Back to Journals » International Journal of Nanomedicine » Volume 12
Renal targeting potential of a polymeric drug carrier, poly-L-glutamic acid, in normal and diabetic rats
Authors Chai HJ, Kiew LV ![]() , Chin Y, Norazit A, Mohd Noor S, Lo YL
, Chin Y, Norazit A, Mohd Noor S, Lo YL ![]() , Looi CY, Lau YS, Lim TM, Wong WF, Abdullah NA
, Looi CY, Lau YS, Lim TM, Wong WF, Abdullah NA ![]() , Abdul Sattar MZ, Johns EJ, Chik Z
, Abdul Sattar MZ, Johns EJ, Chik Z ![]() , Chung LY
, Chung LY
Received 24 April 2016
Accepted for publication 7 September 2016
Published 13 January 2017 Volume 2017:12 Pages 577—591
DOI https://doi.org/10.2147/IJN.S111284
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Hann-Juang Chai,1 Lik-Voon Kiew,1 Yunni Chin,1 Anwar Norazit,2 Suzita Mohd Noor,2 Yoke-Lin Lo,3,4 Chung-Yeng Looi,1 Yeh-Siang Lau,1 Tuck-Meng Lim,5 Won-Fen Wong,6 Nor Azizan Abdullah,1 Munavvar Zubaid Abdul Sattar,7 Edward J Johns,8 Zamri Chik,1 Lip-Yong Chung3
1Department of Pharmacology, 2Department of Biomedical Science, 3Department of Pharmacy, Faculty of Medicine, University of Malaya, 4School of Pharmacy, International Medical University, Kuala Lumpur, 5Department of Chemical Science, Faculty of Science, Universiti Tunku Abdul Rahman, Kampar, 6Department of Medical Microbiology, Faculty of Medicine, University of Malaya, Kuala Lumpur, 7School of Pharmaceutical Sciences, Universiti Sains Malaysia, Minden, Malaysia; 8Department of Physiology, University College Cork, Cork, Republic of Ireland
Background and purpose: Poly-L-glutamic acid (PG) has been used widely as a carrier to deliver anticancer chemotherapeutics. This study evaluates PG as a selective renal drug carrier.
Experimental approach: 3H-deoxycytidine-labeled PGs (17 or 41 kDa) and 3H-deoxycytidine were administered intravenously to normal rats and streptozotocin-induced diabetic rats. The biodistribution of these compounds was determined over 24 h. Accumulation of PG in normal kidneys was also tracked using 5-(aminoacetamido) fluorescein (fluoresceinyl glycine amide)-labeled PG (PG-AF). To evaluate the potential of PGs in ferrying renal protective anti-oxidative stress compounds, the model drug 4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride (AEBSF) was conjugated to 41 kDa PG to form PG-AEBSF. PG-AEBSF was then characterized and evaluated for intracellular anti-oxidative stress efficacy (relative to free AEBSF).
Results: In the normal rat kidneys, 17 kDa radiolabeled PG (PG-Tr) presents a 7-fold higher, while 41 kDa PG-Tr shows a 15-fold higher renal accumulation than the free radiolabel after
24 h post injection. The accumulation of PG-AF was primarily found in the renal tubular tissues at 2 and 6 h after an intravenous administration. In the diabetic (oxidative stress-induced) kidneys, 41 kDa PG-Tr showed the greatest renal accumulation of 8-fold higher than the free compound 24 h post dose. Meanwhile, the synthesized PG-AEBSF was found to inhibit intracellular nicotinamide adenine dinucleotide phosphate oxidase (a reactive oxygen species generator) at an efficiency that is comparable to that of free AEBSF. This indicates the preservation of the anti-oxidative stress properties of AEBSF in the conjugated state.
Conclusion/Implications: The favorable accumulation property of 41 kDa PG in normal and oxidative stress-induced kidneys, along with its capabilities in conserving the pharmacological properties of the conjugated renal protective drugs, supports its role as a potential renal targeting drug carrier.
Keywords: carboxylated polymers, carboxylated polypeptides, carrier, diabetes, renal drug delivery, acute kidney injury, chronic renal failure, end-stage renal failure
Introduction
Renal failure is a serious health problem throughout the world.1,2 Acute kidney injury as a result of abrupt insults to the kidneys carries considerable morbidity and mortality.3–6 Furthermore, chronic renal failure secondary to other chronic diseases, such as type 2 diabetes mellitus, may gradually progress to end-stage renal failure requiring renal replacement therapy. The economic burden placed by renal diseases on the health care system has been escalating in recent years.7
The pathophysiological basis of acute kidney injury or chronic renal failure has drawn great attention. Various renoprotective compounds have been assessed for their ability to improve renal functions.8–10 Poor solubility in plasma, a lack of specific targeting of these compounds to the kidney parenchymal cells, and the dynamic exchange of fluid and solutes along the nephrons can lead to poor renal accumulation and limit the efficacy of these renoprotective compounds. Therefore, new biomaterials are needed to serve as a generic drug carrier to improve plasma solubility, delay renal elimination, improve the renal targeting selectivity, and preserve the efficacy of various classes of renoprotective compounds.
Many pathophysiological states are ones exhibiting oxidative stress, whereby the generation of reactive oxygen species causes free radical damage, which plays a crucial role in causing renal vascular and tubular cell damage, deterioration of renal function, and distortion of renal hemodynamics in both acute and chronic renal failure.11,12 These types of pathological alterations can make a carrier ineffective, even if it has renal targeting potential in a normal kidney.
Accumulation of anionized polymers in the kidneys of normal rats has been reported.13,14 A peak renal tissue localization of a radiolabeled anionized polyvinylpyrrolidone (PVP) derivative, 10 kDa carboxylated PVP, was observed at 3 h after an intravenous (iv) injection.13 This selective renal tissue accumulation suggests that the carboxylated polymers exhibit renal targeting properties. Based on this, we hypothesize that carboxylated polypeptides could be used as a renal targeting carrier for therapeutic agents. Moreover, carboxylated polypeptides, such as poly-L-glutamic acid (PG), possess additional advantages such as excellent water solubility, biocompatibility, nonimmunogenicity, biodegradability, and a high drug loading capability due to their multiple carboxyl side groups.15–18
PG has been utilized as a tumor-targeting drug carrier for various hydrophobic cancer chemotherapeutic agents.19–26 A significant renal accumulation of PG was observed in several studies on the biodistribution of a PG-paclitaxel conjugate in mice.27–29 Studies on other PG-chemotherapeutic conjugates have shown similar findings.30–32 These favorable observations led us to assess the potential use of PG polymers as a renal targeting carrier.
The aim of this study is, therefore, to test the hypothesis that PG polymers selectively accumulate in the kidneys, and these compounds could be candidates for target delivery of renoprotective drugs. This study is divided into several parts. The renal targeting properties of two PG polymers, with molecular weights of one above and one below the renal filtration threshold, were assessed. The ability of these PG polymers in targeting the oxidative-stressed kidneys (streptozotocin [STZ]-induced diabetic rat model) was evaluated. An exploratory synthesis of a PG-renal protecting drug conjugate using 4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride (AEBSF, a nicotinamide adenine dinucleotide phosphate [NADPH] oxidase inhibitor) as a model drug was performed. These experiments form the various portions of a preliminary proof of concept study on the use of PG polymers as a viable renal targeting carrier for the renal protecting drugs.
Materials and methods
Materials
PG (sodium salt, 17 and 41 kDa), deoxycytidine (dCyd), 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride, N-hydroxysuccinimide, and STZ were obtained from Sigma-Aldrich Corporation (St Louis, MO, USA). N, N-dimethylformamide, dicyclohexylcarbodiimide, and dimethylaminopyridine (DMAP) were purchased from Fluka (Buchs, Switzerland). The compounds 2-mercaptoethanol, AEBSF, and Hoechst 33342 were obtained from Promega Cooperation (Madison, WI, USA), and 5-(aminoacetamido) fluorescein (fluoresceinyl glycine amide) (AF) was supplied by Invitrogen (Grand Island, NY, USA). The MicroScint™ 40 scintillation cocktail was obtained from PerkinElmer (Boston, MA, USA).
The [5-3H]-2′-deoxycytidine (3H-dCyd) was supplied by American Radiolabeled Chemicals, Inc. (St Louis, MO, USA). The Snakeskin™ dialysis tube (molecular weight cutoff =10,000 Da) was purchased from Pierce (Rockford, IL, USA). The Amicon Microcon centrifugal filter device (molecular weight cutoff =10,000 Da) was purchased from EMD Millipore (Billerica, MA, USA). All other solvents and reagents were commercially available and were of analytical or HPLC grade.
Preparation of PG-3H-dCyd (radiolabeled PG) and PG-dCyd
The reaction to label PG with 3H-dCyd (American Radiolabeled Chemicals, Inc.) was based on an earlier procedure described for the preparation of PG-gemcitabine33,34 and optimized through reacting PG with nonradioactive dCyd to produce PG-dCyd conjugate (see Supplementary material for details). Two sizes of PGs (41 kDa and 17 kDa, Sigma-Aldrich Corporation) were labeled with 3H-dCyd via carbodiimide reaction to produce 3H-radiolabeled PG derivatives (PG-Tr). Briefly, 10 mg PG (proton form) (0.077 mmol), 0.0455 mg 3H-dCyd (0.2 μmol; 4 mCi), 10 mg dicyclohexylcarbodiimide (DCC, 0.048 mmol), and 0.1 mg DMAP were reacted at room temperature for 24 h in 2 mL dry N, N-dimethylformamide under dry nitrogen stream and stirring. Thereafter, PG-3H-dCyd (in proton form) was precipitated out of the reaction mixture with 2 mL chloroform. The precipitate was then reacted with 5 mL 1 M NaHCO3, dialyzed, and lyophilized to give approximately 8 mg PG-3H-dCyd (sodium salt) (PG-Tr, total radioactivity =875 μCi, determined via scintillation analyzer TRI-CARB 2300TR (Packard Instrument Company, Downers Grove, IL, USA).
Preparation of PG-4-(2-aminoethyl) benzenesulfonyl fluoride
The NADPH oxidase inhibitor AEBSF was conjugated to PG using 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMTMM) as a water-soluble condensing agent.35 Briefly, 20 mg PG (0.13 mmol, sodium salt), 3.2 mg AEBSF (0.013 mmol), and 4.2 mg DMTMM (0.02 mmol) were reacted at room temperature for 3 days in 2 mL distilled water (pH 8–9, stirring). The mixture was dialyzed against distilled water (5×5 L, 1 h per round of dialysis) in Snakeskin™ dialysis tube (molecular weight cutoff =10,000 Da) for 6 h. The dialyzed PG-AEBSF (sodium salt) solution was lyophilized to give 8.5 mg of white powdery product (yield =85.1%, AEBSF loading at 8.5% w/w). The details of the characterization of PG-AEBSF can be found in Supplementary material.
Preparation of PG-AF
AF was conjugated to PG using a similar preparation method for PG-AEBSF.35 Briefly, 20 mg PG (41 kDa) (0.13 mmol), 5.4 mg AF (0.013 mmol), and 4.2 mg DMTMM (0.02 mmol) were reacted at room temperature for 3 days in 2 mL distilled water (pH 8–9, stirring). The mixture was dialyzed against distilled water (5×5 L, 1 h per round of dialysis) in a Snakeskin™ dialysis tube (molecular weight cutoff =10,000 Da) for 6 h. The dialyzed PG-AF (sodium salt) solution was lyophilized to give 8.5 mg of white powdery product (yield =82.3%, AF loading at 7.2% w/w). The details of the characterization of PG-AF can be found in Supplementary material.
PG-dCyd, PG-Tr, PG-AEBSF, and PG-AF were found to be water soluble at up to 100 mg/mL (maximum concentration tested) at pH 7.0. The results coincided with the previous finding on the high water solubility of PG-drug conjugates (with drug loading <10%) at physiological pH.36
Animals
Male Sprague Dawley (SD) rats (body weight 250 g, 7 weeks old) were obtained from Jeffrey Cheah School of Medicine and Health Sciences, Monash University Malaysia. The rats were housed at 25°C in open rat cages at the Department of Pharmacology, Faculty of Medicine, University of Malaya, Malaysia, with a standard pellet diet. Drinking water was provided ad libitum throughout the study. All experiments involving the use of rats were conducted according to the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The animal use protocol was approved by the University of Malaya Institutional Animal Care and Use Committee (protocol approval number: FAR/04/08/2008/NAA(R)). All invasive procedures on rats were performed under anesthesia using an intraperitoneal (ip) injection of ketamine/xylazine cocktail (90 mg/kg and 10 mg/kg, respectively). The rats were subsequently euthanized with an ip injection of 100 mg/kg sodium pentobarbital. All efforts were made to minimize animal suffering. During housing and experiments, all animals were monitored twice daily for health status. No adverse events were observed throughout the experiment.
Establishment of a diabetic rat model
Each SD rat was given a single ip injection dose of 60 mg/kg STZ to induce diabetes, which was verified using a commercially available glucometer (ACCU-CHEK® Advantage II, Roche Diagnostics Corporation, Hague Road, IN, USA) 3 days and 14 days after the STZ administration. Rats with a blood sugar level >18 mM on day 14 post STZ administration, were selected for use.37
On day 14, the cellular oxidative stress level and tissue alteration in kidneys of the diabetic rats (n=6) were examined using the ferric reducing ability of plasma assay38 and kidney-to-body weight ratio determination39 (details of the methods and the results are described in Supplementary material).
The biodistribution of PG-3H-dCyd
Five STZ-induced diabetic rats and five normal rats were used for each predetermined time point of the experiment. PG-Tr (17 or 41 kDa; 10 μCi) or free 3H-deoxycytidine, unbound radiolabels (Free-Tr) (10 μCi) in phosphate-buffered saline (PBS) (0.15 mL) was administered intravenously by tail vein injection. After the injection of a radiotracer, the rats were anesthetized at 0.25, 1, 2, 4, 6, 12, and 24 h using an ip injection of ketamine/xylazine cocktail (90 mg/kg and 10 mg/kg, respectively). A blood sample was then collected via cardiac puncture. The rats were then euthanized with an ip injection of 100 mg/kg pentobarbitone. The tissues of interest, which include the intestine, spleen, stomach, kidneys, liver, lungs, heart, and skeletal muscles, were harvested and weighed. The organs were homogenized in saline at a ratio of 1:4 (w/v). The blood samples were centrifuged at 2,000 ×g for 10 min, and the plasma was collected for analysis.
Aliquots of tissue homogenate or plasma samples (40 μL) were added to MicroScint 40 scintillation fluid (3 mL) (PerkinElmer). The total radioactivity in the mixtures was counted using a PerkinElmer Tri-Carb XT-2300 liquid scintillation counter. The counting efficiency was assessed and corrected by the addition of 3H-dCyd to equivalent tissue homogenates or plasma from untreated rats. The biodistribution data of PG-Tr or Free-Tr were expressed as a mean value of radioactivity obtained from five rats at specified time points in the unit of disintegration per minute per milliliter of plasma or gram of tissue for the selected organs. The values of the area under the radioactivity versus time curve (AUC) of PG-Trs or Free-Tr in plasma and selected organs of both normal and diabetic rats were further estimated using Phoenix WinNonlin version 6.2 (Certara, Princeton, NJ, USA) for the 24 h period.
Renal accumulation of fluorescein-labeled PG/histological analysis of renal tissues
Twelve normal male SD rats were assigned into two groups, with six rats receiving an iv dose of 0.1 mL 41 kDa PG-AF (1 mg AF equivalent/mL) and the remaining six rats receiving free AF (1 mg/mL) in PBS via the tail vein. The rats were then monitored and euthanized at 2 and 6 h with an ip dose of 100 mg/kg pentobarbitone. The kidneys were extracted and drop-fixed immediately in 4% paraformaldehyde. The tissues were embedded in optimal cutting temperature and sectioned at 20 μm using a cryostat. These sections were later mounted using VECTASHIELD® 4′,6-diamidino-2-phenylindole·2HCl mounting media (Vector Laboratories, Burlingame, CA, USA) for fluorescence imaging.
Images were acquired using an Olympus FV1000 spectral confocal with a 60× (oil immersion) UPlanSApo objective (numerical aperture 1.35) (Olympus, Shinjuku, Tokyo, Japan). Images were then captured with Olympus Fluoview™ software. These images were subsequently compiled using Adobe Photoshop 11.1 and Adobe Illustrator 14 (Adobe Systems Incorporated, San Jose, CA, USA). The digitized images were not manipulated except for cropping, sizing, and adjustment of contrast and brightness.
NADPH oxidase inhibitory activity of PG-AEBSF on isolated aortic vessels
PG-AEBSF was subjected to an in vitro lucigenin-enhanced chemiluminescence assay to ascertain the AEBSF-induced inhibition of NADPH oxidase and NAPDH-mediated superoxide production.37 Briefly, multiple aortic rings (from SD rats euthanized with an ip 100 mg/kg pentobarbitone, n=6) were preincubated with AEBSF or PG-AEBSF (0.005–0.01 mM) at 37°C in Krebs-HEPES buffer (composition in mM: NaCl 99.0, NaHCO3 25, KCl 4.7, KH2PO4 1.0, MgSO4·7H2O 1.2, glucose 11.0, CaCl2·2H2O 2.5, and Na-HEPES 20.0) in the presence of diethyldithiocarbamic acid (10 mM, to inactivate endogenous superoxide dismutase) and β-NADPH (0.1 mM) for 45 min. As a positive control, diphenyleneiodonium chloride (DPI, 5 μM, a nonselective NADPH inhibitor) was added to the aortic rings in place of the test compound. At the end of the incubation, the rings were transferred to a 96-well plate containing 300 μL Krebs-HEPES buffer with lucigenin (10 μM) and NADPH (0.1 mM). The plate was immediately loaded into a luminescence reader (Tecan Infinite 200 Pro; Tecan, Männedorf, Switzerland) for 20 min. The rings were then dried for 48 h at 65°C, and the total level of superoxide production was normalized to the dry weight of tissues in milligrams.
Uptake of fluorescein-labeled PG by human umbilical vein endothelial cells
Briefly, 2×104 human umbilical vein endothelial cells (HUVEC; ScienCell Research Laboratories, Carlsbad, CA, USA) were seeded into each well of a 96-well plate and allowed to attach, spread, and proliferate overnight. The medium was then removed and replaced with a new medium supplemented with various concentrations of PG-AF or free AF (1 mg/mL AF equivalent). The HUVEC were further cultured at 37°C in 5% CO2 for 1 h to permit uptake. The medium was then removed and washed twice with PBS before fixing with 4% formaldehyde. Fixed cells were washed with PBS and stained with nucleic dye Hoechst 33342 for another 30 min. The cells were then observed for intracellular fluorescence at excitation/emission wavelength(Ex/Em) =350/461 nm and Ex/Em =488/519 nm for Hoechst 33342 and AF, respectively. The plates were scanned using an ArrayScan high content screening system (Cellomics Inc., Pittsburgh, PA, USA), and the fluorescence intensity was determined with the Target Activation BioApplication software (Cellomics Inc.).
Data processing and statistical analysis
Tables and Figures were generated, and all statistical analyses were performed using Student’s t-test or one-way/two-way analysis of variance (ANOVA) with Tukey/Bonferroni post hoc tests where indicated. The difference between the sample means was considered statistically significant at P<0.05.
Results
Plasma clearance of PG
In Figure 1, the decline in the plasma radioactivity of the 17 kDa (∆) or 41 kDa PG-Tr (▾) and that of Free-Tr (X) was compared in normal rats (Figure 1A) and diabetic rats (Figure 1B) over 24 h. The plasma radioactivities of both the Free-Tr and 17 kDa PG-Tr-treated rats (normal and diabetic) were found to decline in a similar fashion (P>0.05 for all time points, using two-way ANOVA with Bonferroni post hoc tests). These compounds were cleared from the plasma within 2 h after they were given to the rats. By contrast, 41 kDa PG-Tr was retained in the plasma of the normal rats for at least 12 h post iv injection (P<0.0001 for the time interval from 0.25 h to 12 h, using two-way ANOVA with Bonferroni post hoc tests). This prolonged plasma retention of 41 kDa PG-Tr, however, was not observed in diabetic rats.
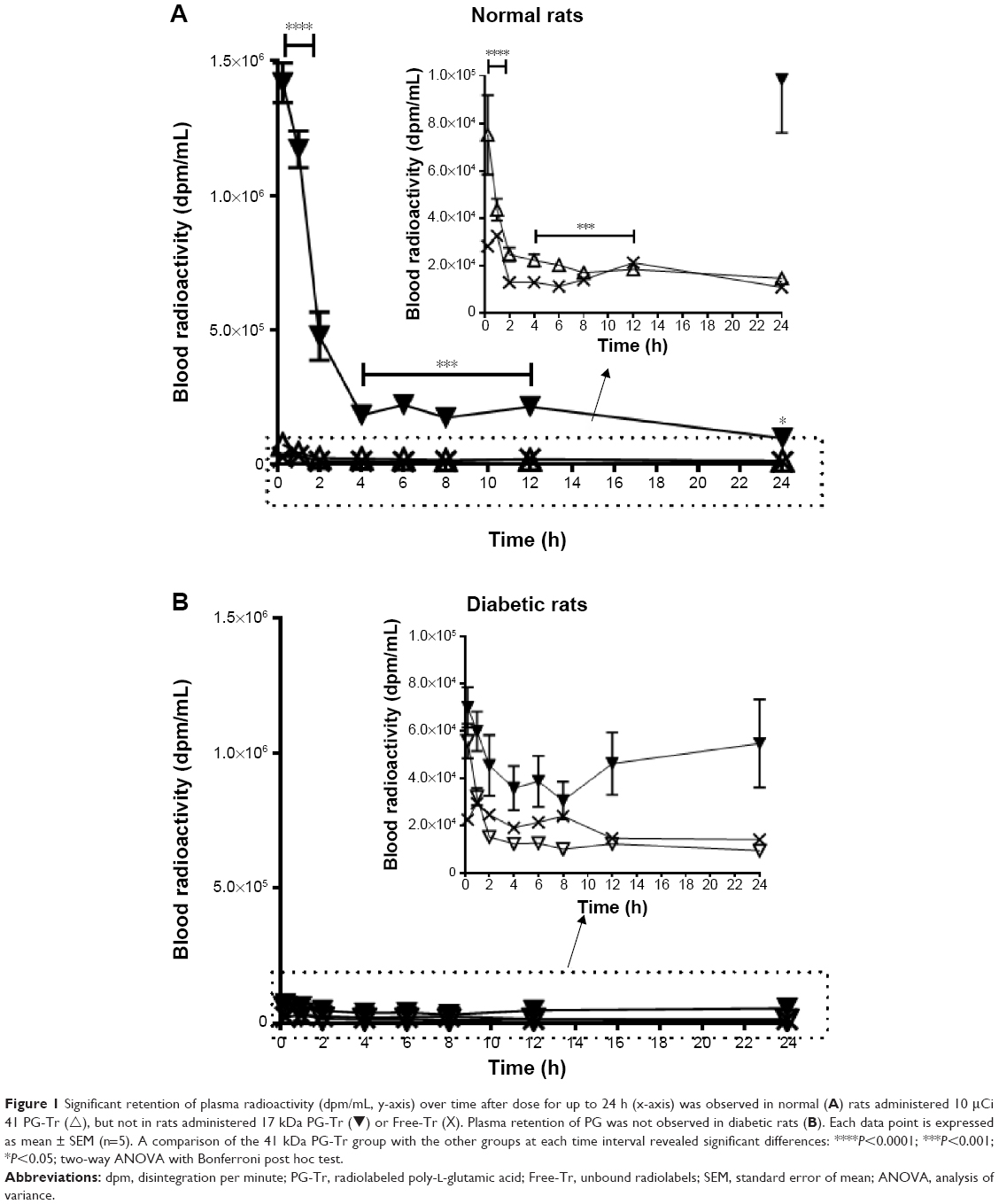 | Figure 1 Significant retention of plasma radioactivity (dpm/mL, y-axis) over time after dose for up to 24 h (x-axis) was observed in normal (A) rats administered 10 μCi 41 PG-Tr (∆), but not in rats administered 17 kDa PG-Tr (▾) or Free-Tr (X). Plasma retention of PG was not observed in diabetic rats (B). Each data point is expressed as mean ± SEM (n=5). A comparison of the 41 kDa PG-Tr group with the other groups at each time interval revealed significant differences: ****P<0.0001; ***P<0.001; *P<0.05; two-way ANOVA with Bonferroni post hoc test. |
Biodistribution of PG
The biodistribution of Free-Tr, 17 kDa, and 41 kDa PG-Tr were measured at predetermined time points after an iv injection (Table 1). While Free-Tr exhibited a swift decline in the rats without any specific tissue accumulation over the observed time frame, 17 kDa PG-Tr and 41 kDa PG-Tr exhibited a selective and prolonged localization in the kidneys to various degrees.
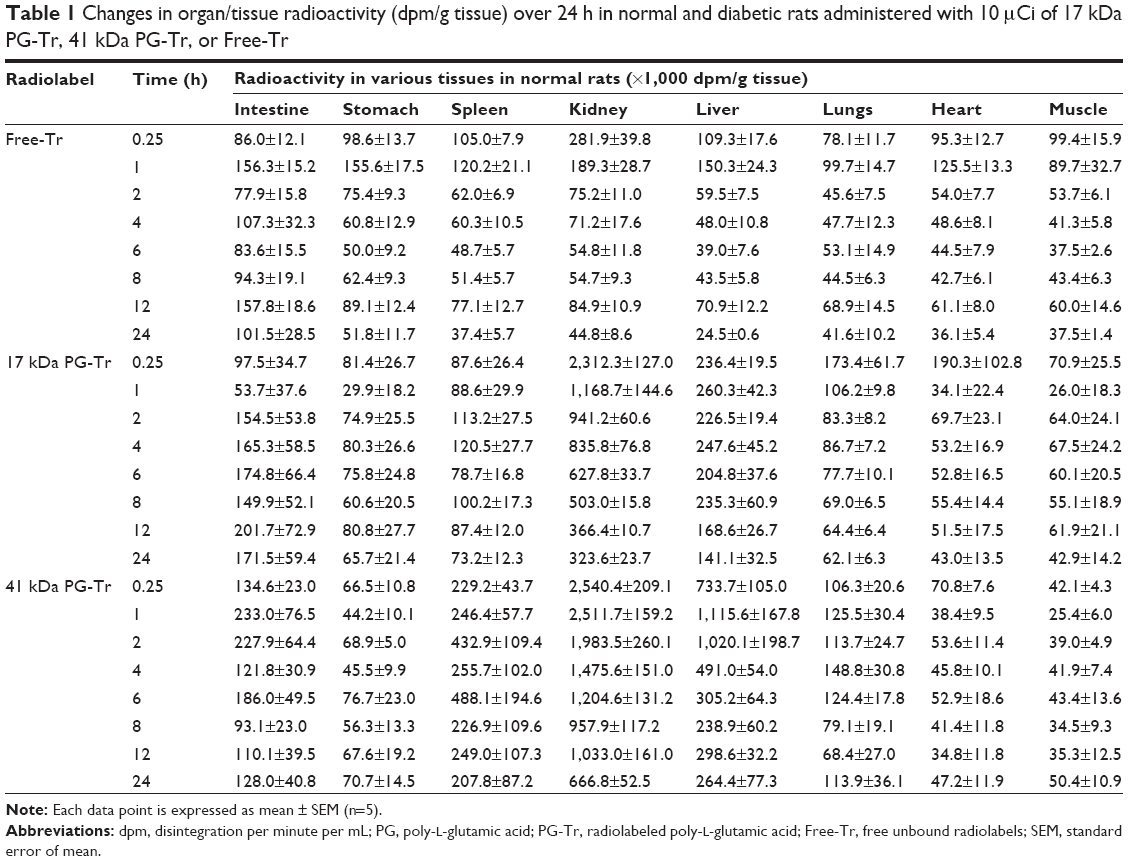 | Table 1 Changes in organ/tissue radioactivity (dpm/g tissue) over 24 h in normal and diabetic rats administered with 10 μCi of 17 kDa PG-Tr, 41 kDa PG-Tr, or Free-Tr |
Specifically, the rats receiving 41 kDa PG-Tr showed a higher renal accumulation (mean values of 22-, 12-, and 15-fold higher than that of Free-Tr at 6, 12, and 24 h post injection, respectively; all P<0.0001) when compared with the 17 kDa PG-Tr group (mean values of 11-, 8-, and 7-fold higher than that of the Free-Tr group at 6, 12, and 24 h post injection, respectively; all P<0.0001) throughout the monitoring period. The difference between the extents of renal accumulation of 41 kDa PG-Tr and that of 17 kDa PG-Tr was statistically significant with P<0.001 for all time points. Approximately 17% of the administered dose of 41 kDa PG-Tr was accumulated in the kidneys at 1 h post injection and 7% at 12 h post injection, compared to 12% at 1 h and 1% at 12 h post injection for 17 kDa PG-Tr (Figure 2A).
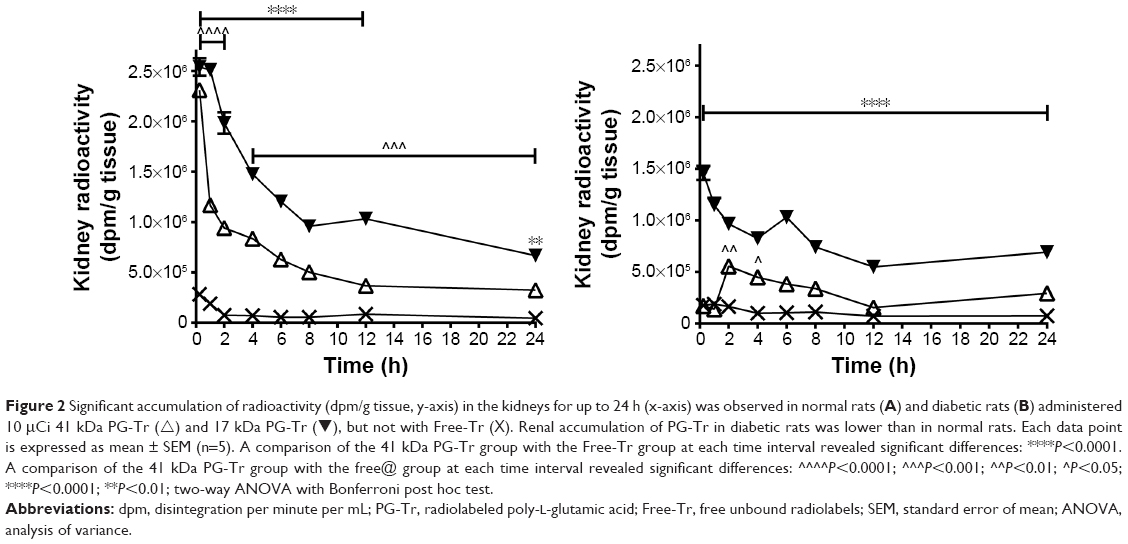 | Figure 2 Significant accumulation of radioactivity (dpm/g tissue, y-axis) in the kidneys for up to 24 h (x-axis) was observed in normal rats (A) and diabetic rats (B) administered 10 μCi 41 kDa PG-Tr (∆) and 17 kDa PG-Tr (▾), but not with Free-Tr (X). Renal accumulation of PG-Tr in diabetic rats was lower than in normal rats. Each data point is expressed as mean ± SEM (n=5). A comparison of the 41 kDa PG-Tr group with the Free-Tr group at each time interval revealed significant differences: ****P<0.0001. A comparison of the 41 kDa PG-Tr group with the free@ group at each time interval revealed significant differences: ^^^^P<0.0001; ^^^P<0.001; ^^P<0.01; ^P<0.05; ****P<0.0001; **P<0.01; two-way ANOVA with Bonferroni post hoc test. |
Similar selective renal retention profiles of 41 kDa PG-Tr (10-, 8-, and 9-fold higher than that of Free-Tr at 6, 12, and 24 h post injection, respectively; all P<0.0001) and renal retention profiles of 17 kDa PG-Tr (4-, 2-, and 4-fold higher than that of Free-Tr at 6 [P<0.0001], 12 [P>0.05], and 24 [P<0.001] h post injection, respectively) were observed in kidneys of the diabetic rats under oxidative stress (Table 2 and Figure 2B). The apparent exposure of 17 kDa PG-Tr and 41 kDa PG-Tr that accumulated in the diabetic kidneys as reflected in the AUC, however, was generally lower than that of the normal kidneys. More 41 kDa PG-Tr than 17 kDa PG-Tr accumulated in the diabetic kidneys (P<0.001 for all time points) (Figure 2B).
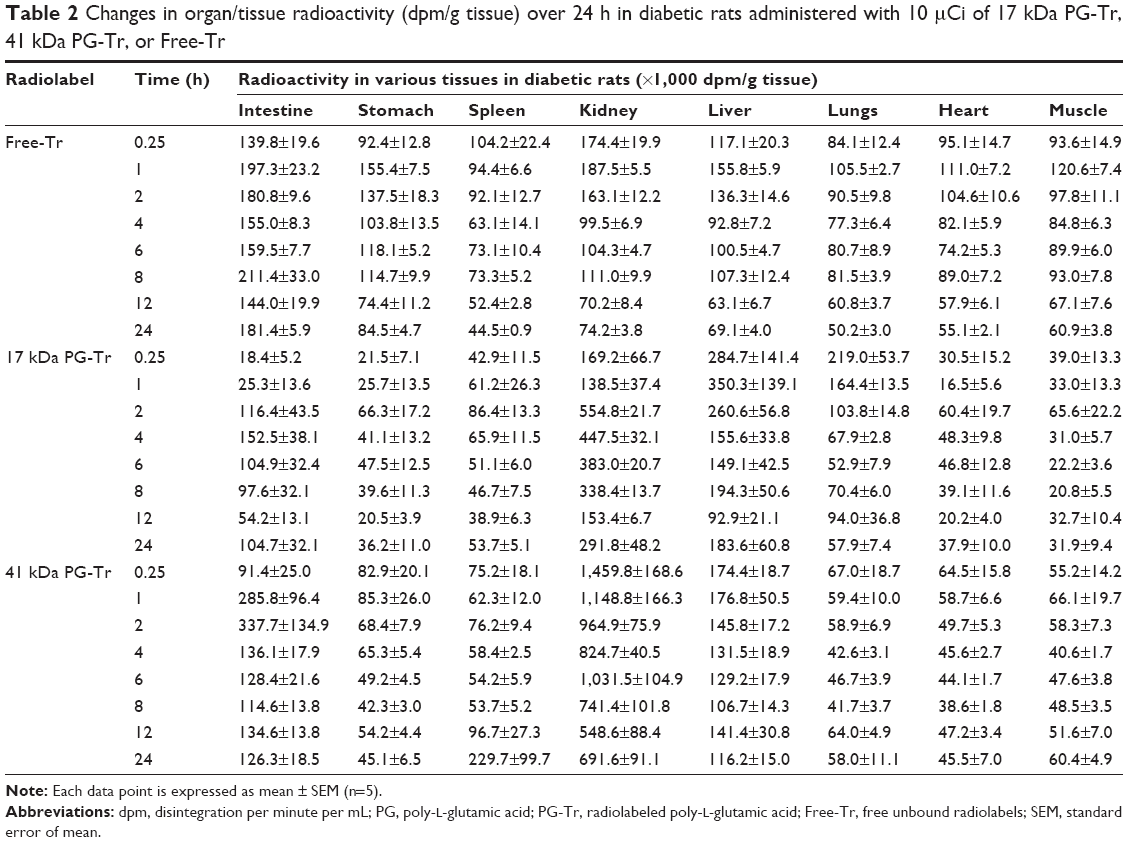 | Table 2 Changes in organ/tissue radioactivity (dpm/g tissue) over 24 h in diabetic rats administered with 10 μCi of 17 kDa PG-Tr, |
The AUC plots that represent the renal radioactivity changes over time post PG* or Free-Tr administration to the normal or diabetic rats were constructed. The AUC value of the Free-Tr plot was low in both normal and diabetic rats. By contrast, the AUC values of both 17 kDa PG-Tr and 41 kDa PG-Tr were high in both normal and diabetic rats. This indicates a much higher renal exposure of PG-Tr, with 41 kDa PG-Tr being the highest in normal rats, followed by diabetic rats (Figure 2). Generally, the AUCs of 17 kDa PG-Tr in both normal rats and diabetic rats are approximately one-half of those of 41 kDa PG-Tr, suggesting a possible selective renal retention property of 41 kDa PG-Tr (Figure 3).
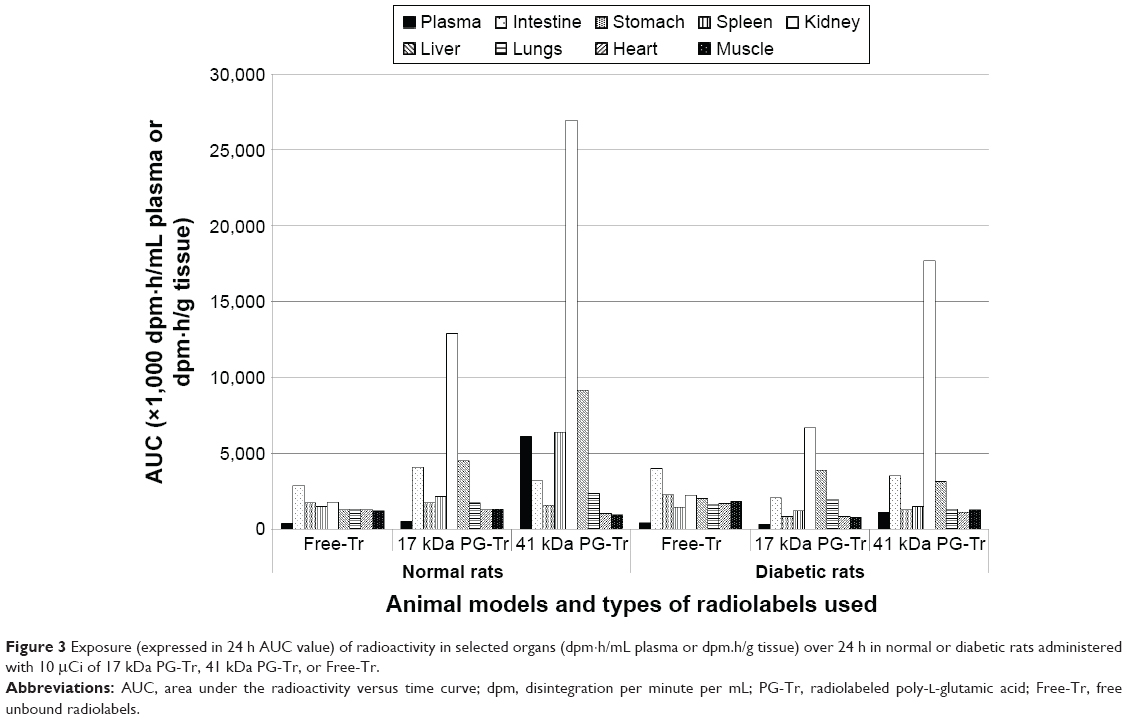 | Figure 3 Exposure (expressed in 24 h AUC value) of radioactivity in selected organs (dpm·h/mL plasma or dpm.h/g tissue) over 24 h in normal or diabetic rats administered with 10 μCi of 17 kDa PG-Tr, 41 kDa PG-Tr, or Free-Tr. |
Selective uptake and accumulation of PG-AF by the epithelial cells of kidney tubules of normal rats
An accumulation of 41 kDa PG-AF was detected in the epithelial cells of the kidney tubules at 2 and 6 h posttreatment (Figure 4). No evidence of fluorescence was detected in the glomeruli of PG-AF-treated animals, or in any part of the parenchymal cells in the kidneys of the AF-only-treated animals.
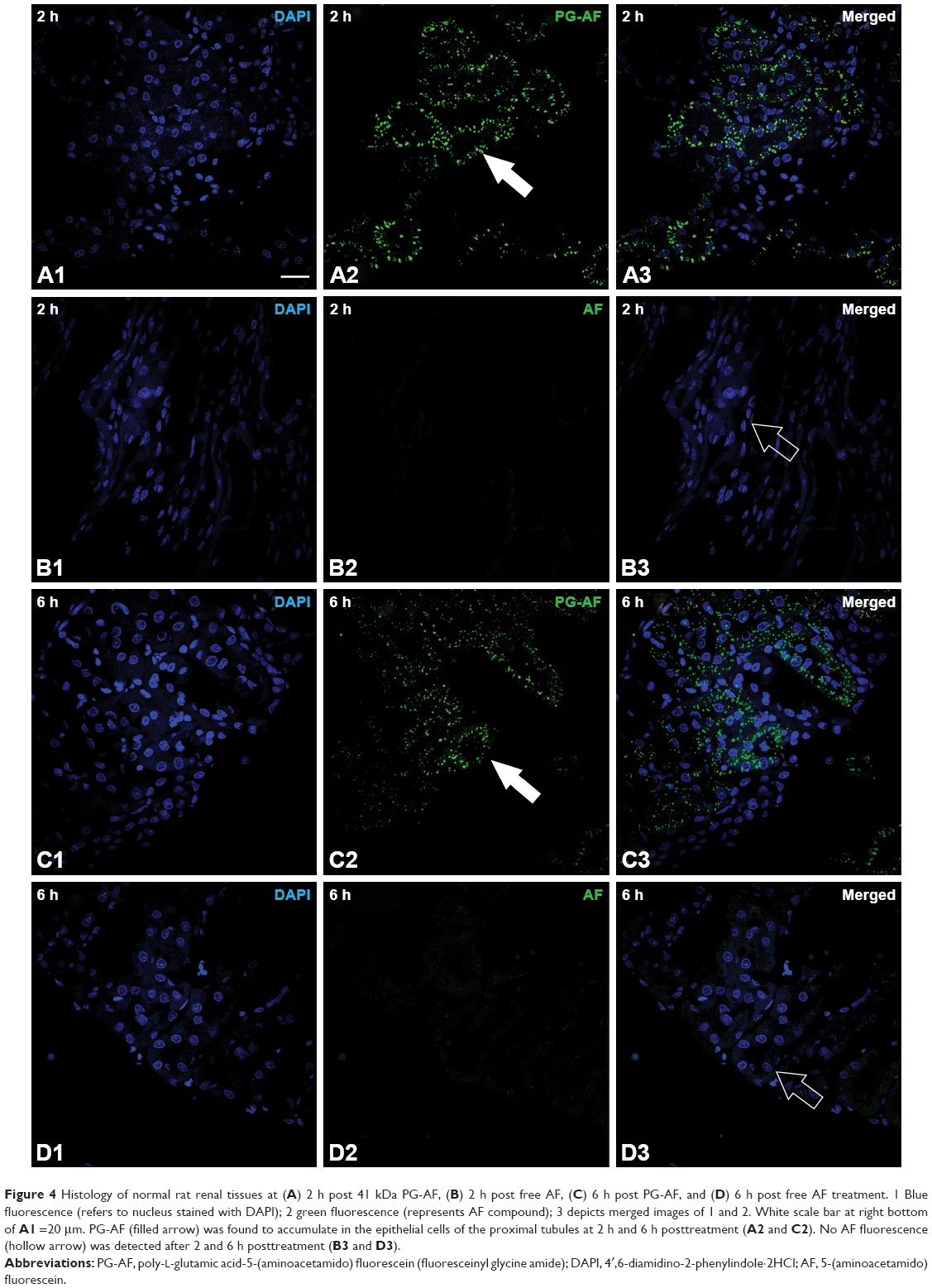 | Figure 4 Histology of normal rat renal tissues at (A) 2 h post 41 kDa PG-AF, (B) 2 h post free AF, (C) 6 h post PG-AF, and (D) 6 h post free AF treatment. 1 Blue fluorescence (refers to nucleus stained with DAPI); 2 green fluorescence (represents AF compound); 3 depicts merged images of 1 and 2. White scale bar at right bottom of A1 =20 μm. PG-AF (filled arrow) was found to accumulate in the epithelial cells of the proximal tubules at 2 h and 6 h posttreatment (A2 and C2). No AF fluorescence (hollow arrow) was detected after 2 and 6 h posttreatment (B3 and D3). |
Inhibition of NADPH oxidase by PG-AEBSF in isolated rat aortic vessels and the uptake of PG-AF by HUVEC
The activity of the PG-conjugated drug AEBSF (an NADPH oxidase inhibitor) was evaluated in vitro using rat isolated aortic vessels and a lucigenin-enhanced chemiluminescence assay. Both AEBSF and PG-AEBSF were found to reduce NADPH-mediated superoxide production. This dose-dependent inhibition by NADPH oxidase in the aortic endothelium (P<0.001 at AEBSF concentration ≥0.05 mM when compared to the control) is indicated by a reduction in chemiluminescence detected in in vitro NADPH oxidase inhibition studies (Figure 5). PG-AEBSF shows a similar NADPH oxidase inhibitory activity to that of AEBSF at all concentrations tested, indicating the activity preservation of bound AEBSF. The NADPH oxidase inhibitor DPI (5 μM) significantly reduced superoxide anion generation.
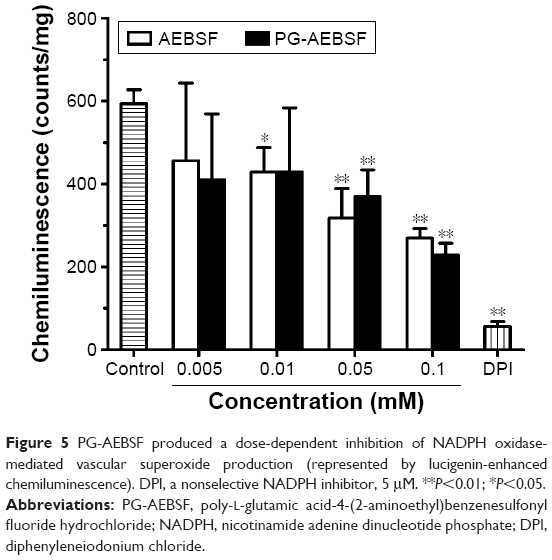 | Figure 5 PG-AEBSF produced a dose-dependent inhibition of NADPH oxidase-mediated vascular superoxide production (represented by lucigenin-enhanced chemiluminescence). DPI, a nonselective NADPH inhibitor, 5 μM. **P<0.01; *P<0.05. |
PG-AEBSF has a comparable NADPH oxidase inhibitory activity to free AEBSF at all concentrations tested. This result indicates the preservation of NADPH oxidase inhibitory properties of AEBSF after conjugation with a PG carrier. Subsequent observation of an in vitro PG-AF uptake by the HUVEC (Figure 6) shows that following a 1 h incubation, only PG-AF, but not free AF, accumulates in the cells in a dose-dependent manner. This finding has further suggested that PG-AEBSF is taken up by the endothelium before AEBSF is released from PG to exert inhibition on the intracellular NADPH oxidase.
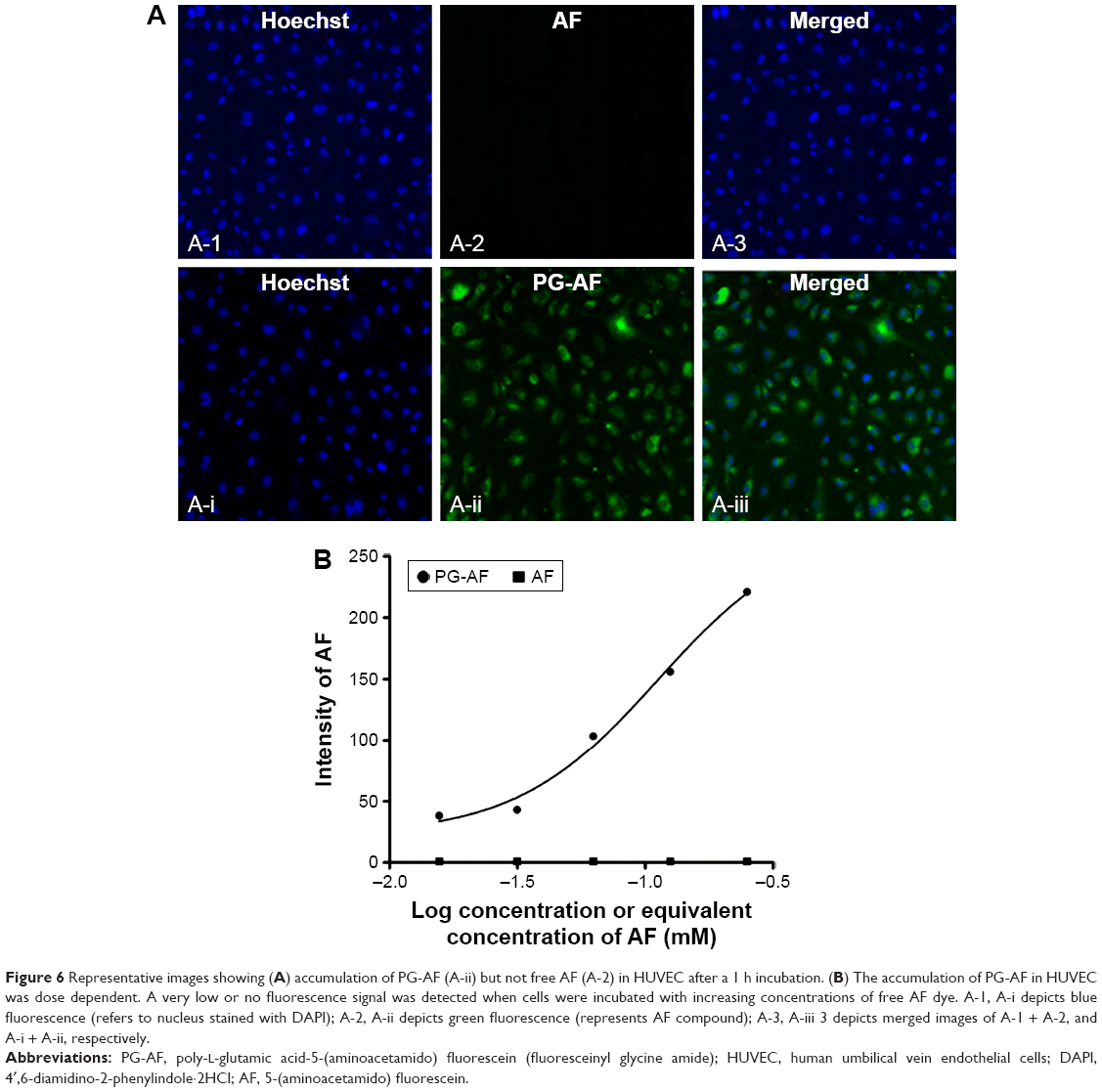 | Figure 6 Representative images showing (A) accumulation of PG-AF (A-ii) but not free AF (A-2) in HUVEC after a 1 h incubation. (B) The accumulation of PG-AF in HUVEC was dose dependent. A very low or no fluorescence signal was detected when cells were incubated with increasing concentrations of free AF dye. A-1, A-i depicts blue fluorescence (refers to nucleus stained with DAPI); A-2, A-ii depicts green fluorescence (represents AF compound); A-3, A-iii 3 depicts merged images of A-1 + A-2, and A-i + A-ii, respectively. |
Discussion
This study aims to evaluate if PG polymers accumulate effectively in the kidneys, and whether these polymers could be used to ferry renal protective anti-oxidative stress compounds (in the current study, an NADPH oxidase inhibitor) without distorting their pharmacological properties. Several novel findings are forthcoming. In this 24 h period animal study, the 41 kDa PG polymer with a size above the renal filtration threshold accumulated within the kidneys to a greater extent than the smaller 17 kDa polymer, whose size was below the renal filtration threshold. Importantly, there was little accumulation of either polymer in any other tissues studied. In the STZ-induced diabetic rat model, where renal oxidative stress, hyperplasia, and hyperfiltration were present, a preferential renal accumulation of 41 kDa PG polymer was observed. The renal accumulation of 41 kDa PG polymer, however, was slightly lower than that in the normal rats. Moreover, the conjugation of 41 kDa PG polymer to an NADPH oxidase inhibitor, AEBSF, did not affect the ability of AEBSF to suppress NADPH oxidase-based superoxide anion generation in the aortic endothelial cells. Together, these observations strongly support the PG polymer as a potentially effective carrier to selectively deliver drugs into the kidneys either in a normal or a diseased state.
There is a possibility that hydrolytic or enzymatic degradation of PGs in the blood40 may potentially detach the radiotracers from the polymer backbone and distort the radiotracing results. To evaluate the stability of PG-Tr in plasma, PG-dCyd (a PG conjugated with nonradioactive dCyd) was prepared and the plasma degradation of PG-dCyd was tracked over time. A detachment of dCyd from PG-dCyd was detected at 24 h after PG-dCyd was incubated in the plasma (see Supplementary material). Thus, the biodistribution of PG-Tr in normal and diabetic rats was not monitored beyond 24 h to avoid inaccuracy.
In normal rats, both 17 kDa PG-Tr and 41 kDa PG-Tr exhibited a significant renal accumulation during the first 8–12 h post dose, while no significant accumulation of Free-Tr was observed in any other organs. These results concur with the hypothesis of Kodaira et al13 on the renal targeting potential of anionic carboxylated polymers. Of the two polymers, 41 kDa PG-Tr exhibited a better renal retention profile than 17 kDa PG-Tr, as shown by a higher and longer accumulation in the kidneys (Figure 2). The glomerular filtration threshold for solutes was at 30 kDa.41–43 Therefore, glomerular filtration could be the main mechanism attributing to the relatively swift loss of 17 kDa PG-Tr from the kidneys and plasma. The renal retention of 17 kDa PG-Tr observed in this study may indicate an involvement of post-glomeruli process such as active reuptake of small-sized anionic carboxylated polymers by the proximal tubules.13 Meanwhile, the favorable renal retention of 41 kDa PG-Tr suggests an extravasation of PG-Tr from the renal vasculature and the accumulation of PG-Tr in the renal parenchymal tissues. Our study on the renal accumulation pattern of fluorescence-labeled 41 kDa PG (PG-AF) shows an uptake and localization of PGs in the epithelial cells of the proximal tubules at 2 and 6 h after an iv administration to the normal rats.
The AUC plots of the biodistribution studies show a high renal accumulation of either 17 kDa PG-Tr or 41 kDa PG-Tr compared to Free-Tr in normal rats (Figure 3). The renal accumulation of 41 kDa PG-Tr is 2-fold compared to 17 kDa PG-Tr in normal rats and the difference is nearly 3-fold in diabetic rats. This finding suggests that 41 kDa PG-Tr is capable of accumulating in the kidneys at a relatively high extent even in a diabetic state. This further supports the suitability of 41 kDa PG-Tr as a renal carrier even in kidneys under oxidative stress.
Failure of using a 100% carboxylated PVP in a renal targeting study was reported previously.13 The authors proposed that the efficiency of renal targeting of anionic carboxylated polymers may be significantly influenced by the degree of carboxylation of the polymer body. Contradicting findings were observed in the present study. An effective renal accumulation of 100% carboxylated PG polymers in both normal and diseased kidneys may suggest that the aforementioned hypothesis could be material-dependent and may not be applicable to all anionic carboxylated carriers. Furthermore, we have demonstrated that 41 kDa PG exhibits a better renal accumulation than 17 kDa PG (Figure 2). This observation indicates that polymer size may play an important role in determining the renal targeting performance of carboxylated polymers. A similar observation of an extensive and prolonged renal accumulation has been noted for another large-sized carrier, 43 kDa RGDfK-(hydroxypropyl)methacrylamide (HPMA).44
The renal targeting performance of PGs in altered renal physiology resembling that of acute kidney injury or chronic renal failure was also studied in the current study. As oxidative stress plays a crucial role in the alteration of renal physiology, hemodynamics, and function over the course of renal failure,11,12,45 we investigated the renal accumulation pattern of PGs in STZ-induced diabetic rats. Under the diabetic condition, an increased glucose supply to the kidneys due to hyperglycemia, coupled with a high metabolic rate of the kidneys, would elevate renal oxidative stress and cause accelerated renal tissue and vascular damage.11,12 Liu and Barac-Nieto reported similar renal pathological changes as early as 2 weeks post STZ administration in rats.39 We found an elevated renal oxidative stress level and an increased kidney-to-body weight ratio in rats 2 weeks after STZ administration (results are described in Supplementary material). An elevation in kidney-to-body weight ratio is indicative of renal pathophysiological changes. Nonetheless, an increase in kidney mass in these rats could also be due to an increase in protein synthesis in the proximal tubules of diseased kidneys as early as 5 days post STZ administration.39
In this study, there was no significant accumulation of free radiotracers in any other organs or tissues in the diabetic rats, while 41 kDa PG showed a selective prolonged renal accumulation during the first 12 h post injection. The renal accumulation of 17 kDa PG, however, was significantly reduced in the first 4 h after dose administration. A 1 h delay in 17 kDa PG renal accumulation was also observed.
Although prolonged renal accumulation of 41 kDa PG was evident, the extent of the accumulation of PGs in the first 6 h was lower in the normal rats than in the diabetic rats (Figure 2). This phenomenon could be due to a compromised glomerular filtration function, which in turn could lead to a rapid loss of PGs from the blood circulation (as depicted in Figure 1). It has been reported that the glomerular filtration functionality may be altered in the diabetic state due to an epithelial dysfunction,46,47 which may lead to renal filtration of macromolecules above the filtration threshold.46,48 Despite a lower initial accumulation, the retention pattern and the levels of 41 kDa PG in the diabetic kidneys were comparable to those in the normal kidneys at later time intervals. This result indicates that a portion of PGs was being retained in the diabetic kidneys over time, possibly via active uptake or passive retention mechanisms. The data in this study support the use of 41 kDa PG as a renal targeting drug carrier in renal pathological states, particularly in the diabetic state.
A low and delayed 17 kDa PG renal accumulation could be correlated to the impairment of an active proximal tubular reuptake process in the diabetic kidneys. As previously suggested,13 an active proximal tubular reuptake process may be crucial in the accumulation of small-sized, anionized carboxylated polymeric carriers in the kidneys. Our results also support an earlier finding13 that altered renal functions and hemodynamics in the diabetic state may affect the active proximal tubular reuptake process and the renal accumulation of renal targeting agents. Consequently, the performance of small-sized carboxylated carriers may be affected by the pathological state of the kidneys. A reduction in the proximal tubular reuptake of a 14 kDa lysozyme-based renal targeting drug carrier after being administered to an adriamycin model of advanced renal disease in rats was also reported.49 This further supports the importance of having a functional renal reuptake mechanism when a tubular reuptake-based renal targeting drug carrier is used.
In addition, the renal accumulation of 17 kDa PG was found to be very much lower at the 0.25 and 1 h time points in the diabetic rats than that in the normal rats receiving similar treatments. Such a reduced accumulation may be correlated to the concurrent increase in the distribution of 17 kDa PG to other organs, such as the liver and lungs with approximately 20%–35% increase in the accumulation at the 0.25 and 1 h time points in the diabetic rats (Table 1). A decreased accumulation of 17 kDa PG in the liver and lungs beyond the 1 h time point, which corresponds to an increase in renal accumulation of 17 kDa PG, may suggest a redistribution of 17 kDa PG from other organs to the kidneys.
NADPH oxidase in the endothelial or epithelial cells of the renal vasculature or parenchymal cells plays an important role in the generation of reactive oxygen species. These oxygen radicals may cause vascular and parenchymal damage that results in acute kidney injury or chronic renal failure. Developing an effective delivery strategy for NADPH oxidase inhibitors to reduce the generation of reactive oxygen species could aid in minimizing vascular injury and slowing down renal dysfunction, thereby reversing or preventing further renal damage.
AEBSF, a relatively nontoxic NADPH oxidase inhibitor,50 was selected as a model drug because of its aminoalkyl moiety. The moiety could conjugate with the –COOH side chain of the PG carrier without affecting the sulfonyl fluoride group that is responsible for the NADPH oxidase inhibition. The synthesis yield of PG-AEBSF was 70%, and the synthesized PG-AEBSF exhibited about 10% drug loading. PG-AEBSF is stable in aqueous solution and plasma for up to 72 h. In vitro chemiluminescence assays have shown that PG-AEBSF (0.005–0.01 mM AEBSF equivalent) has an NADPH oxidase inhibitory activity comparable to that of free AEBSF (0.005–0.01 mM). This finding indicates that the pharmacological properties of AEBSF are preserved after conjugation, as shown in Figure 5. Subsequent in vitro fluorescently labeled PG cellular uptake studies have shown cellular uptake of the PG drug into the intracellular matrix as a whole, instead of the pre-uptake extracellular release of the drug from the PG. These findings are further supported by a subsequent observation of PG-AF uptake by HUVEC, whereby PG-AF but not free AF accumulates in the cells in a dose-dependent manner following a 1 h incubation (Figure 6). These findings also suggest that PG can protect AEBSF from premature extracellular release, and ensure the payload is available intracellularly as a pharmacologically active agent.
Targeted drug delivery has become one of the major interests of the research community in the past three decades, particularly in the area of tumor drug delivery. Nevertheless, the growth in renal targeting delivery is relatively slow. Thus far, only two low-molecular-weight proteins, namely lysozymes and streptavidin, and three low-molecular-weight synthetic polymeric carriers, namely 10 kDa anionic (carboxylated) N-vinylpyrrolidone, 6–8 kDa poly(vinylpyrrolidone-co-dimethyl maleic acid), and 19 kDa acetylated low-molecular-weight chitosan, have been suggested as potential carriers for renal targeting drug delivery.51 All these carriers possess a molecular size that is below the renal filtration threshold, and these compounds depend on the functional proximal tubular reuptake mechanism for their effective renal accumulation. In the current study, we propose 41 kDa PG polymer as a new member of this category of compounds. At the same time, we also point out the possible compromise of renal reuptake or targeting of this class of carrier in a diseased kidney (for example, oxidative-stressed kidney in our study). Compared to using a small-sized renal targeting drug carrier, the feasibility of employing a drug carrier having a molecular size above the renal filtration threshold for renal targeting drug was not previously reported, although the renal accumulation of tumor-targeting drug carriers of such size range (for example, N-[2-hydroxypropyl]methacrylamide, polymer [pHPMA]52 and PG27–32) has been described.
In the current study, we have performed some assessments on the renal accumulation patterns of 41 kDa PG and we would like to propose 41 kDa PG as a renal drug carrier that is potentially capable of delivering drug payload to the renal tissues in both normal and compromised renal anatomical or physiological states (oxidative stress-induced kidney in the current study as an example). Although further studies are required to elucidate the underlying mechanisms, 41 kDa PG has appeared to be one among few (if none) renal targeting carriers that approach the proximal tubular tissues from the basolateral end (as compared to the reported low-molecular-weight renal targeting carriers that approach the proximal tubular tissues from the apical end via tubular reuptake).51 This characteristic may be beneficial in terms of renal drug accumulation, as the carrier-drug complex needs not risk being excreted by the kidney should tubular reuptake mechanism fail to capture the compound in a renal pathological state. Furthermore, a fully biodegradable characteristic of PG carriers would ensure a full release of the drug payload to the renal tissues, thus maximizing the exposure of renal tissues to the drug for a good therapeutic effect.
Conclusion
Organ-specific drug delivery plays an increasingly important role in pharmaceutical development and formulation either by enhancing the efficacy or reducing the adverse effects of new therapeutics. We have shown that PG polymers, particularly PG polymers with a molecular weight above the renal filtration threshold, exhibit a selective and prolonged accumulation in normal or oxidative stress-induced kidneys. We have also demonstrated that anti-oxidative stress drugs can be grafted to PG polymers without affecting the pharmacological properties of the drug. These unique features of PG polymers enhance their potential as a renal targeting drug carrier. Our findings on the selective accumulation of PG in the kidney tubular epithelium also suggest other potential applications for PG in delivering renal protective compounds to the renal tissues during detrimental conditions, such as renal ischemia.
Acknowledgments
This work was supported by grants from University of Malaya (UMRG: RG035/09HTM) and the Ministry of Higher Education, Malaysia (MoHE-HIR grants: UM.C/625/1/HIR/MoHE/MED/17, UM.C/625/1/HIR/MoHE/MED/33, and FRGS: FP068/2007C). The funders had no role in the study design, data collection, and analysis; decision to publish; or preparation of the manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
Eknoyan G, Lameire N, Barsoum R, et al. The burden of kidney disease: improving global outcomes. Kidney Int.2004;66(4):1310–1314. | ||
Zhang Q-L, Rothenbacher D. Prevalence of chronic kidney disease in population-based studies: systematic review. BMC Public Health.2008;8(1):117. | ||
Thadhani R, Pascual M, Bonventre JV. Acute renal failure. N Engl J Med. 1996;334(22):1448–1460. | ||
Star RA. Treatment of acute renal failure. Kidney Int.1998;54(6):1817–1831. | ||
Sheridan AM, Bonventre JV. Pathophysiology of ischemic acute renal failure. In: Ronco C, Bellomo R, La Greca G, editors. Blood Purification in Intensive Care. Basel: Karger; 2001:7–21. | ||
Fede G, D’Amico G, Arvaniti V, et al. Renal failure and cirrhosis: a systematic review of mortality and prognosis. J Hepatol.2011;56(4):810–818. | ||
Meguid El Nahas A, Bello AK. Chronic kidney disease: the global challenge. Lancet.2005;365(9456):331–340. | ||
Ayodele OE, Alebiosu CO, Salako BL. Diabetic nephropathy – a review of the natural history, burden, risk factors and treatment. J Natl Med Assoc.2004;96(11):1445–1454. | ||
Chatterjee PK. Novel pharmacological approaches to the treatment of renal ischemia-reperfusion injury: a comprehensive review. Naunyn Schmiedebergs Arch Pharmacol.2007;376(1–2):1–43. | ||
He L, Shen P, Fu Q, et al. Nephro-protective effect of Kangqianling decoction on chronic renal failure rats. J Ethnopharmacol.2009;122(2):367–373. | ||
Greene DA, Stevens MJ, Obrosova I, Feldman EL. Glucose-induced oxidative stress and programmed cell death in diabetic neuropathy. Eur J Pharmacol.1999;375(1–3):217–223. | ||
Vasavada N, Agarwal R. Role of oxidative stress in diabetic nephropathy. Adv Chronic Kidney Dis.2005;12(2):146–154. | ||
Kodaira H, Tsutsumi Y, Yoshioka Y, et al. The targeting of anionized polyvinylpyrrolidone to the renal system. Biomaterials.2004;25(18):4309–4315. | ||
Kamada H, Tsutsumi Y, Sato-Kamada K, et al. Synthesis of a poly(vinylpyrrolidone-co-dimethyl maleic anhydride) co-polymer and its application for renal drug targeting. Nat Biotech.2003;21(4):399–404. | ||
McCormick-Thomson LA, Sgouras D, Duncan R. Poly(amino acid) copolymers as a potential soluble drug delivery system. 2. Body distribution and preliminary biocompatibility testing in vitro and in vivo. J Bioact Compat Polym.1989;4(3):252–268. | ||
Sumi H, Kawabe K, Nakajima N. Effect of various polyamino acids and D- and L-amino acids on the blood fibrinolytic system. Comp Biochem Physiol B.1992;102(1):159–162. | ||
Li C. Poly(L-glutamic acid) – anticancer drug conjugates. Adv Drug Deliv Rev.2002;54(5):695–713. | ||
Kenny AD. Evaluation of sodium poly-alpha, L-glutamate as a plasma expander. Proc Soc Exp Biol Med.1959;100(4):778–780. | ||
Batz HG, Ringsdorf H, Ritter H. Pharmacologically active polymers, 7. Cyclophosphamide- and steroid hormone containing polymers as potential anticancer compounds. Die Makromolekulare Chemie.1974;175(8):2229–2239. | ||
Hurwitz E, Wilchek M, Pitha J. Soluble macromolecules as carriers for daunorubicin. J Appl Biochem.1980;2(1):25–35. | ||
Kato Y, Saito M, Fukushima H, Takeda Y, Hara T. Antitumor activity of 1-beta-D-arabinofuranosylcytosine conjugated with polyglutamic acid and its derivative. Cancer Res.1984;44(1):25–30. | ||
Morimoto Y, Sugibayashi K, Sugihara S. Antitumor agent poly(amino acid) conjugates as a drug carrier in cancer chemotherapy. J Pharmacobiodyn.1984;7(9):688–698. | ||
Hoes CJT, Potman W, van Heeswijk WAR, et al. Optimization of macromolecular prodrugs of the antitumor antibiotic adriamycin. J Control Release.1985;2:205–213. | ||
Mochizuki E, Inaki Y, Takemoto K. Synthesis of poly-L-glutamates containing 5-substituted uracil moieties. Nucleic acids Symp Ser.1985;16(16):121–124. | ||
Zunino F, Pratesi G, Micheloni A. Poly(carboxylic acid) polymers as carriers for anthracyclines. J Control Release.1989;10(1):65–73. | ||
Hoes CJT, Grootoonk J, Duncan R, et al. Biological properties of adriamycin bound to biodegradable polymeric carriers. J Control Release.1993;23(1):37–53. | ||
Li C, Yu DF, Newman RA, et al. Complete regression of well-established tumors using a novel water-soluble poly(L-glutamic acid)-paclitaxel conjugate. Cancer Res.1998;58(11):2404–2409. | ||
Li C, Price JE, Milas L, et al. Antitumor activity of poly(L-glutamic acid)-paclitaxel on syngeneic and xenografted tumors. Clin Cancer Res. 1999;5(4):891–897. | ||
Li C, Newman RA, Wallace S. Reformulating paclitaxel. Sci Med.1999;6(1):32–41. | ||
Singer JW, De Vries P, Bhatt R, et al. Conjugation of camptothecins to poly-(L-glutamic acid). Ann N Y Acad Sci. 2000;922:136–150. | ||
Singer JW, Bhatt R, Tulinsky J, et al. Water-soluble poly-(L-glutamic acid)-Gly-camptothecin conjugates enhance camptothecin stability and efficacy in vivo. J Control Release.2001;74(1–3):243–247. | ||
Zou Y, Wu QP, Tansey W, et al. Effectiveness of water soluble poly(L-glutamic acid)-camptothecin conjugate against resistant human lung cancer xenografted in nude mice. Int J Oncol.2001;18(2):331–336. | ||
Kiew L-V, Cheong S-K, Sidik K, Chung L-Y. Improved plasma stability and sustained release profile of gemcitabine via polypeptide conjugation. Int J Pharm.2010;391(1–2):212–220. | ||
Kiew LV, Cheong SK, Ramli E, Sidik K, Lim TM, Chung LY. Efficacy of a poly-L-glutamic acid-gemcitabine conjugate in tumor-bearing mice. Drug Dev Res.2012;73(3):120–129. | ||
Pelet JM, Putnam D. An in-depth analysis of polymer-analogous conjugation using DMTMM. Bioconjug Chem.2011;22(3):329–337. | ||
Kiew LV, Cheong SK, Sidik K, Chung LY. Improved plasma stability and sustained release profile of gemcitabine via polypeptide conjugation. Int J Pharm.2010;391(1–2):212–220. | ||
Lau YS, Tian XY, Huang Y, Murugan D, Achike FI, Mustafa MR. Boldine protects endothelial function in hyperglycemia-induced oxidative stress through an antioxidant mechanism. Biochem Pharmacol.2013;85(3):367–375. | ||
Benzie IFF, Strain JJ. The ferric reducing ability of plasma (FRAP) as a measure of “antioxidant power”: the FRAP assay. Anal Biochem.1996;239(1):70–76. | ||
Liu S, Barac-Nieto M. Renal protein degradation in streptozotocin diabetic mice. Diabetes Res Clin Pract.1997;34(3):143–148. | ||
Salmaso S, Caliceti P. Self assembling nanocomposites for protein delivery: supramolecular interactions of soluble polymers with protein drugs. Int J Pharm.2013;440(1):111–123. | ||
Duncan R, Spreafico F. Polymer conjugates. Pharmacokinetic considerations for design and development. Clin Pharmacokinet.1994;27(4):290–306. | ||
Maeda H, Matsumura Y. Tumoritropic and lymphotropic principles of macromolecular drugs. Crit Rev Ther Drug Carrier Syst.1989;6(3):193–210. | ||
Reynolds T. Polymers help guide cancer drugs to tumor targets – and keep them there. J Natl Cancer Inst.1995;87(21):1582–1584. | ||
Dolman MEM, Fretz MM, Segers GJW, et al. Renal targeting of kinase inhibitors. Int J Pharm.2008;364(2):249–257. | ||
Kuyvenhoven JP, Meinders AE. Oxidative stress and diabetes mellitus: pathogenesis of long-term complications. Eur J Intern Med.1999;10(1):9–19. | ||
Rodriguez-Manas L, Angulo J, Peiro C, et al. Endothelial dysfunction and metabolic control in streptozotocin-induced diabetic rats. Br J Pharmacol.1998;123(8):1495–1502. | ||
Kathryn T. Dyslipidaemia, inflammation and endothelial dysfunction in diabetes mellitus. Int Congr Ser.2004;1262:511–514. | ||
Marcello C. Renal glomerular permselectivity and vascular endothelium. Biomed Pharmacother.2005;59(1–2):30–37. | ||
Prakash J, van Loenen-Weemaes AM, Haas M, et al. Renal-selective delivery and angiotensin-converting enzyme inhibition by subcutaneously administered captopril-lysozyme. Drug Metab Dispos.2005;33(5):683–688. | ||
Diatchuk V, Lotan O, Koshkin V, Wikstroem P, Pick E. Inhibition of NADPH oxidase activation by 4-(2-aminoethyl)-benzenesulfonyl fluoride and related compounds. J Biol Chem.1997;272(20):13292–13301. | ||
Dolman ME, Harmsen S, Storm G, Hennink WE, Kok RJ. Drug targeting to the kidney: advances in the active targeting of therapeutics to proximal tubular cells. Adv Drug Deliv Rev.2010;62(14):1344–1357. | ||
Borgman MP, Coleman T, Kolhatkar RB, Geyser-Stoops S, Line BR, Ghandehari H. Tumor-targeted HPMA copolymer-(RGDfK)-(CHX-A”-DTPA) conjugates show increased kidney accumulation. J Control Release.2008;132(3):193–199. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.