Back to Journals » Open Access Journal of Clinical Trials » Volume 12
Remdesivir is Effective for Moderately Severe Patients: A Re-Analysis of the First Double-Blind, Placebo-Controlled, Randomized Trial on Remdesivir for Treatment of Severe COVID-19 Patients Conducted in Wuhan City
Authors Shih WJ ![]() , Shen X
, Shen X ![]() , Zhang P
, Zhang P ![]() , Xie T
, Xie T ![]()
Received 16 May 2020
Accepted for publication 16 July 2020
Published 30 July 2020 Volume 2020:12 Pages 15—21
DOI https://doi.org/10.2147/OAJCT.S262606
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Arthur E. Frankel
Weichung J Shih,1 Xin Shen,2 Peng Zhang,2 Tai Xie2
1Rutgers University School of Public Health, Piscataway, NJ 08854, USA; 2CIMS Global, Somerset, NJ 08873, USA
Correspondence: Weichung J Shih Email [email protected]
Introduction: The first clinical trial on remdesivir for treatment of severe COVID-19 conducted in China was terminated prematurely due to limited patient enrollment, which rendered the findings inconclusive. We re-analyzed the efficacy with a statistically more powerful and clinically meaningful method based on published data using the 6-point ordinal scale of patient’s disease severity.
Methods: We defined response as patient’s point reached, either 2 (hospitalized, no requirement for supplementary oxygen therapy) or 1 (discharged or met discharge criterion), and then analyzed with logistic regression with baseline score, day of assessment, treatment group, baseline by treatment interaction, and day by treatment interaction as covariates. The binary endpoint was supported by the recent FDA’s guidance on COVID-19.
Results: Eighty-two percent (82%) of the patients were in the disease severity point=3 (hospitalized, required supplemental oxygen (but not NIV/HFNC)) – the moderately severe category. The response rate was 85% for remdesivir-treated patients with baseline disease point=3 versus 70% response rate for likewise placebo-treated patients on Day 28 (OR=2.38, P=0.0012). On Day 14, the response rate for these patients was 43% for remdesivir versus 33% for placebo (OR=1.53, P=0.0022). For patients with baseline disease point=4 (critically severe category), no similar comparisons were statistically significant.
Conclusion and Discussion: The Chinese trial was not really under-powered as previously perceived or portrayed by many opinions. This result supports the preliminary findings of ACTT that remdesivir is effective for patients who were not critically severe. This result also suggests that remdesivir should be given to hospitalized COVID-19 patients as soon as possible. There is no race difference in the treatment effect.
Keywords: COVID-19, novel coronavirus, remdesivir
Introduction
The first double-blind, placebo-controlled, randomized trial on intravenous remdesivir for the treatment of severe COVID-19 patients conducted in Wuhan, China1 was highly watched during the pandemic crisis. The main results2 received global attention immediately. In the paper,2 the authors reported that the study stopped early after 237 of the planned 453 patients were enrolled, owing to the fact that the outbreak of COVID-19 was brought under control in China in the midst of the trial. The message that no statistically significant benefits were observed for remdesivir beyond those of standard care treatment was highlighted in the paper. A commentary followed the paper,3 and the general opinion thereafter, attributed the disappointing primary endpoint result to the reduced sample size, leading to the study being underpowered to detect clinically meaningful differences between the two treatment groups.
Specifically, the paper reported that remdesivir treatment was not associated with a difference in time to clinical improvement (TTCI), expressed by a hazard ratio of 1.23 [95% CI: 0.87–1.75]. The median TTCI was 21 days in the remdesivir group vs 23 days in the control group, for the 28-day trial. TTCI was the primary endpoint defined in the study protocol as a two-point reduction in patients’ admission status on a six-point ordinal scale, or live discharge from the hospital, whichever came first. The six-point scale was 6=death; 5=hospitalization, requiring extracorporeal membrane oxygenation (ECMO) and/or invasive mechanical ventilation (IMV); 4=hospitalization, requiring non-invasive ventilation (NIV) and/or high-flow oxygen therapy (HFNC); 3=hospitalization, requiring supplemental oxygen (but not NIV/HFNC); 2=hospitalization, but not requiring supplemental oxygen; 1=hospital discharge or meets discharge criteria (discharge criteria are defined as clinical recovery, ie, fever, respiratory rate, oxygen saturation return to normal, and cough relief, all maintained for at least 72 hours); see Table 1. Scale=3 represents moderately severe and scale=4 and 5 represent critically severe categories.
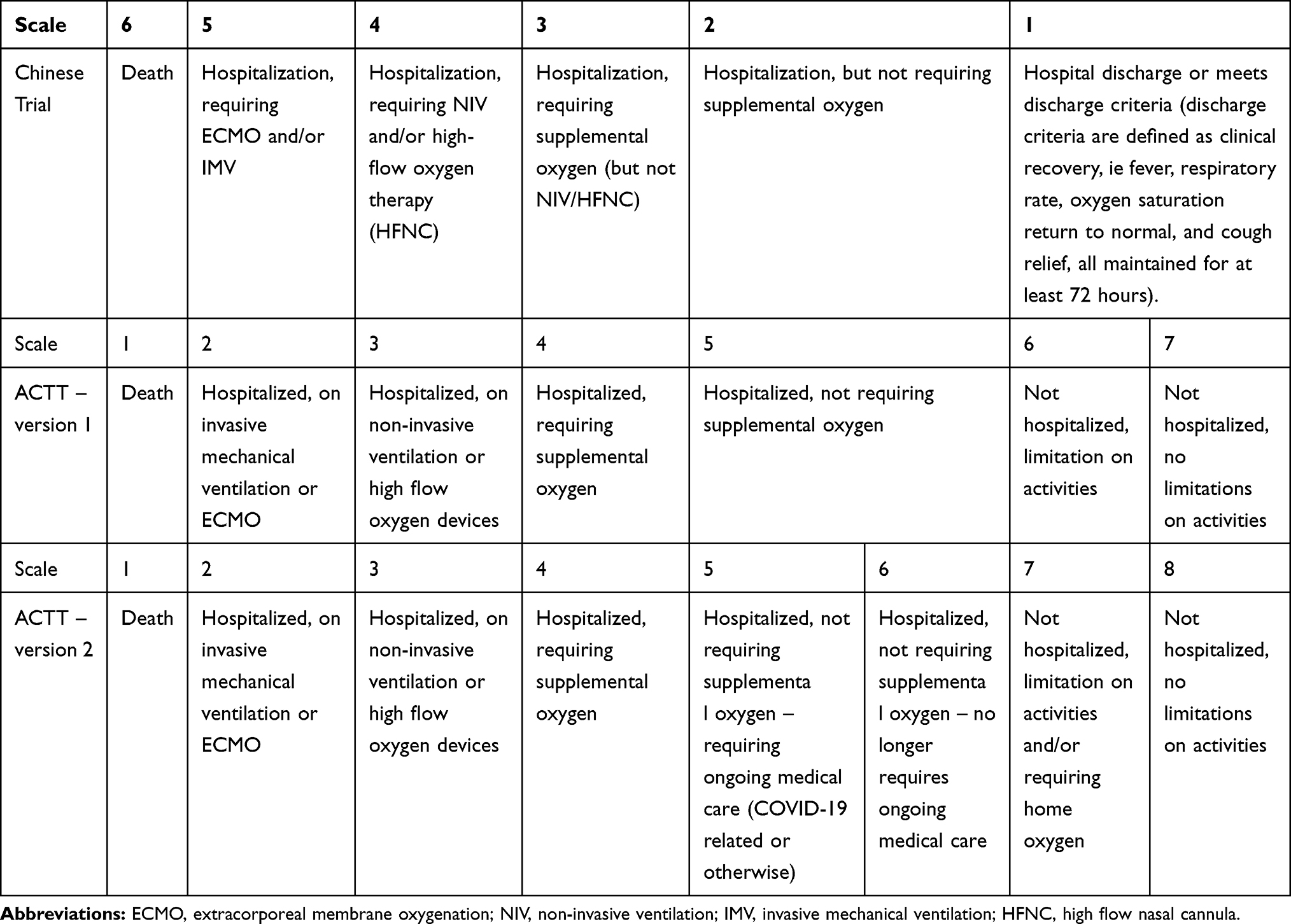 |
Table 1 COVID-19 Disease Ordinal Scale Categories in Chinese Remdesivir Trial and in NIAID’s ACTT Versions 1 and 2 |
In contrast, the preliminary results of the Adaptive COVID-19 Treatment Trial (ACTT)4,5 showed that remdesivir led a 31% faster recovery than the standard care treatment. Specifically, the median time to recovery was 11 days for patients treated with remdesivir compared with 15 days for those who received placebo (p<0.001).5 With the high statistical significance, the trial was stopped early and was renamed “ACTT-1,” as remdesivir became the “standard of care” for the rest of the trial as part of the adaptive design.7,8 Contrary to the Chinese trial, this preliminary result from interim data suggests a possible “over-power” scenario for ACTT-1.
To mitigate the difference between a seemingly “under-powered” study, on one hand, and a possible “over-powered” study on another, we first examine the difference and similarity between the two trials in their primary and secondary endpoints. Motivated by the definition of “recovery” used in ACTT, we then form a binary endpoint of a properly defined “response” – an idea first suggested by Shih et al6 and also listed as one of the three endpoints in a recent Guidance for Industry9 issued by the US Food and Drug Administration (FDA) for COVID-19. We then re-analyze the data from the Chinese remdesivir trial by performing landmark logistic regression analyses with the newly defined binary endpoint. The findings derived from this re-analysis effort should shed some light on the efficacy of remdesivir in the Chinese trial – whether it was really an underpowered study or not, to what extent and on which patient population remdesivir is effective. Our re-analysis should also help in the further assessment of the ACTT data and other future (more than 140) clinical trials currently under development for the treatment of the novel coronavirus globally.
Methods
Ordinal Scale of COVID-19 Severity and Endpoints
Both the Chinese and the US trials used an ordinal scale of categories to indicate a patient’s disease severity status on a specific day, which was based on a blueprint of the World Health Organization (WHO) in treating COVID-19.10 We have reviewed the 6-point scale used in the Chinese trial in the Introduction section. For NIAID’s ACTT, however, there was first a version #1 of a 7-point scale, then revised to a version 2 of an 8-point scale (revision date: March 20, 2020).4 Table 1 displays the details. Aside from the reversed order of points, in essence, ACTT refined the “Live discharge from hospital” in the Chinese trial scale into two more categories. Furthermore, the 8-point scale in ACTT version #2 refined the point=5 category in version #1 into point 5 and point 6 categories. We note that the point=5 category of ACTT version #1 corresponds to the Chinese trial’s scale point=2 category exactly. These all indicate a “mildly severe” status, where the patient was hospitalized, but not requiring supplemental oxygen.
As shown in ClinicalTrials.gov, prior to March 20, the primary endpoint of ACTT was “percentage of subjects reporting each severity rating on the 7-point ordinal scale”; between March 20 and April 20, the primary endpoint was changed to “percentage of subjects reporting each severity rating on the 8-point ordinal scale.” After April 20, the primary endpoint was switched to “time to recovery by Day 29.” Day of recovery is defined as the first day on which the subject satisfies one of the following three categories from the ordinal scale: 1) Hospitalized, not requiring supplemental oxygen – no longer requires ongoing medical care (Point=6); 2) Not hospitalized, limitation on activities, and/or requiring home oxygen (Point=7); 3) Not hospitalized, no limitations on activities (Point=8). In a time of dealing with the complex situation of a pandemic where it is difficult to know exactly what appropriate endpoint would be designated as the primary outcome, these changes seemed to be understood and accepted by the regulatory agency.8
In contrast, in the Chinese trial, the primary endpoint TTCI was defined in the protocol as “time to a 2-point reduction in patients’ admission status on the 6-point ordinal scale, or live discharge from the hospital, whichever came first.” The percentage of subjects reporting each severity rating on the 6-point ordinal scale was a key secondary endpoint. This key secondary endpoint was used by the trial’s data safety monitoring board (DSMB) to monitor the Chinese trial.6 Another endpoint was time to a 1-point reduction, which is also included in the NIAID trial as a secondary endpoint.
The endpoint of time to recovery or clinical improvement, whether defined by 1- or 2-point improvement (TTCI) in the Chinese trial, or as defined in the NIAID’s ACTT, seemed to have escaped the difficulty of “hazard ratio” interpretation and enjoyed a simpler understanding of “median day to response” for clinicians and journalists. However, this kind of time-to-response endpoint has some technical limitations. First, the scores might fluctuate, especially when the scale was refined into more categories. Thus, the “time to response” really meant a time to the first response, ignoring the possibility of sequential worsening on a later day. Second, time-to-improvement does not make clinical sense for patients who died during the study. For the severe COVID-19 cases, the 28-day mortality rate was about 13–14% in the Chinese trial and 8–12% in the NIAID trial. For the dead, the time to recovery or clinical improvement (TTCI) is infinite or undefined, but has been censored at day 28 or 29. The censoring is obviously an unfair accounting to patients who were alive without reaching the recovery or improvement criterion by the end of the study. We therefore explore the following alternative analysis.
Alternative Data Analysis for the Chinese Trial
Based on the NIAID trial, in which the “recovery” criterion was defined by reaching categories with point=6, 7, or 8, we seek the corresponding categories in the Chinese trial and determine similarly the “recovery” criterion as reaching the clinical status with point=2 or 1 in the 6-category (reversed) scale. See Table 1. As expressed by clinical experts,5,7 sparing severely ill patients from requiring supplemental oxygen in the midst of the pandemic crisis is a clinically meaningful event to the patients as well as to the health-care providers, as the supplemental oxygen equipment may then be cleared to other patients who are in need.
We therefore classify each outcome in the Chinese trial a “response” or “non-response” at each assessment day by examining the 6-point scale status: Point=2 or 1 being a response; otherwise a nonresponse. We then analyze the binary response data with the method of logistic regression. Our analysis is based on the summary data shown in Shih et al’s study6 at the last DSMB meeting on March 29, 2020, which is close to the completion of the trial’s final data lock on April 1, 2020 reported by Wang et al.2 The logistic regression model includes the baseline disease status, treatment group, assessment day, treatment by day interaction, and treatment by baseline status interaction. Notice that this model will obtain the treatment effect adjusted for the baseline status and assessment day in the study. Our main aim is to assess the treatment effect on Day 28 while controlling for baseline status. We also test the treatment effect on Day 14 to see if there is an early treatment effect 4 days after the 10-day intravenous regimen of remdesivir. Given that the two analyses at the two different days are correlated, we use the Hochberg’s step-wise procedure to control the overall type-I error rate:11 test the hypothesis associated with the smaller p-value against alpha=0.025 and that associated with the larger p-value against alpha=0.05 level. We express the treatment effect of remdesivir in terms of the odds ratio of response (with 95% confidence interval) relative to the placebo.
Results
The dataset included 231 patients (153 remdesivir, 78 placebo) for the 6-point ordinal scale at baseline and 225 patients (149 remdesivir, 76 placebo) on Day 28. The baseline score distribution (%) is summarized in Table 2: (0, 0, 81.0, 17.6, 0.7, 0.7) for the remdesivir group and (0, 3.8, 83.3, 11.5, 1.3, 0) for the placebo group, for point=1 (discharged or met discharge criteria) to 6 (death). As seen, the majority (81–83%) were point=3 patients, who were hospitalized, required supplemental oxygen (but not NIV/HFNC) – the moderately severe category. About 12–18% were point=4 patients, who were hospitalized and required non-invasive ventilation (NIV) and/or high-flow oxygen therapy (HFNC). Very few were in category 5, who required extracorporeal membrane oxygenation (ECMO) and/or invasive mechanical ventilation (IMV). The proportions of responders (defined as point≤2), uncontrolled for baseline status, are displayed in Figure 1 by treatment groups at each study assessment day. The increasing trend of response is obvious for both treatment groups. Table 3 shows the main results of the logistic regression analysis. The response rate was 85% for remdesivir-treated patients with baseline status point=3 (moderately severe category) versus 70% response rate for likewise placebo-treated patients on Day 28 (OR=2.38, p=0.0012). On Day 14, the response rate for these patients was 43% for remdesivir versus 33% for placebo (OR=1.53, p=0.0022). Both were statistically significant with the multiple test adjustment. For patients with baseline status point=4 (critically severe category), which was a much smaller cohort in the study, no similar comparisons were statistically significant, although the placebo group had a higher response rate numerically.
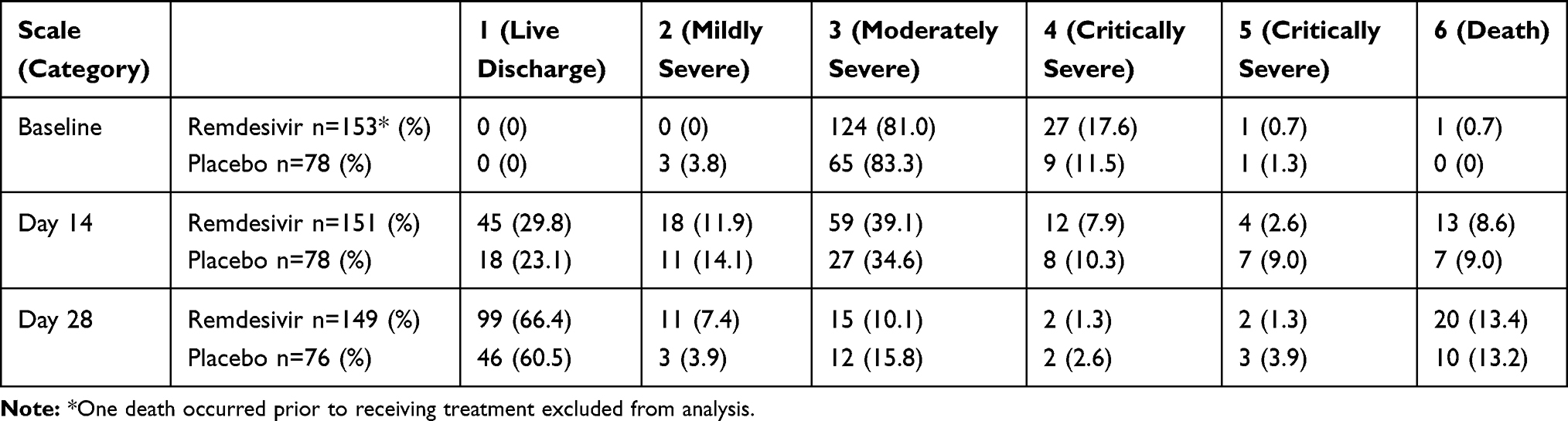 |
Table 2 Distribution of Categorical Scale at Baseline, Day 14 and Day 28 by Treatment Group |
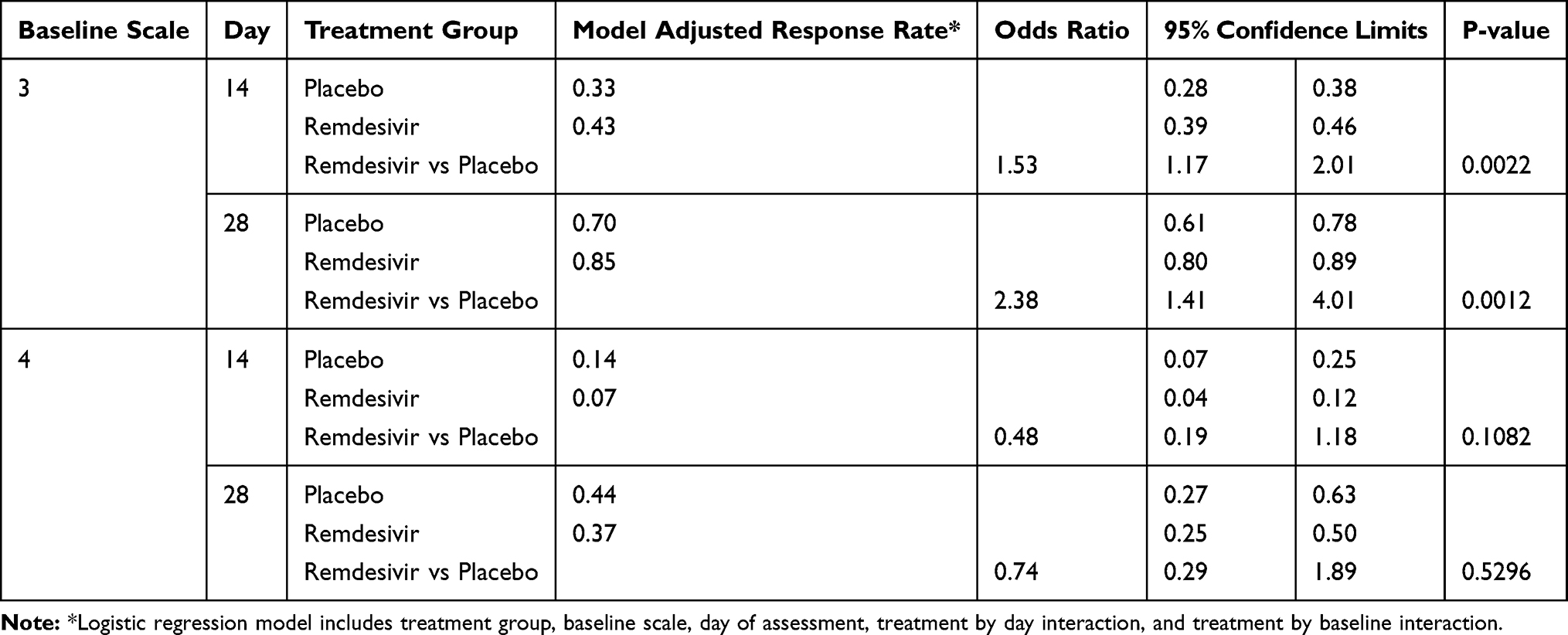 |
Table 3 Treatment Effect Controlled by Baseline Scale and Day of Assessment |
 |
Figure 1 Response rate (%) by day: remdesivir vs control. |
Conclusion and Discussion
It is clear that the logistic regression analysis of the binary endpoint provides more statistical power for the data, and shows that the remdesivir IV 10-day regimen is effective for moderately severe COVID-19 patients in improving the odds of response by 2.4-fold on Day 28 and 1.5-fold on Day 14 since the start of treatment, with high statistical significance. Thus, the Chinese study was not really “under-powered” as it was previously perceived, despite its early end of patient enrollment. But why and how is this logistic regression analysis statistically valid and clinically sound? For these questions, we offer the following points:
The binary endpoint that pools the scale=2 and 1 together as “response” has been suggested by the trial’s data safety monitoring board (DSMB) prior to the final data analysis as an alternative to the time-to-clinical improvement (TTCI) endpoint,6 and it is recommended recently by the FDA.9 It may not have been chosen as the pre-specified primary endpoint at an urgent time when there is so much unknown about COVID-19 (eg, ACTT made several adaptations regarding endpoints and sample sizes during the course of the trial, as its study title properly indicated), but the binary response is well justifiable. It is similar to oncology Phase II trials, where we usually pool the complete response (CR) and partial response (PR) together as “response,” and the rest stable disease (SD) and disease progression (DP) as “non-response” for an ORR (objective response rate) analysis. The dichotomization of a multi-level scale aggregates more events on both sides of “response” versus “non-response”; hence sharpening the comparison and strengthening the signal. This process makes the analysis more powerful than using the original multi-level scale. The landmark analysis at Day 28 – the end of the follow-up day is also simple and clear for interpretation. On the contrary, the time-to-recovery or time-to-clinical improvement (TTCI) has an intrinsic problem for the dead whose time measure would be infinite or undefined. The binary endpoint also makes sense to clinicians; after all, their decision is always of a binary nature: Do I use this drug to treat my patient or not? The binary endpoint is also clinically meaningful on the ground that, when patients no longer require supplementary oxygen (scale=2) or are discharged from hospital (scale=1), the disease burden is released from the patients as well as from the health-care facilities in the pandemic situation.
In conclusion, our re-analysis demonstrated that good response rates were achieved with strong statistical significance for remdesivir for the moderately severe patients; valid conclusions can still be made despite the early termination and reduced sample size. The re-analysis supports the preliminary finding of ACTT that remdesivir is effective, but we qualify that the efficacy applies only to patients whose COVID-19 condition at enrollment was not critically severe, which is the majority of hospitalized patients with COVID-19. We also echo the decision of making remdesivir available as a part of standard care in the hospital setting in recognition of the urgent need, and agree that the FDA’s issuance of EUA is an important step toward developing more effective therapies for all range of COVID-19 patients.
Acknowledgment
Thanks to Chen Yao, M.D., who was a member of the trial’s DSMB, at Peking University Clinical Research Institute, Beijing, China, Yeming Wang, M.D. and Bin Cao, M.D., who were Principal Investigators of the Chinese Remdeivir trial and authors of the trial report in references,1,2 for their communications and/or previews of an early abstract of this manuscript.
Disclosure
Weichung J. Shih was a member of the DSMB of the Chinese remdesivir trial. The opinion expressed here is that of the authors, not of the DSMB. The authors declare no other conflicting interests in this work.
References
1. A Phase 3 randomized, double-blind, placebo-controlled, multicenter study to evaluate the efficacy and safety of remdesivir in hospitalized adult patients with severe 2019- nCoVRespiratory disease. PI: Cao Bin. (ClinicalTrials.gov Identifier: NCT04257656)
2. Wang Y, Zhang D, Du G, et al. Remdesivir in adults with severe COVID-19: a randomised, double-blind, placebo-controlled, multicentre trial. Lancet. 2020;395(10236):1569–1578. doi:10.1016/S0140-6736(20)31022-9
3. Norrie JD. Remdesivir for COVID-19: challenges of underpowered studies. Lancet. 2020;395(10236):1525–1527. doi:10.1016/S0140-6736(20)31023-0
4. A multicenter, adaptive, randomized blinded controlled trial of the safety and efficacy of investigational therapeutics for the treatment of COVID-19 in hospitalized adults. National Institute of Allergy and Infectious Diseases (NIAID). ClinicalTrials.gov Identifier: NCT04280705.
5. Beigel JH, Tomashek KM, Dodd LE, et al. Remdesivir for the treatment of Covid-19 —preliminary report. N Engl J Med. 2020. doi:10.1056/NEJMoa2007764
6. Shih WJ, Yao C, Xie T Data monitoring for the chinese clinical trials of remdesivir in treating patients with COVID-19 during the pandemic crisis. Therapeutic innovation & regulatory science; May 16, 2020. Available from: https://link.springer.com/article/10.1007/s43441-020-00159-7?wt_mc=Internal.Event.1.SEM.ArticleAuthorOnlineFirst.
7. Hughes S Remdesivir now ‘Standard of care’ for COVID-19, Fauci says – multiple trials release data, some in partial form. Medscape; April 29, 2020. Available from: https://www.medscape.com/viewarticle/929685.
8. Frellick M FDA authorizes emergency use of remdesivir for COVID-19. Medscape; May 01, 2020. Available from: https://www.medscape.com/viewarticle/929836.
9. COVID-19: developing Drugs and Biological Products for Treatment or Prevention, Guidance for Industry, U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER); May, 2020.
10. World Health Organization. WHO R&D Blueprint Novel Coronavirus: Outline of Trial Designs for Experimental Therapeutics; 2020.
11. Hochberg Y. A sharper Bonferroni procedure for multiple tests of significance. Biometrika. 1988;75(4):800–802. doi:10.1093/biomet/75.4.800
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.