wang Y, Jin L, et al. Bile acid-mediated activation of brown fat protects from alcohol-induced steatosis and liver injury in mice. Cell Mol Gastroenterol Hepatol. 2022;13(3):809–826. doi:10.1016/j.jcmgh.2021.12.001
52. Gopoju R, Wang J, Pan X, et al. Hepatic FOXA3 overexpression prevents Western diet-induced obesity and MASH through TGR5. J Lipid Res. 2024;65(4):100527. doi:10.1016/j.jlr.2024.100527
53. Carino A, Marchianò S, Biagioli M, et al. Agonism for the bile acid receptor GPBAR1 reverses liver and vascular damage in a mouse model of steatohepatitis. FASEB J. 2019;33(2):2809–2822. doi:10.1096/fj.201801373RR
54. Wang XX, Xie C, Libby AE, et al. The role of FXR and TGR5 in reversing and preventing progression of Western diet-induced hepatic steatosis, inflammation, and fibrosis in mice. J Biol Chem. 2022;298(11):102530. doi:10.1016/j.jbc.2022.102530
55. Marchianò S, Biagioli M, Bordoni M, et al. Defective bile acid signaling promotes vascular dysfunction, supporting a role for g-protein bile acid receptor 1/farnesoid x receptor agonism and statins in the treatment of nonalcoholic fatty liver disease. J Am Heart Assoc. 2023;12(23):e031241. doi:10.1161/JAHA.123.031241
56. Marchianò S, Biagioli M, Morretta E, et al. Combinatorial therapy with BAR502 and UDCA resets FXR and GPBAR1signaling and reverses liver histopathology in a model of NASH. Sci Rep. 2023;13(1):1602. doi:10.1038/s41598-023-28647-4
57. Fiorucci S, Rapacciuolo P, Fiorillo B, et al. Discovery of a potent and orally active dual gpbar1/cyslt(1)r modulator for the treatment of metabolic fatty liver disease. Front Pharmacol. 2022;13:858137. doi:10.3389/fphar.2022.858137
58. Biagioli M, Marchianò S, Di Giorgio C, et al. Combinatorial targeting of G-protein-coupled bile acid receptor 1 and cysteinyl leukotriene receptor 1 reveals a mechanistic role for bile acids and leukotrienes in drug-induced liver injury. Hepatology. 2023;78(1):26–44. doi:10.1002/hep.32787
59. Marchianò S, Biagioli M, Giorgio CD, et al. Allo-lithocholic acid, a microbiome derived secondary bile acid, attenuates liverfibrosis. Biochem Pharmacol. 2025;236:116883. doi:10.1016/j.bcp.2025.116883
60. Burchat N, Vidola J, Pfreundschuh S, et al. Intestinal stearoyl-coa desaturase-1 regulates energy balance via alterations in bile acid homeostasis. Cell Mol Gastroenterol Hepatol. 2024;18(6):101403. doi:10.1016/j.jcmgh.2024.101403
61. Yang H, Cai Q, Xin J, et al. SEMA6B induces macrophage-mediated inflammation and hepatocyte apoptosis in hepatitis B virus-related acute-on-chronic liver failure. Theranostics. 2024;14(13):5200–5218. doi:10.7150/thno.97007
62. Li Q, Wang J, Lu M, Qiu Y, Lu H. Acute-on-chronic liver failure from chronic-hepatitis-b, who is the behind scenes. Front Microbiol. 2020;11:583423. doi:10.3389/fmicb.2020.583423
63. Patel NH, Chen AY, Osiowy C, et al. US Acute Liver Failure Study Group. Hepatitis B virus variants and cytokine patterns in acute liver failure and transplant-free survival. Liver Int. 2025;45(7):e70175. doi:10.1111/liv.70175
64. Lin H, Zhang Y, Yan H, et al. Sargassum fusiforme fucoidan ameliorates obesity-associated metabolic dysfunction via a tauroursodeoxycholic acid-mediated TGR5-cAMP-PKA signaling pathway. J Agric Food Chem. 2025;73(32):20235–20253. doi:10.1021/acs.jafc.5c06044
65. Wang YD, Chen WD, Yu D, Forman BM, Huang W. The G-protein-coupled bile acid receptor, Gpbar1 (TGR5), negatively regulates hepatic inflammatory response through antagonizing nuclear factor kappa light-chain enhancer of activated B cells (NF-kappaB) in mice. Hepatology. 2011;54(4):1421–1432. doi:10.1002/hep.24525
66. Renga B, Bucci M, Cipriani S, et al. Cystathionine gamma-lyase, a H2S-generating enzyme, is a GPBAR1-regulated gene and contributes to vasodilation caused by secondary bile acids. Am J Physiol Heart Circ Physiol. 2015;309(1):H114–26. doi:10.1152/ajpheart.00087.2015
67. Bélanger M, Côté J, Butterworth RF. Neurobiological characterization of an azoxymethane mouse model of acute liver failure. Neurochem Int. 2006;48(6–7):434–440. doi:10.1016/j.neuint.2005.11.022
68. Gilbert MC, Setayesh T, Wan YY. The contributions of bacteria metabolites to the development of hepatic encephalopathy. Liver Res. 2023;7(4):296–303. doi:10.1016/j.livres.2022.11.005
69. Zhuang L, Ding W, Zhang Q, et al. TGR5 attenuated liver ischemia-reperfusion injury by activating the keap1-nrf2 signaling pathway in mice. Inflammation. 2021;44(3):859–872. doi:10.1007/s10753-020-01382-y
70. Zhou H, Zhou S, Shi Y, et al. TGR5/Cathepsin E signaling regulates macrophage innate immune activation in liver ischemia and reperfusion injury. Am J Transplant. 2021;21(4):1453–1464. doi:10.1111/ajt.16327
71. Zhao Q, Chen DP, Chen HD, et al. NK-cell-elicited gasdermin-D-dependent hepatocyte pyroptosis induces neutrophil extracellular traps that facilitate HBV-related acute-on-chronic liverfailure. Hepatology. 2025;81(3):917–931. doi:10.1097/HEP.0000000000000868
72. Liu GZ, Xu XW, Tao SH, Gao MJ, Hou ZH. HBx facilitates ferroptosis in acute liver failure via EZH2 mediated SLC7A11 suppression. J Biomed Sci. 2021;28(1):67. doi:10.1186/s12929-021-00762-2
73. Wu BK, Li CC, Chen HJ, et al. Blocking of G1/S transition and cell death in the regenerating liver of Hepatitis Bvirus X protein transgenic mice. Biochem Biophys Res Commun. 2006;340(3):916–928. doi:10.1016/j.bbrc.2005.12.089
74. Terradillos O, de La Coste A, Pollicino T, et al. The hepatitis B virus X protein abrogates Bcl-2-mediated protection against Fas apoptosis in the liver. Oncogene. 2002;21(3):377–386. doi:10.1038/sj.onc.1205110
75. Zhang X, Hu Y, Wang W, et al. IRGM/Irgm1 increases autophagy to inhibit activation of NLRP3 inflammasome in inflammatory injury induced acute liver failure. Cell Death Discov. 2024;10(1):272. doi:10.1038/s41420-024-02052-w
76. De Gregorio R, Moraca F, Rapacciuolo P, et al. Harnessing the estradienone scaffold to develop dual GPBAR1 and LIFR modulators for liver fibrosis. J Med Chem. 2025;68(19):20037–20059. doi:10.1021/acs.jmedchem.5c00705
77. He Q, Li X, Li H, et al. TGR5 activation by dietary bioactives and related improvement in mitochondrial function for alleviating diabetes and associated complications. J Agric Food Chem. 2025;73(11):6293–6314. doi:10.1021/acs.jafc.4c10395
78. Li J, Cheng R, Wan H. Overexpression of TGR5 alleviates myocardial ischemia/reperfusion injury via AKT/GSK-3β mediated inflammation and mitochondrial pathway. Biosci Rep. 2020;40(1):BSR20193482. doi:10.1042/BSR20193482
79. Hu J, Zhang Y, Yi S, et al. Lithocholic acid inhibits dendritic cell activation by reducing intracellular glutathione via TGR5 signaling. Int J Biol Sci. 2022;18(11):4545–4559. doi:10.7150/ijbs.71287
80. Ni C, Wang L, Bai Y, et al. Taurochenodeoxycholic acid activates autophagy and suppresses inflammatory responses in microglia of MPTP-induced Parkinson’s disease mice via AMPK/mTOR, AKT/NFκB and Pink1/Parkin signaling pathways mediated by Takeda G protein-coupled receptor 5. Free Radic Biol Med. 2025;235:347–363. doi:10.1016/j.freeradbiomed.2025.04.053
81. Xu N, Bai Y, Han X, et al. Taurochenodeoxycholic acid reduces astrocytic neuroinflammation and alleviates experimental autoimmune encephalomyelitis in mice. Immunobiology. 2023;228(3):152388. doi:10.1016/j.imbio.2023.152388
82. Carino A, Marchianò S, Biagioli M, et al. The bile acid activated receptors GPBAR1 and FXR exert antagonistic effects on autophagy. FASEB J. 2021;35(1):e21271. doi:10.1096/fj.202001386R
83. Gertzen CGW, Gohlke H, Häussinger D, et al. Schmitt L. The many facets of bile acids in the physiology and pathophysiology of the human liver. Biol Chem. 2021;402(9):1047–1062. doi:10.1515/hsz-2021-0156
84. Jia H, He X, Jiang T, Kong F. Roles of bile acid-activated receptors in monocytes-macrophages and dendritic cells. Cells. 2025;14(12):920. doi:10.3390/cells14120920
85. Chen I, Cassaro S. Physiology, Bile Acids. In: StatPearl. Treasure Island (FL): StatPearls Publishing; 2025.
86. Marchianò S, Biagioli M, Roselli R, et al. Atorvastatin protects against liver and vascular damage in a model of diet induced steatohepatitis by resetting FXR and GPBAR1 signaling. FASEB J. 2022;36(1):e22060. doi:10.1096/fj.202101397R
87. Zhou L, Yan M, Luo Q, et al. Elevated bile acids induce circadian rhythm sleep disorders in chronic liverDiseases. Cell Mol Gastroenterol Hepatol. 2025;19(3):101439. doi:10.1016/j.jcmgh.2024.101439
88. Li C, Xing X, Li M, et al. Bile acids produced by gut microbiota activate TGR5 to promote colorectal livermetastasis progression by inducing MDSCs infiltration in liver. Int Immunopharmacol. 2025;158:114829. doi:10.1016/j.intimp.2025.114829
89. Lian S, Lu M, Jiajing L, et al. Conjugated lithocholic acid activates hepatic TGR5 to promote lipotoxicity and masld-mash transition by disrupting carnitine biosynthesis. Adv Sci. 2025;12(20):e2410602. doi:10.1002/advs.202410602
90. Bidault-Jourdainne V, Merlen G, Glénisson M, et al. TGR5 controls bile acid composition and gallbladder function to protect the liverfrom bile acid overload. JHEP Rep. 2020;3(2):100214. doi:10.1016/j.jhepr.2020.100214
91. Li G, Xu X, Chai L, Guo Q, Wu W. Increase in bile acids after sleeve gastrectomy improves metabolism by activating GPBAR1 to increase cAMP in mice with nonalcoholic fatty liverdisease. Immun Inflamm Dis. 2024;12(7):e1149. doi:10.1002/iid3.1149
92. Leonhardt J, Dorresteijn MJ, Neugebauer S, et al. Immunosuppressive effects of circulating bile acids in human endotoxemia and septic shock: patients with liver failure are at risk. Crit Care. 2023;27(1):372. doi:10.1186/s13054-023-04620-5
93. Keitel V, Häussinger D. Role of TGR5 (GPBAR1) in Liver Disease. Semin Liver Dis. 2018;38(4):333–339. doi:10.1055/s-0038-1669940
94. Biagioli M, Carino A, Fiorucci C, et al. GPBAR1 functions as gatekeeper for liver nkt cells and provides counterregulatory signals in mouse models of immune-mediated hepatitis. Cell Mol Gastroenterol Hepatol. 2019;8(3):447–473. doi:10.1016/j.jcmgh.2019.06.003
95. Zhang F, Xiao X, Li Y, et al. Therapeutic Opportunities of GPBAR1 in Cholestatic Diseases. Front Pharmacol. 2022;12:805269. doi:10.3389/fphar.2021.805269
96. Di Giorgio C, Urbani G, Marchianò S, et al. Liver GPBAR1 associates with immune dysfunction in primary sclerosing cholangitis and its activation attenuates cholestasis in abcb4-/- mice. Liver Int. 2025;45(2):e16235. doi:10.1111/liv.16235
97. Biagioli M, Carino A, Fiorucci C, et al. The bile acid receptor GPBAR1 modulates CCL2/CCR2 signaling at the liversinusoidal/macrophage interface and reverses acetaminophen-induced livertoxicity. J Immunol. 2020;204(9):2535–2551. doi:10.4049/jimmunol.1901427
98. Klindt C, Reich M, Hellwig B, et al. The G protein-coupled bile acid receptor TGR5 (Gpbar1) modulates endothelin-1 signaling in liver. Cells. 2019;8(11):1467. doi:10.3390/cells8111467
99. Zhang J, Zhou J, He Z, et al. Salidroside attenuates NASH through regulating bile acid-FXR/TGR5 signaling pathway via targeting gut microbiota. Int J Biol Macromol. 2025;307(Pt 4):142276. doi:10.1016/j.ijbiomac.2025.142276
100. Biagioli M, Marchianò S, Di Giorgio C, et al. Activation of GPBAR1 attenuates vascular inflammation and atherosclerosis in a mouse model of NAFLD-related cardiovascular disease. Biochem Pharmacol. 2023;218:115900. doi:10.1016/j.bcp.2023.115900
101. Jin W, Zheng M, Chen Y, Xiong H. Update on the development of TGR5 agonists for human diseases. Eur J Med Chem. 2024;271:116462. doi:10.1016/j.ejmech.2024.116462
102. Qi Y, Duan G, Wei D, Zhao C, Ma Y. The bile acid membrane receptor tgr5 in cancer: friend or foe? Molecules. 2022;27(16):5292. doi:10.3390/molecules27165292
103. Wang J, Xu H, Liu Z, et al. Bile acid-microbiota crosstalk in hepatitis B virus infection. J Gastroenterol Hepatol. 2024;39(8):1509–1516. doi:10.1111/jgh.16604
104. Romain B, Inés VN, Vincent L, David D, Christophe R. Farnesoid X receptor agonists as broad-acting antivirals: evidence from hepatitis viruses and beyond? Antiviral Res. 2025;241:106223. doi:10.1016/j.antiviral.2025.106223
105. Godoy-Lugo JA, Mendez DA, Rodriguez R, et al. Improved lipogenesis gene expression in liver is associated with elevated plasma angiotensin 1-7 after AT1 receptor blockade in insulin-resistant OLETF rats. Mol Cell Endocrinol. 2022;555:111729. doi:10.1016/j.mce.2022.111729
106. Yang J, Yang Y, Tan X, et al. Unlocking the potential of the ACE2/Ang-(1-7)/Mas Axis in liver diseases: from molecular mechanisms to translational applications. Diabetes Obes Metab. 2025;27(8):4069–4082. doi:10.1111/dom.16435
107. Yang H, Park M, Lee JH, et al. New peripherally-restricted CB1 receptor antagonists, PMG-505-010 and −013 ameliorate obesity-associated NAFLD and fibrosis. Biomed Pharmacother. 2024;180:117501. doi:10.1016/j.biopha.2024.117501
108. Parfieniuk-Kowerda A, Martonik D, Andrzejuk A, Tarasik A, Flisiak R. Cannabinoids and the endocannabinoid system in liver diseases. Clin Exp Hepatol. 2024;10(4):211–217. doi:10.5114/ceh.2024.145358
109. AlShahrani AN, Al-Khlaiwi T, Meo SA, Siddiqui IA, Alghanem B, Almourfi F. Endocannabinoid and hematological responses to pre- and post-therapeutic exercises in liver transplant patients. Am J Clin Exp Immunol. 2024;13(6):259–271. doi:10.62347/FNLX9490
110. Bader Eddin L, Mahgoub A, Almarzooqi S, Adeghate E, Subramanya SB, Ojha S. beta-caryophyllene ameliorates thioacetamide-induced liver fibrosis in rats: a preventative approach. Int J Mol Sci. 2025;26(17):8493. doi:10.3390/ijms26178493
111. Biagioli M, Di Giorgio C, Massa C, et al. Microbial-derived bile acid reverses inflammation in IBD via GPBAR1 agonism and RORgammat inverse agonism. Biomed Pharmacother. 2024;181:117731. doi:10.1016/j.biopha.2024.117731
112. Chiang JYL, Ferrell JM. Bile acid receptors FXR and TGR5 signaling in fatty liver diseases and therapy. Am J Physiol Gastrointest Liver Physiol. 2020;318(3):G554–G573. doi:10.1152/ajpgi.00223.2019
113. Ito K, Okumura A, Takeuchi JS, et al. Dual agonist of farnesoid x receptor and takeda g protein-coupled receptor 5 inhibits hepatitis b virus infection in vitro and in vivo. Hepatology. 2021;74(1):83–98. doi:10.1002/hep.31712
114. Seong MS, Jang JA, Jeong YR, et al. Fibroblast growth factor 11 inhibits hepatitis b virus gene expression through fxrα suppression. J Microbiol. 2023;61(7):693–702. doi:10.1007/s12275-023-00065-1
115. Chen W, Xu H, Guo L, Zheng F, Yao J, Wang L. Role of ACSL4 in modulating farnesoid X receptor expression and M2 macrophage polarization in HBV-induced hepatocellular carcinoma. MedComm. 2024;5(9):e706. doi:10.1002/mco2.706
116. Fouillé R, Verrier ER, De Meyer A, et al. A novel in vitro system for simultaneous infections with hepatitis B, C, D and E viruses. JHEP Rep. 2025;7(5):101383. doi:10.1016/j.jhepr.2025.101383
117. Erken R, Andre P, Roy E, et al. Farnesoid X receptor agonist for the treatment of chronic hepatitis B: a safety study. J Viral Hepat. 2021;28(12):1690–1698. doi:10.1111/jvh.13608
118. Wu X, Ni Z, Song T, et al. C-terminal truncated hbx facilitates oncogenesis by modulating cell cycle and glucose metabolism in fxr-deficient hepatocellular carcinoma. Int J Mol Sci. 2023;24(6):5174. doi:10.3390/ijms24065174
119. Niu Y, Chen L, Wu M, et al. Partial abrogation of FXR-KNG1 signaling by carboxyl-terminal truncated HBx-C30 in hepatitis B virus-associated hepatocellular carcinoma. Virus Res. 2021;293:198264. doi:10.1016/j.virusres.2020.198264
120. Pan B, Chen S, Zhang Z, et al. New mechanistic understanding of FXR agonist Vonafexor: inducing sublethal damage of HBV-positive liver cancer cells via promoting anti-tumor immunity. Br J Cancer. 2025;133(5):697–708. doi:10.1038/s41416-025-03089-z
121. Wiczkowski A. The influence of hypervolemia on the secretion of atrial natriuretic peptide, the renin-angiotensin-aldosterone system’s activity and concentration of vasopressin, parathormone and calcitonin in hepatitis B virus infected patients with chronic liver diseases. Przegl Epidemiol. 1994;48(4):433–440.
122. Chen R, Zhou S, Liu J, et al. Renin-angiotensin system inhibitors and risk of hepatocellular carcinoma among patients with hepatitis B virus infection. CMAJ. 2024;196(27):E931–E939. doi:10.1503/cmaj.240003
123. Li H, Wu HL, Lv H, Wei HS, Wang HB, Wang PL. The association of TGF beta1 and AT1R gene polymorphisms with hereditarysusceptibility and clinical phenotype of HBV-induced liver cirrhosis. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2007;24(3):298–301.
124. Montalbano R, Honrath B, Wissniowski TT, et al. Exogenous hepatitis B virus envelope proteins induce endoplasmic reticulum stress: involvement of cannabinoid axis in liver cancer cells. Oncotarget. 2016;7(15):20312–20323. doi:10.18632/oncotarget.7950
125. Moudi B, Mohades MR, Mahmoudzadeh-Sagheb H, Heidari Z. Immunohistochemical expression of CB1 receptors in the liver of patients with HBV related-HCC. Arab J Gastroenterol. 2023;24(1):34–39. doi:10.1016/j.ajg.2022.10.002
126. Coppola N, Zampino R, Bellini G, et al. The impact of the CB2-63 polymorphism on the histological presentation of chronic hepatitis B. Clin Microbiol Infect. 2015;21(6):609.e1–4. doi:10.1016/j.cmi.2015.02.021
127. Lowe HI, Toyang NJ, McLaughlin W. Potential of Cannabidiol for the Treatment of Viral Hepatitis. Pharmacogn Res. 2017;9(1):116–118. doi:10.4103/0974-8490.199780
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.