Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Reprogramming fibroblasts to pluripotency using arginine-terminated polyamidoamine nanoparticles based non-viral gene delivery system
Authors Zhu K, Li J, Lai H, Yang C, Guo C, Wang C
Received 8 September 2014
Accepted for publication 25 October 2014
Published 12 December 2014 Volume 2014:9(1) Pages 5837—5847
DOI https://doi.org/10.2147/IJN.S73961
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Kai Zhu,1,2,* Jun Li,1,2,* Hao Lai,1,2 Cheng Yang,1,2 Changfa Guo,1,2 Chunsheng Wang1,2
1Department of Cardiac Surgery, Zhongshan Hospital, Fudan University, 2Shanghai Institute of Cardiovascular Disease, Shanghai, People’s Republic of China
*These authors contributed equally to this article
Abstract: Induced pluripotent stem cells (iPSCs) have attracted keen interest in regenerative medicine. The generation of iPSCs from somatic cells can be achieved by the delivery of defined transcription factor (Oct4, Sox2, Klf4, and c-Myc[OSKM]). However, most instances of iPSC-generation have been achieved by potentially harmful genome-integrating viral vectors. Here we report the generation of iPSCs from mouse embryonic fibroblasts (MEFs) using arginine-terminated generation 4 polyamidoamine (G4Arg) nanoparticles as a nonviral transfection vector for the delivery of a single plasmid construct carrying OSKM (pOSKM). Our results showed that G4Arg nanoparticles delivered pOSKM into MEFs at a significantly higher transfection efficiency than did conventional transfection reagents. After serial transfections of pOSKM-encapsulated G4Arg nanoparticles, we successfully generated iPSCs from MEFs. Our study demonstrates that G4Arg nanoparticles may be a promising candidate for generating of virus-free iPSCs that have great potential for clinical application.
Keywords: mouse embryonic fibroblasts, induced pluripotent stem cells
Introduction
The reprogramming of somatic cells into induced pluripotent stem cells (iPSCs) by exogenous transcription factors has attracted keen interest because of the potential use in regenerative medicine.1,2 Without the concerns of ethical issues and immune rejection, iPSCs have characteristics similar to those of embryonic stem cells (ESCs), such as morphology, self-renewal, pluripotency, and epigenetic status. iPSCs were first generated from mouse embryonic fibroblasts (MEFs) by retroviral transduction of Yamanaka transcription factors (Oct4, Sox2, Klf4, and c-Myc[OSKM]).2 To date, although reprogramming has been also performed by reprogramming proteins, microRNAs, small molecule compounds, and environmental stress stimulation, the most commonly used and simplest approach for iPSC-generation is delivery of OSKM transcription factors.3–7 Previous studies have demonstrated that iPSCs can be generated by serial transfection of a single plasmid construct carrying OSKM (pOSKM) for transient expression as a single polycistronic transcript. OSKM are translated into separate proteins via a ribosomal skipping mechanism, mediated by insertion of the 2A peptide sequence of picornavirus between the complementary (c)DNAs encoding each transcription factor.8,9
Most instances of OSKM delivery have been achieved using retroviral or lentiviral vectors, which may result in unpredictable and permanent alterations in the genome from viral vector integration.10 Although iPSCs can be obtained through nonintegrative adenoviral delivery of OSKM, the efficiency of reprogramming is extremely low, and a considerable percentage of clones are tetraploid.11,12 Therefore, a novel nonviral delivery vector is necessary for clinical applications of iPSCs. Many nonviral vectors have been developed for delivery of transcription factors, including the piggyBac transposon, nonintegrating episomal vectors, and nanoscale vectors.13–16 However, the limitation of these vectors is optimization of their biocompatibility and transfection efficiency, which critically influences reprogramming efficiency.17
Among nanoscale vectors, the poly(amidoamine) (PAMAM) dendrimer represents a promising nanoscale gene carrier because of its precisely controlled radial and symmetrical structure, intriguing multivalency, high cargo payload, and excellent biocompatibility. Positive charges on the terminal amine groups of the PAMAM dendrimer promote gene loading, cell penetration, and endosomal escape during transfection, resulting in functional gene transfection.18 High-generation dendrimers carry highly positive charges on their surface, which confer a high gene transfection efficiency.19,20 However, large-scale synthesis of high-generation PAMAM is technically complicated and economically costly, and hardly meets the high manufacturing grade required for clinical application.21 Therefore, it is worthwhile to develop a terminal-modified low-generation PAMAM that carries terminal charges similar to those of a high-generation PAMAM. It has been demonstrated that arginine residues in cell penetration peptides enable highly effective cell membrane penetration. In a recent study, conjunction of arginine-rich motifs to gene delivery vectors has been beneficial to improve cellular uptake and gene delivery.22 Additionally, an arginine-terminated low-generation PAMAM has been constructed and shown to be a good candidate as a gene delivery vector.21,23,24 Therefore, an arginine-terminated low-generation PAMAM may be a promising carrier for OSKM delivery, to generate virus-free iPSCs.
In this study, arginine-terminated generation 4 PAMAM (G4Arg) nanoparticles were used as nonviral vectors for delivery of pOSKM into MEFs to induce pluripotency. A schematic representation of this novel strategy is shown in Figure 1. The findings of this study provide a potential nanotechnological method for generating virus-free iPSCs.
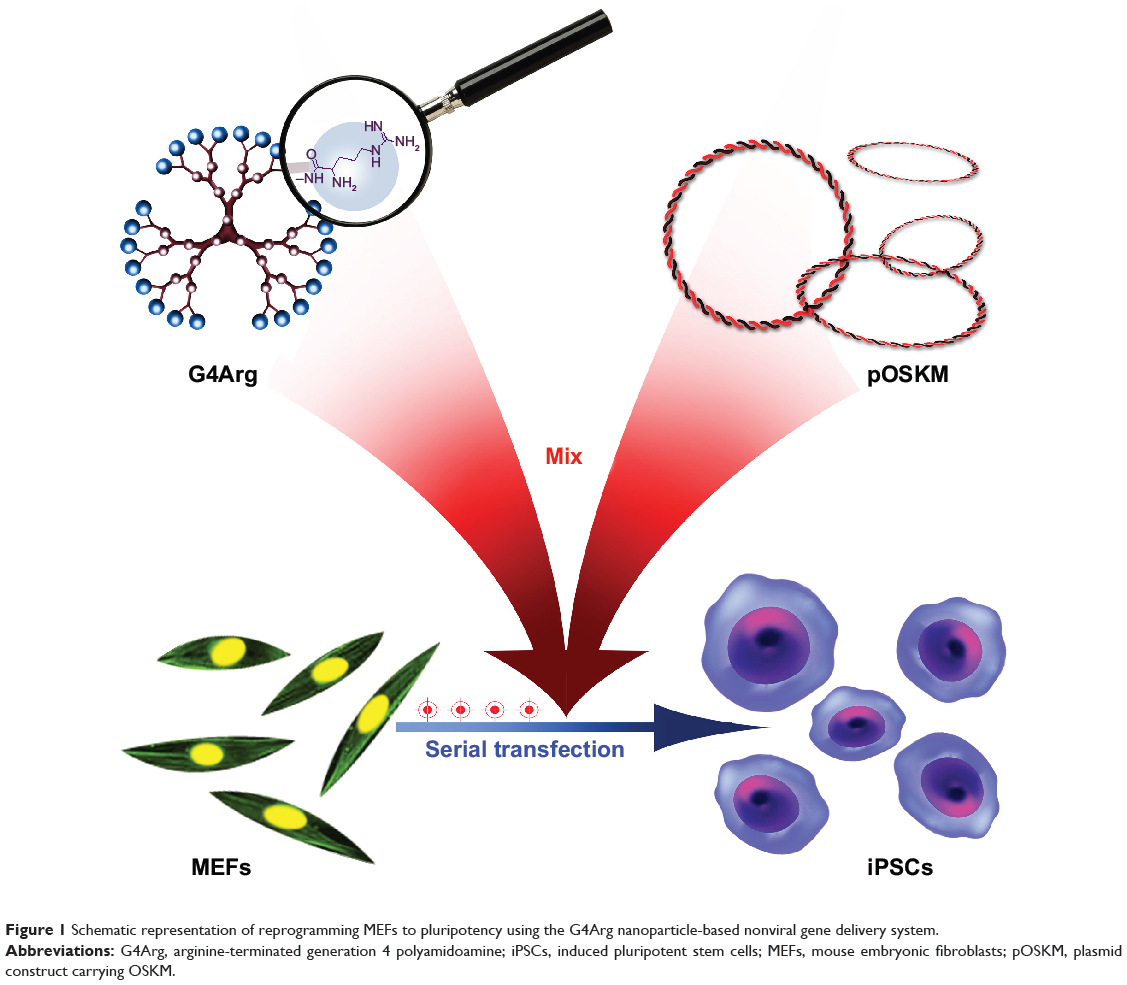
|
Figure 1 Schematic representation of reprogramming MEFs to pluripotency using the G4Arg nanoparticle-based nonviral gene delivery system. |
Materials and methods
Synthesis of G4Arg nanoparticles
G4Arg nanoparticles were synthesized by conjugating Na-Fmoc-Ng-2,2,4,6,7-pentamethyl-dihydrobenzofuran-5-sulfonyl-L-arginine (Fmoc-Arg(Pbf)-OH) to the periphery of G4Arg dendrimers (G4) using the NHydroxybenzotriazole/O(7Azabenzotriazol1yl)NNN’N’tetramethyluronium hexafluophosphate (HOBt/HBTU)-coupling method.21 Briefly, four equivalents of 1-hydroxybenzotriazole (Anaspec, San Jose, CA, USA), four equivalents of O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (Anaspec), four equivalents of Fmoc-Arg(pbf)-OH (Novabiochem, San Diego, CA, USA), and eight equivalents of N,N-diisopropylethylamine (Sigma-Aldrich Corp, St Louis, MO, USA) were dissolved in 5 mL anhydrous N,N-dimethylformamide (DMF) (Sigma-Aldrich Corp). Then, 50 mg G4 nanoparticles (Sigma-Aldrich) in 5 mL DMF was added to the above solution, and the mixture was stirred at 25°C for 48 hours. The resulting product was precipitated with ethyl ether at 4°C for 12 hours. After three rounds of precipitation, the precipitate was dissolved in 3 mL DMF and then mixed with a solution of 3 mL of 30% piperidine in DMF (v/v). The solution was then stirred at 25°C for 2 hours to deprotect the Fmoc groups. The resulting mixture was then subjected to precipitation. To remove the Pbf groups, a solution of 7.6 mL trifluoroacetic acid, 2 mL triisopropylsilane, and 2 mL water (95:2.5:2.5, v/v/v) was added to the resulting precipitate at 25°C. After 1 hour, the product was solubilized in water and dialyzed against pure water at 4°C. Nanoparticles were obtained by lyophilizing the dialyzed product. G4Arg nanoparticles were dissolved in tris(hydroxymethyl)aminomethane Tris)-HCl buffer (50 mM) for the following experiments.
Preparation and characteristics of G4Arg-DNA complexes
pOSKM was prepared by incorporating Oct4, Sox2, Klf4, and c-Myc genes as described previously.8 To evaluate the transfection efficiency in vitro, the green fluorescent protein (GFP) gene was incorporated to produce pOSKMG.9 The plasmids, in diethylpyrocarbonate (DEPC) water, were added to the G4Arg solution at various nanoparticle to DNA nitrogen-phosphorus (N/P) ratios (1:1, 5:1, 10:1, 15:1, 20:1, 35:1, and 50:1). After 10 minutes of mixing by vibration and then 20 minutes of sedimentation at room temperature, we obtained G4Arg-DNA complexes. The zeta potential and particle size of the G4Arg-DNA complexes in phosphate-buffered saline (PBS) were determined by a Zetasizer Nano (Malvern Instruments, Malvern, UK). Transmission electron microscopy (JEOL, Tokyo, Japan) was used for morphological characterization of the G4Arg-DNA complexes.
Cell culture
All animal experiments were approved by the Institutional Review Board and Institutional Animal Care and Use Committee of Fudan University.
The preparation of feeder cells, 1×105 STO cells (a mouse embryonic fibroblast cell line) (American Type Culture Collection, Manassas, VA, USA) were seeded on 6 cm gelatin-coated dishes and incubated in fibroblast culture medium (Cyagen, Guangzhou, People’s Republic of China) at 37°C for 12 hours, to reach ~80% confluence. The medium was replaced with fresh medium, followed by addition of mitomycin C (final concentration: 10 μg mL−1). After 3 hours of incubation at 37°C, the medium was removed, and the cells were washed three times with PBS (pH 7.4). The cells were harvested and reseeded on a new 6 cm gelatin-coated dish, in fibroblast medium. The mitotically inactivated STO cells were maintained in a 5% CO2 incubator at 37°C for 12 hours before use.
To isolate primary MEFs, uteri were removed from 13.5 days–pregnant C57/BL6 mice (Department of Laboratory Animal Science, Fudan University, Shanghai, People’s Republic of China) under aseptic conditions and washed with PBS. The heads, limbs, and visceral tissues of the fetuses were removed, and the remaining tissues were washed three times with PBS. The tissues were then transferred into TrypLE (Gibco®, Life Technologies Corp, Carlsbad, CA, USA) and incubated at 37°C for 30 minutes. After incubation, fibroblast culture medium was added to the digested tissues, and the cells were dissociated by pipetting. The dissociated cells were cultured in a 5% CO2 incubator at 37°C.
MTT assay
The cytotoxicity of G4Arg nanoparticles was assessed by examining MEFs viability using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Cells seeded in 96-well plates at a density of 1×105 cells per well were cultured for 24 hours and then treated with various concentrations of nanoparticles (5–320 μg/mL) for 24 hours or 48 hours. Then, 10 μL of an MTT solution (5 mg/mL) (Sigma-Aldrich Corp) was added to each well, followed by incubation at 37°C for 4 hours. The cells were then treated with dimethyl sulfoxide (DMSO) (40 μL/well) (Boster, Wuhan, People’s Republic of China) at 37°C for 1 hour. The absorbance of each well was determined at 490 nm using a microplate reader (model 680, Bio-Rad Laboratories, Hercules, CA, USA). Each result was the average of six wells, and untreated cells were considered to be 100% viable.
Optimization of transfection process
MEFs were seeded in 6-well plates at a destiny of 2×105 cells per well. The following day, the culture medium was replaced with solutions of G4Arg-DNA complexes at various N/P ratios with 5 μg of plasmid. Lipofectamine® 2000 (Invitrogen®, Life Technologies Corp) -based gene transfection was performed as a positive control, according to the manufacturer’s protocol. After incubation at 37°C for 4 hours, the transfection medium was replaced with fresh fibroblast medium. After 1 day, GFP expression in transfected MEFs was observed under a fluorescence microscope (BX51; Olympus, Tokyo, Japan). Transfected cells were collected in PBS, and the transfection efficiency was determined as the fluorescence intensity per well by a flow cytometer (FACSAriaII; BD Biosciences, Franklin Lakes, NJ, USA). Untransfected cells were used to set the autofluorescence baseline. Data were analyzed using Flowjo 7.6 software (Treestar, Ashland, OR, USA), with gating at 1%. To optimize the transfection conditions, various plasmid dosages (1, 2.5, 5, and 10 μg), and transfection durations (1, 2, 3, 4, and 5 hours) were applied to transfection for optimizing the transfection conditions.
Reprogramming of MEFs
MEFs (2×105 per well) were seeded in 6-well plates and transfected with G4Arg-pOSKM complexes using the optimized plasmid dosage, transfection time, and N/P ratio. After the last transfection, the culture medium was changed to ESC culture medium (Cyagen). Cells were maintained for 1 week with daily changes of ESC medium. After 1 week, the cells were transferred onto STO feeder cells at a 1:6 split ratio. iPSC colonies had formed by 23 days posttransfection (day 30), based on ESC-like colony morphology. To establish iPSC lines, the colonies were mechanically passaged and maintained on STO feeder cells in ESC medium.
Alkaline phosphatase staining
For alkaline phosphate (AP) staining, the cells were washed three times with PBS, fixed in 4% paraformaldehyde for 2 minutes at 4°C, and washed again with PBS. AP staining was performed with an Alkaline Phosphatase Detection Kit (Merck Millipore, Billerica, MA, USA) according to the manufacturer’s protocol. Images were acquired by microscopy (Olympus).
Determining of reprogramming efficiency
The reprogramming efficiency was calculated as the number of iPSC colonies formed per number of cells seeded for transfection. iPSC colonies were identified by ESC-like morphology and positive AP staining. We characterized two iPSC lines (C1 and C2) derived from these colonies.
Immunofluorescence staining
For immunostaining, iPSCs (passage 5) were fixed in 4% paraformaldehyde for 20 minutes at 4°C and then washed three times with PBS. The cells were then permeabilized with 0.1% Triton X-100 for 10 minutes. After washing with PBS, the cells were incubated in 4% bovine serum albumin for 1 hour to block nonspecific binding. The cells were incubated with primary antibodies against SSEA1, Nanog, Oct4, and Sox2 (Santa Cruz Biotechnology Inc, Dallas, TX, USA) overnight at 4°C. After washing with PBS, the cells wree incubated with Cy-3-labeled secondary antibodies (Jackson ImmunoResearch Inc., West Grove, PA, USA) for 1 hour. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (1:2,000 dilution) (Sigma-Aldrich Corp).
Karyotype analysis
C1 iPSCs, at passage 8, were treated with 0.4 μg/mL Colcemid™ for 2 hours at 37°C and then collected by trypsinization. Hypotonization was performed with prewarmed KCl (75 mM). A 3:1 mixture of methanol and acetic acid was used for cell fixation. Slides were prepared and analyzed for 3 days by trypsin banding and Giemsa staining. Karyotyping was performed using the MetaScan Karyotyping System (IMSTAR, Paris, France).
Teratoma formation
C1 and C2 iPSCs were harvested and resuspended in ESC medium. Nonobese diabetic-severe combined immunodeficient (NOD-SCID) mice (Slac Laboratory Animal Center, Shanghai, People’s Republic of China), at approximately 8 weeks of age, were injected intramuscularly with 1×106 of iPSCs in their right hind leg. At 5 weeks postinjection, teratomas were collected and processed for hematoxylin-eosin staining.
Statistics
Data were analyzed with SPSS 17.0 software (SPSS Inc., Chicago, IL, USA). All values are expressed as the mean ± standard error of the mean. One-way analysis of variance with the post hoc Bonferroni test was performed to assess significant differences among multiple groups. Significant differences between two groups were evaluated by the Student’s t-test.
Results
Characteristics of G4Arg-DNA complexes
The particle size and zeta potential of G4Arg-DNA complexes are shown in Figure 2A and B. At an N/P ratio of 10, G4Arg-DNA complexes exhibited a particle size of 168.4±20.1 nm and zeta potential of 26.4±1.2 mV. The morphologies of Arg-G4-DNA complexes at an N/P ratio of 10 are shown in Figure 2C.
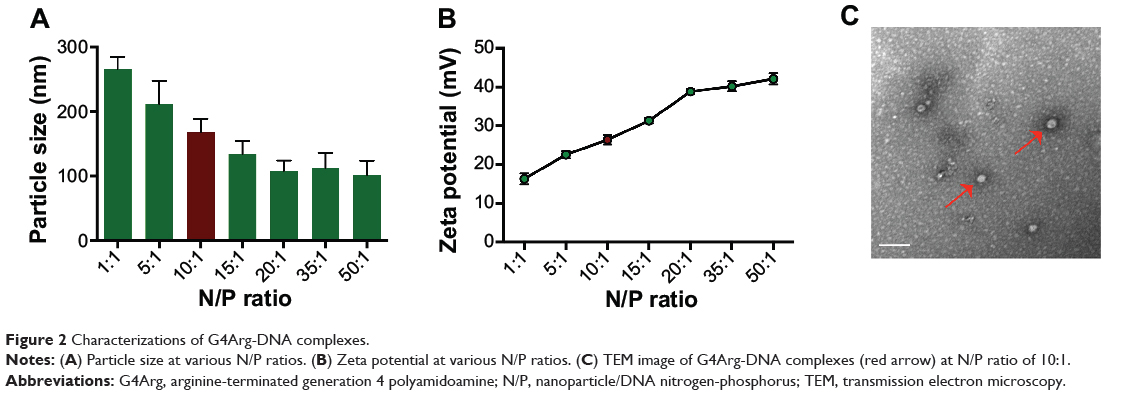
|
Figure 2 Characterizations of G4Arg-DNA complexes. |
Evaluation of G4Arg-based transfection in MEFs
After incubation with 5–320 μg/mL G4Arg nanoparticles for 24 or 48 hours, MEFs exhibited extremely low cell apoptosis (>80% cell viability) (Figure 3A). The fluorescence of G4Arg-pOSKMG-transfected MEFs is shown in Figure 3B. Lipofectamine 2000- and FuGENE® HD-based pOSKMG transfection of MEFs were used as controls. The maximum transfection efficiency was achieved at an N/P ratio of 10:1 (Figure 3C). Additionally, 5 μg/well of DNA and 3 hours of transfection were optimal for transfection (Figure 3D and E). Using the optimized conditions, G4Arg exhibited a higher transfection efficiency (14.25%±2.11%) than did Lipofectamine 2000 (5.81%±0.84%) or FuGENE HD (8.02%±0.71%) for delivery of pOSKMG (P<0.05) (Figure 3F).
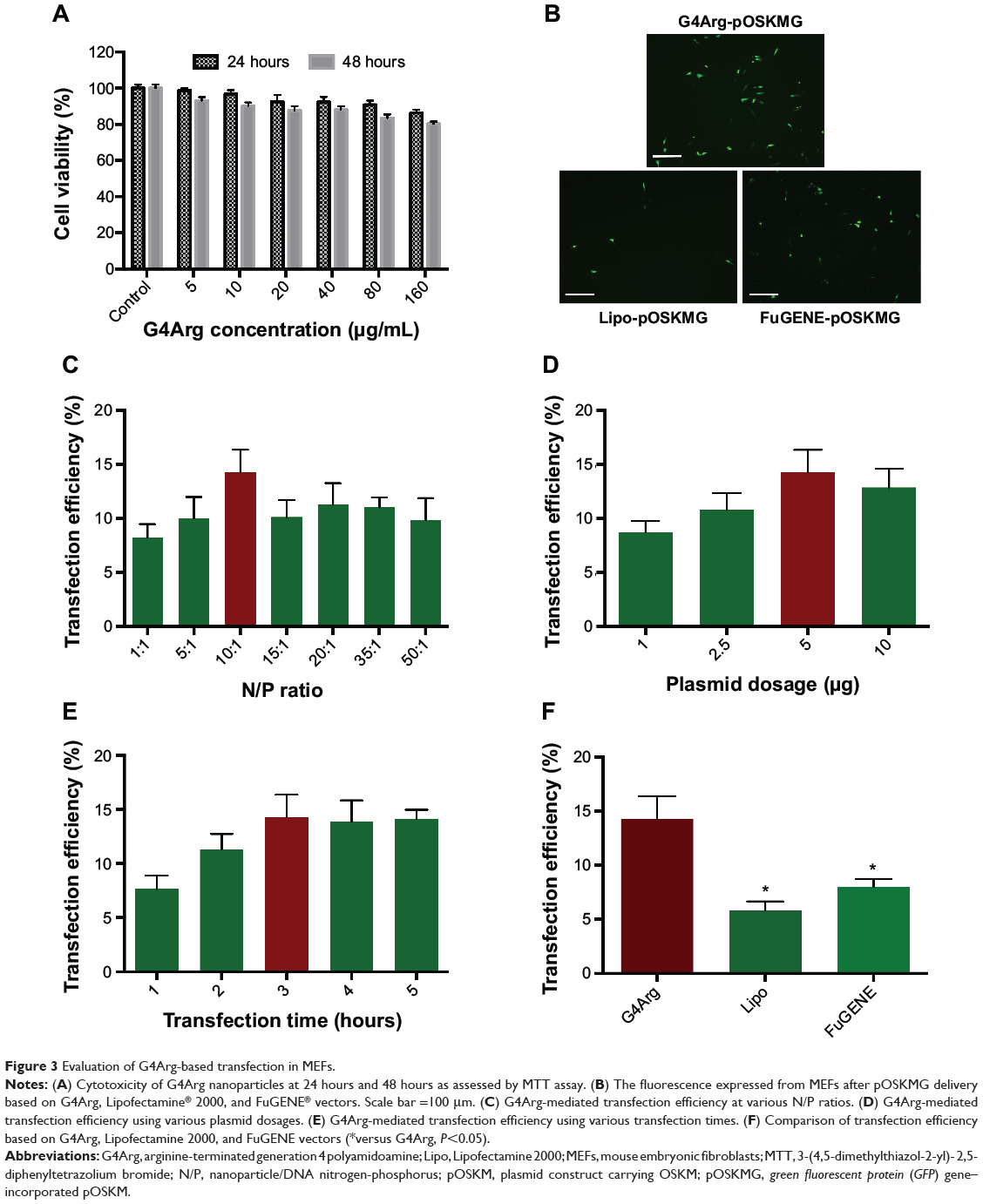
|
Figure 3 Evaluation of G4Arg-based transfection in MEFs. |
Generation of iPSCs
Figure 4 illustrates the timeline of the reprogramming protocol, with three consecutive transfections of the G4Arg-OSKM complexes. ESC-like colonies appeared at approximately 23 days posttransfection (day 30).
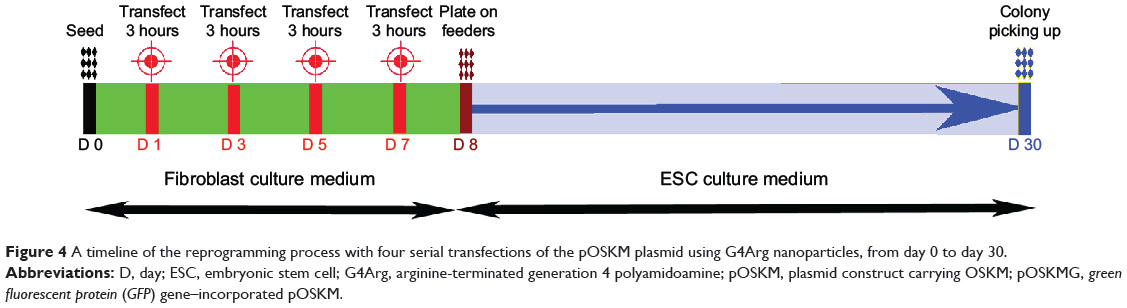
|
Figure 4 A timeline of the reprogramming process with four serial transfections of the pOSKM plasmid using G4Arg nanoparticles, from day 0 to day 30. |
Characteristics of induced iPSCs
The picked colonies grew with an ESC-like morphology when being maintained on feeder cells. Typical positive AP staining was observed in some of these colonies (Figure 5A). Eventually, we obtained one to seven AP-positive colonies from the initially seeded 2×105 cells. Reprogramming efficiency was up to 0.0035% according to the colony count per initially seeded cell number. Immunofluorescence staining confirmed that the iPSC colonies expressed pluripotency markers, including Oct4, SSEA-1, Sox2, and Nanog (Figure 5B and C). Additionally, high-resolution, G-band karyotyping of the colonies revealed a normal, diploid, male chromosomal content (Figure 5D).
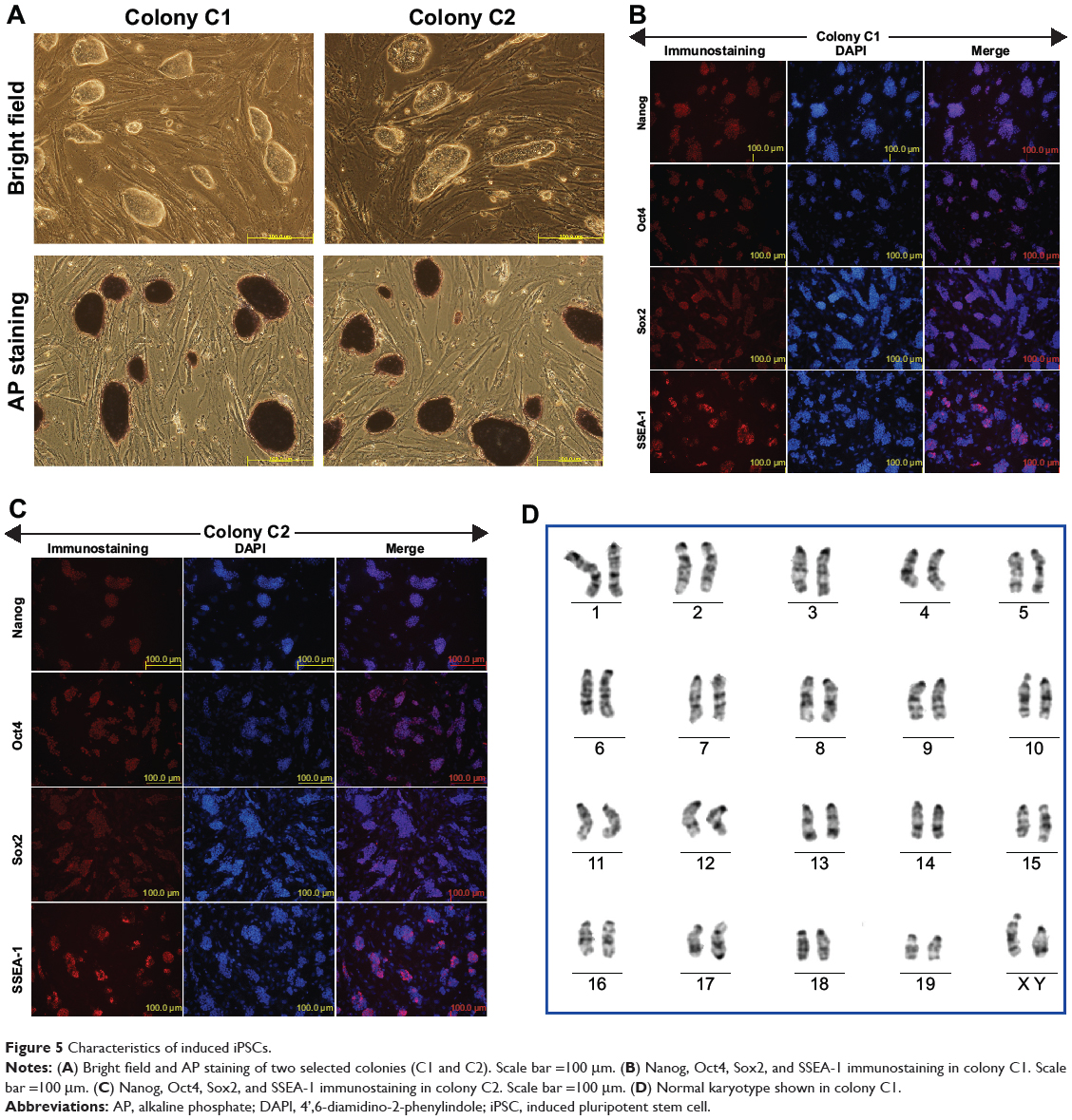
|
Figure 5 Characteristics of induced iPSCs. |
Injection of the induced iPSCs into NOD-SCID mice resulted in the formation of teratomas (Figure 6A). Hematoxylin-eosin staining confirmed the presence of tissues derived from the three germ layers: endoderm (gut epithelium and secretory epithelium), mesoderm (cartilage, muscle fibers, bone, and adipose), and ectoderm (epidermis and neural rosettes) (Figure 6B).

|
Figure 6 In vivo teratoma formation by injection of induced iPSCs. |
Discussion
Nonviral gene delivery systems can mediate the delivery of reprogramming factors and the generation of virus-free iPSCs to reduce the risks associated with retroviral vectors and other viral-based platforms. Here, a G4Arg nanoparticle was developed as a simple and biocompatible gene transfection platform for pOSKM delivery. Moreover, we achieved successful generation of virus-free iPSCs from MEFs using this novel platform.
We found that pOSKM could be loaded and condensed by G4Arg nanoparticles effectively. G4Arg nanoparticles were constructed by conjugating Fmoc-Arg(pbf)-OH to the periphery of amine-terminated G4 dendrimers. Compared with PAMAM, we assumed that arginine-terminated PAMAM would facilitate more effective interactions with negatively charged DNA by the simultaneous positive charges of the primary amine and guanidine groups of PAMAM and the arginine residue. Therefore, arginine-terminated PAMAM enabled higher gene delivery efficiency than did PAMAM.21 Additionally, previous studies demonstrate that the arginine-terminated PAMAM of lower generations, from 1 to 3, have relatively low gene delivery efficiency. This observation can be explained by the structural features associated with the lower generation, which cannot provide the sufficient multivalency and cooperativity required to efficiently deliver genes into cells.21,25 Therefore, we chose G4Arg nanoparticles as a novel platform for the pOSKM delivery. Furthermore, after simple mixing at room temperature, G4Arg-pOSKM complexes existed as uniform nanoparticles with an average particle size of 168.4 nm and a positive zeta potential value of 26.4 mV, suggesting sustained colloidal stability of the G4Arg-DNA complexes. Uniform packing of pOSKM into nanoparticles may be very important for transmembrane delivery of genes.
G4Arg nanoparticles could efficiently deliver pOSKM into MEFs. A previous study has reported that the efficiency of Lipofectamine- and FuGENE-based transfection of pOSKMG is extremely low.9 In the present study, the optimized transfection conditions were an N/P ratio of 10:1, 5 μg/well DNA, and 3 hours of transfection. Using these transfection conditions, G4Arg nanoparticle transfected pOSKMG at an efficiency of 14.25%±2.11%, which was much higher than that of the commercial transfection vectors Lipofectamine 2000 (5.81%±0.84%) and FuGENE HD (8.02%±0.71%) (P<0.05). It is assumed that one of the mechanisms associated with cellular uptake of nanosized particles is endocytosis that is limited to a positive zeta potential, and size ranges of approximately 20–200 nm in diameter.26 G4Arg-DNA complexes exhibited a suitable zeta potential (26.4±1.2 mV) and particle size (168.4±20.1 nm). These properties can greatly increase the possibility of endocytosis-mediated cellular uptake, leading to a relatively higher transfection efficiency. After confirming the optimized conditions, we transfected pOSKM into MEFs. It has been previously reported that transfection of large plasmids packaged in nanoparticles results in less exogenous gene expression than does transfection of smaller plasmids at a similar N/P ratio. This phenomenon can be explained by the fact that the plasmid length influences DNA release from the vector and intracellular migration of the DNA from the cytoplasm to the nucleus.9 In our study, pOSKMG was used as a reporter plasmid to investigate optimization of the N/P ratio, whereas pOSKM was ultimately transfected into MEFs to generate iPSCs. Therefore, we assumed that the pOSKM could be transfected at a higher efficiency than pOSKMG (14.25%±2.11%) to generate iPSCs because pOSKM was smaller than pOSKMG. Biocompatibility is another important factor for nanoparticle-based transfection systems.27,28 Our study demonstrated that G4Arg nanoparticle-based transfection resulted in low cytotoxicity (>80% cell viability). Collectively, these results demonstrate that G4Arg nanoparticles are indeed an effective and safe delivery system for pOSKM and have a great potential for further applications in virus-free iPSC-generation.
We developed a simple nonviral protocol to generate iPSCs by delivery of the G4Arg-pOSKM complexes into MEFs. There is a high demand for the generation of virus-free and transgene-free iPSCs for clinical application.14 Our nanoparticle-based strategy for Yamanaka factors delivery provides a promising approach for this end point. In this study, G4Arg nanoparticles mediated four serial deliveries of pOSKM into MEFs to maintain exogenous gene expression for a sufficient period to generate iPSCs. The resulting seven iPSC colonies were morphologically similar to ESC colonies and positively stained for AP activity. In the characterization of two iPSC lines established from these colonies, we observed a normal karyotype and the expression of a typical set of pluripotency marker genes. Additionally, the iPSCs were capable of differentiation into derivatives of the three germ layers (endoderm, mesoderm, and ectoderm) in immunodeficient mice. Nevertheless, it was unknown whether the other primary colonies had the typical characteristics of iPSCs. Further studies are needed to investigate potential karyotype abnormalities and the pluripotency characteristics of the other colonies.
The efficiency of G4Arg nanoparticle-based reprogramming efficiency was up to 0.0035% according to the colony count per initially seeded cell number (one to seven colonies derived from 2×105 cells). In previous studies, nonviral systems showed reprogramming efficiencies of 0.001%–0.015%.2–16,29 Although the G4Arg nanoparticle system did not show an obviously superior reprogramming efficiency over other nonviral systems, it still constitutes an effective and biocompatible approach to deliver transcription factor genes without the labor-intensive, time-consuming, and potentially harmful factors of viral systems. Further optimization of our transfection protocol, such as the seeded cell density and transfection timeline, will be investigated to improve the reprogramming efficiency. Moreover, studies have shown that the reprogramming efficiency can be enhanced by modifying cell signaling, drug treatment, and by using adult stem cells as the starting cells.16,30–33 By employing these strategies, we believe that the reprogramming efficiency of ArgG4 nanoparticle-based pOSKM delivery may be enhanced further.
One of the concerns with use of of iPSCs is whether the cells are transgene free. Although nanoparticle-based transfection systems have a lower risk of transgene integration than do viral-based systems, previous studies have reported integration into the genome. Therefore, further investigation needs to be performed to evaluate the risk of transgene integration in our nanoparticle-based transfection system.9,34 Furthermore, after optimizing our G4Arg-based system, chimeric mice will be produced to assess the full potency of the generated iPSCs.
It is noteworthy that human fibroblasts are harder to reprogram than MEFs.9 Although we generated iPSCs from MEFs, the efficiency of G4Arg nanoparticle-based reprogramming efficiency of human fibroblasts is uncertain. Therefore, further studies will be performed to investigate the efficiency of our reprogramming system in human fibroblasts.
Conclusion
We demonstrate for the first time that G4Arg nanoparticles can be used to deliver a single polycistronic reprogramming plasmid into MEFs, thereby achieving significantly higher transfection efficiency than with conventional transfection reagents. By serial transfections using G4Arg nanoparticles, we have developed a novel methodology to generate iPSCs from MEFs, while avoiding the use of viral vectors.
Acknowledgments
We thank Mr Gaohua Li for help with schematic representations. This study was supported by the National Science Foundation of China (grant numbers 81301312 and 81270326), Project of Research Ability Promotion for Young Teachers in Fudan University (grant number 20520133290), and the Youth Foundation of Zhongshan Hospital (grant number 2013ZSQN15).
Author contributions
CG and CW designed the study. KZ, JL, CY, and HL performed the reprogramming experiments and pluripotency analysis. KZ, JL, and CG performed the nanoparticle synthesis and characterization experiments. KZ and JL wrote and edited the paper. All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest.
References
Deng W. Induced pluripotent stem cells: paths to new medicines. A catalyst for disease modelling, drug discovery and regenerative therapy. EMBO Rep. 2010;11(3):161–165. | ||
Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–676. | ||
Kim D, Kim CH, Moon JI, et al. Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins. Cell Stem Cell. 2009;4(6):472–476. | ||
Warren L, Manos PD, Ahfeldt T, et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell. 2010;7(5):618–630. | ||
Hou P, Li Y, Zhang X, et al. Pluripotent stem cells induced from mouse somatic cells by small-molecule compounds. Science. 2013;341(6146):651–654. | ||
Xu X, Wang Q, Long Y, et al. Stress-mediated p38 activation promotes somatic cell reprogramming. Cell Res. 2013;23(1):131–141. | ||
Okita K, Ichisaka T, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature. 2007;448(7151):313–317. | ||
Gonzalez F, Barragan Monasterio M, Tiscornia G, et al. Generation of mouse-induced pluripotent stem cells by transient expression of a single nonviral polycistronic vector. Proc Natl Acad Sci U S A. 2009;106(22):8918–8922. | ||
Montserrat N, Garreta E, González F, et al. Simple generation of human induced pluripotent stem cells using poly-beta-amino esters as the non-viral gene delivery system. J Biol Chem. 2011;286(14):12417–12428. | ||
Nakagawa M, Koyanagi M, Tanabe K, et al. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat Biotechnol. 2008;26(1):101–106. | ||
Zhou W, Freed CR. Adenoviral gene delivery can reprogram human fibroblasts to induced pluripotent stem cells. Stem Cells. 2009;27(11):2667–2674. | ||
Stadtfeld M, Nagaya M, Utikal J, Weir G, Hochedlinger K. Induced pluripotent stem cells generated without viral integration. Science. 2008;322(5903):945–949. | ||
Woltjen K, Michael IP, Mohseni P, et al. piggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature. 2009;458(7239):766–770. | ||
Yu J, Hu K, Smuga-Otto K, et al. Human induced pluripotent stem cells free of vector and transgene sequences. Science. 2009;324(5928):797–801. | ||
Lee CH, Kim JH, Lee HJ, et al. The generation of iPS cells using non-viral magnetic nanoparticle based transfection. Biomaterials. 2011;32(28):6683–6691. | ||
Cao X, Deng W, Qu R, et al. Non-viral co-delivery of the four Yamanaka factors for generation of human induced pluripotent stem cells via calcium phosphate nanocomposite particles. Adv Funct Mater. 2013;23(43):5403–5411. | ||
Patel M, Yang S. Advances in reprogramming somatic cells to induced pluripotent stem cells. Stem Cell Rev. 2010;6(3):367–380. | ||
Astruc D, Boisselier E, Ornelas C. Dendrimers designed for functions: from physical, photophysical, and supramolecular properties to applications in sensing, catalysis, molecular electronics, photonics, and nanomedicine. Chem Rev. 2010;110(4):1857–1959. | ||
Shcharbin D, Shakhbazau A, Bryszewska M. Poly(amidoamine) dendrimer complexes as a platform for gene delivery. Expert Opin Drug Deliv. 2013;10(12):1687–1698. | ||
Conti DS, Brewer D, Grashik J, Avasarala S, da Rocha SR. Poly(amidoamine) dendrimer nanocarriers and their aerosol formulations for siRNA delivery to the lung epithelium. Mol Pharm. 2014;11(6):1808–1822. | ||
Liu C, Liu X, Rocchi P, Qu F, Iovanna JL, Peng L. Arginine-terminated generation 4 PAMAM dendrimer as an effective nanovector for functional siRNA delivery in vitro and in vivo. Bioconjug Chem. 2014;25(3):521–532. | ||
Nakase I, Akita H, Kogure K, et al. Efficient intracellular delivery of nucleic acid pharmaceuticals using cell-penetrating peptides. Acc Chem Res. 2012;45(7):1132–1139. | ||
Kim TI, Bai CZ, Nam K, Park JS. Comparison between arginine conjugated PAMAM dendrimers with structural diversity for gene delivery systems. J Control Release. 2009;136(2):132–139. | ||
Bai CZ, Choi S, Nam K, An S, Park JS. Arginine modified PAMAM dendrimer for interferon beta gene delivery to malignant glioma. Int J Pharm. 2013;445(1–2):79–87. | ||
An M, Parkin SR, DeRouchey JE. Intermolecular forces between low generation PAMAM dendrimer condensed DNA helices: role of cation architecture. Soft Matter. 2014;10(4):590–599. | ||
Ma D. Enhancing endosomal escape for nanoparticle mediated siRNA delivery. Nanoscale. 2014;6(12):6415–6425. | ||
Marin E, Briceño MI, Caballero-George C. Critical evaluation of biodegradable polymers used in nanodrugs. Int J Nanomedicine. 2013;8:3071–3090. | ||
Liu C, Zhang N. Nanoparticles in gene therapy principles, prospects, and challenges. Prog Mol Biol Transl Sci. 2011;104:509–562. | ||
Okita K, Nakagawa M, Hyenjong H, Ichisaka T, Yamanaka S. Generation of mouse induced pluripotent stem cells without viral vectors. Science. 2008;322(5903):949–953. | ||
Huangfu D, Osafune K, Maehr R, et al. Induction of pluripotent stem cells from primary human fibroblasts with only Oct4 and Sox2. Nat Biotechnol. 2008;26(11):1269–1275. | ||
Marson A, Foreman R, Chevalier B, et al. Wnt signaling promotes reprogramming of somatic cells to pluripotency. Cell Stem Cell. 2008;3(2):132–135. | ||
Eminli S, Utikal J, Arnold K, Jaenisch R, Hochedlinger K. Reprogramming of neural progenitor cells into induced pluripotent stem cells in the absence of exogenous Sox2 expression. Stem Cells. 2008;26(10):2467–2474. | ||
Giorgetti A, Montserrat N, Aasen T, et al. Generation of induced pluripotent stem cells from human cord blood using OCT4 and SOX2. Cell Stem Cell. 2009;5(4):353–357. | ||
Bhise NS, Wahlin KJ, Zack DJ, Green JJ. Evaluating the potential of poly(beta-amino ester) nanoparticles for reprogramming human fibroblasts to become induced pluripotent stem cells. Int J Nanomedicine. 2013;8:4641–4658. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.