Back to Journals » International Journal of Nanomedicine » Volume 14
Remarkable Boron Delivery Of iRGD-Modified Polymeric Nanoparticles For Boron Neutron Capture Therapy
Authors Chen J, Yang Q, Liu M, Lin M, Wang T, Zhang Z, Zhong X, Guo N, Lu Y, Xu J, Wang C, Han M, Wei Q ![]()
Received 2 May 2019
Accepted for publication 11 September 2019
Published 8 October 2019 Volume 2019:14 Pages 8161—8177
DOI https://doi.org/10.2147/IJN.S214224
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Linlin Sun
Jiejian Chen,1,2 Qiyao Yang,1,2 Minchen Liu,3 Mengting Lin,2 Tiantian Wang,2 Zhentao Zhang,2 Xincheng Zhong,2 Ningning Guo,2 Yiying Lu,2 Jing Xu,1 Changsheng Wang,4 Min Han,2 Qichun Wei1
1Department of Radiation Oncology, Ministry of Education Key Laboratory of Cancer Prevention and Intervention, The Second Affiliated Hospital, College of Medicine, Zhejiang University, Hangzhou 310009, People’s Republic of China; 2Institute of Pharmaceutics, College of Pharmaceutical Sciences, Zhejiang University, Hangzhou 310058, People’s Republic of China; 3Engineering Research Center of Modern Preparation Technology of TCM, Shanghai University of Traditional Chinese Medicine, Shanghai 201203, People’s Republic of China; 4Department of Spinal Surgery, The First Affiliated Hospital, Fujian Medical University, Fuzhou 350005, People’s Republic of China
Correspondence: Qichun Wei
Department of Radiation Oncology, Ministry of Education Key Laboratory of Cancer Prevention and Intervention, The Second Affiliated Hospital, College of Medicine, Zhejiang University, Hangzhou 310009, People’s Republic of China
Tel +86 571 8778 3521
Fax +86 571 8721 4404
Email [email protected]
Changsheng Wang
Department of Spinal Surgery, The First Affiliated Hospital, Fujian Medical University, Fuzhou 350005, People’s Republic of China
Email [email protected]
Purpose: Boron neutron capture therapy (BNCT) is an emerging binary radiotherapy, which is limited for application due to the challenge of targeted delivery into tumor nowadays. Here, we propose the use of iRGD-modified polymeric nanoparticles for active targeted delivery of boron and doxorubicin (DOX) in BNCT.
Methods: 10B-enriched BSH was covalently grafted to PEG-PCCL to prepare 10B-polymer, then surface-modified with iRGD. And, DOX was physically incorporated into polymers afterwards. Characterization of prepared polymers and in vitro release profile of DOX from polymers were determined by several methods. Cellular uptake of DOX was observed by confocal microscope. Accumulation of boron in cells and tissues was analyzed by ICP-MS. Biodistribution of DOX was studied by ex vivo fluorescence imaging and quantitative measurement. Tumor vascular normalization of Endostar for promoting delivery efficiency of boron on refractory B16F10 tumor was also studied.
Results: The polymers were monodisperse and spheroidal in water with an average diameter of 24.97 nm, which were relatively stable at physiological pH and showed a sustained release of DOX, especially at endolysosomal pH. Enhanced cellular delivery of DOX was found in iRGD-modified polymer group. Cellular boron uptake of iRGD-modified polymers in A549 cells was remarkably raised fivefold (209.83 ng 10B/106 cells) compared with BSH. The polymers represented prolonged blood circulation, enhanced tumor accumulation of 10B against BSH, and favorable tumor:normal tissue boron concentration ratios (tumor:blood = 14.11, tumor:muscle = 19.49) in A549 tumor-bearing mice 24 hrs after injection. Both fluorescence imaging and quantitative measurement showed the highest tumor accumulation of DOX at 24 hrs after injecting of iRGD-modified polymers. Improvement of vascular integrity and reduction of vascular mimicries were found after Endostar injection, and raised tumor accumulation of boron as well.
Conclusion: The developed nanoparticle is an inspiring candidate for the safe clinical application for BNCT.
Keywords: BNCT, drug delivery, polymer–drug conjugate, BSH, doxorubicin
Introduction
Boron neutron capture therapy (BNCT) was widely verified as a superior therapeutic strategy for neoplasms during recent decades.1 It is a binary radio-therapeutic management consisting of the nuclear capture and fission reactions that eventuate when non-radioactive element boron-10 (10B) is irradiated with neutrons of appropriate energy to yield excited boron-11 (11B*). This undergoes prompt nuclear fission to produce high-linear energy transfer (LET) alpha particles (4He) and recoiling lithium-7 (7Li) nuclei. The reaction mechanism is: 10B + nth → [11B] * → 4He + 7Li + 2.31 MeV. Both the 4He and the 7Li ions provoke closely spaced ionizations in the instant vicinity of the reaction, with a short path-lengths (5~9 µm), which accounts for the limited destructive effects to boron-containing cells. BNCT, therefore, is considered as both biologically and physically targeted radiotherapy.
The success of BNCT is dependent on the favorable thermal neutrons and the targeted delivery of enough 10B to tumor tissues (~109 atoms/cell, tumor:blood and tumor:normal tissue boron concentration ratios of >3:1).2,3 Gratifyingly, the advancements of neutron-producing accelerators installed in hospitals make BNCT be more realistic to clinical application.4,5 As for BNCT agent, sodium mercaptoundecahydro-closo-dodecaborate (BSH) is currently used in clinical trials. However, the unfavorable cell membrane penetration of BSH would cause the failure of BNCT.6,7 Herein, there has been an imperative requirement to develop novel boron agents.
Taking advantage of the enhanced permeability and retention (EPR) effect of tumor tissues, nanomaterial-based drug delivery systems (liposomes, polymers, dendrimers, etc.) allow the accumulation of drugs into the targeted lesions.8–11 There are two main types of approaches for the incorporation of boron into delivery systems, either by physical encapsulation or covalent attachment. Theoretically, active targeting domains (molecule, peptide, and monoclonal antibody) conjugated into carriers will further strengthen the drug delivery efficiency.12
Ring-opening polymerization of ε-caprolactone with ethylene glycol (PEG) as initiator and stannous octoate as catalyst is usually used to prepare PEG-PCL. Through chemical engineering of the structure ε-caprolactone with benzyl chloroformate, and ring-opening polymerizing with ethylene glycol (PEG), and then deoxidizing of benzyl carboxylate to carboxyl group, desired PEG-PCCL with functional groups (carboxyl) are achieved. In this work, BSH was covalently grafted to the PEG-PCCL to prepare PEGylated 10B-polymers, then surface-modified with the promising tumor-penetrating peptides (iRGDs).13 It is demonstrated that the developed 10B-polymers (iRGD-PEG-PCCL-B) are challenged against 10B-polymers without modification of iRGDs (mPEG-PCCL-B) and intact BSH on their performances of intracellular uptake, prolonged blood circulation and tumor accumulation. As for some refractory cancer with dreadful tumor microenvironment, we applied recombinant human endostatin (Endostar) for tumor vascular normalization, aiming to modulate tumor microenvironment for tumor reaccumulation of therapeutic agents. Encouragingly, indications for optimization of the tumor environment like increased tumor vessels encased compactly with α-SMA+ pericytes and reduced vasculogenic mimicry (VM) were observed in B16F10 tumor tissues after Endostar procedure.
Combined-modality therapy is increasingly recommended in management to achieve a better outcome for patients.14,15 Our previous studies16,17 documented a better response to combination Doxorubicin (DOX) and radiotherapy than that of single modality, for that doxorubicin can enhance radiosensitivity in tumor cells. We suppose, hypothetically, combined BNCT with DOX can be more sensitive to neoplasms. Accordingly, DOX was physically incorporated into the hydrophobic core of polymers. The resulting drug delivery system (iRGD-PEG-PCCL-B/DOX) for combined BNCT and chemotherapy could not only achieve sufficient tumor boron accumulation for successful BNCT, but also reduce systemic toxicity of DOX for chemotherapy (Figure 1A and B).
 |
Figure 1 Schematic illustration of polymers' self-assembly for targeting delivery of BSH and DOX into cancer cells. (A) Synthetic scheme of iRGD-PEG-PCCL-B/DOX. (B) The working mechanism of the iRGD-mediated transport pathway. |
Materials And Methods
Materials And Reagents
Monomethoxy-poly (ethylene glycol) (mPEG, Mw 5000 g/mol), ε-caprolactone, lithium diisopropylamide 3(LDA), benzyl chloroformate, triethylamine (TEA), N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloridetrimethylamine (EDCI), N-Hydroxysuccinimide (NHS), pyrene, tetrahydrofuran (THF), and dimethyl sulfoxide (DMSO) were purchased from Aladdin Chemistry Co., Ltd. (Shanghai, China). Nitric acid (65%) and hydrogen peroxide (30%) were purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). N-(2-aminoethyl) maleimide and indocyanine green (ICG) were purchased from TCI Co., Ltd. (Shanghai, China). BSH (99.5 atoms% 10B) was purchased from Katchem Co., Ltd. (Prague, Czechoslovakia). Doxorubicin hydrochloride was purchased from Dalian Meilun Biotechnology Co., Ltd. (Dalian, China). iRGD(CRGDKGPDC) were customized from GL Biochem Co., Ltd. (Shanghai, China). Recombinant human endostatin (Endostar) was kindly provided from Simcere Pharmaceutical Co., Ltd. (Nanjing, China). DAPI and Lyso-Tracker Red were purchased from Beyotime Co., Ltd. (Shanghai, China). Acetal-PEG was synthesized according to the reference.18 All other materials and solvents were of reagent grade and used as received.
Preparation Of mPEG-PCCL And Acetal-PEG-PCCL
mPEG-PCCL was prepared in three steps. Firstly, the monomer (α-benzylcarboxylate-ε-caprolactone) was synthesized in terms of the reported protocol.18 Then, we employed ring-opening polymerization of α-benzylcarboxylate-ε-caprolactone using mPEG as initiator and stannous octoate as catalyst to synthetize mPEG-PBCL. mPEG-PCCL was obtained by removal of the benzyl groups of mPEG-PBCL in the presence of charcoal-coated with palladium and hydrogen in the end. Similarly, Acetal-PEG-PCCL was also synthesized by the same method.
Preparation Of mPEG-PCCL-B And Acetal-PEG-PCCL-B
mPEG-PCCL-B were prepared using combination reaction. BSH (92 mg, 20 eq.) and N-(2-aminoethyl) maleimide (58 mg, 30 eq.) were dissolved in 8 mL anhydrous dimethyl sulfoxide containing 92 μL trimethylamine to be continuously stirred for 48 hrs under nitrogen atmosphere at room temperature. Next, mPEG-PCCL (137 mg, 1 eq.) dissolved into 5 mL anhydrous dimethyl sulfoxide was activated using EDCI/NHS (76 mg/46 mg, 18 eq.) and dropped into BSH solution to react for another 48 hrs in the presence of nitrogen at room temperature. After reaction, the solution was extensively dialyzed (Spectrum, MW cut-off 3500) against deionized water to remove the unreactant for three days, and freeze-dried. Acetal-PEG-PCCL-B was obtained in this method analogously.
Preparation Of iRGD-PEG-PCCL And iRGD-PEG-PCCL-B
The iRGD peptide was conjugated to the micellar surface by the functional acetal groups. Acetal-PEG-PCCL or Acetal-PEG-PCCL-B was assembled to polymers by dispersing the polymers (162.5 mg, 40 mol% of Acetal-PEG-PCCL or Acetal-PEG-PCCL-B, and 60 mol% of mPEG-PCCL) in component solvent including acetone (3 mL) and methanol (3 mL), and dropwise addition of polymer solution to sodium phosphate buffer solution (20 mL, pH = 7.0, ionic strength 0.2 M) under stirring at room temperature, followed by rotary evaporation of organic solvent. The polymer solution was then acidified to pH 2.0 with diluted HCl (0.5 M) and kept stirring for 3 hrs at room temperature to generate aldehyde-polymeric polymers. The resultant solution was then neutralized with NaOH (0.5 M). iRGD (2.2 mg, 0.8 eq.) was dissolved in micellar solution under stirring for 2 hrs. Finally, sodium cyanoborohydride (1.5 mg, 10 eq.) was added to the polymer solution to react for 4 days, then unreacted peptide and reducing reagent were removed by dialysis against deionized water. The conjugation efficiency of iRGD to Acetal-polymers was determined by the HPLC method as reported.18
Preparation Of DOX-Encapsulated Polymers
Chemotherapeutic drug DOX was physically encapsulated in polymers (mPEG-PCCL-B and iRGD-PEG-PCCL-B) to produce DOX-loaded polymers. Briefly, DOX·HCl (0.25 mg, 1 eq.) and TEA (0.2 μL, 3 eq.) were dissolved in component solvent containing acetone (1 mL) and methanol (1 mL) in a round-bottom flask followed by the addition of block copolymers (10 mg). The solution was added dropwise to PBS (4 mL, pH 7.4) under gentle stirring at room temperature. After stirring for 24 hrs, the solution was shifted to a dialysis bag (Spectrum, MW cut-off 3500) and dialysed for 12 hrs to get rid of organic solvent and unloaded DOX.
Characterization Of Polymers
To figure out the structures and average molecular weights of the polymers, 1H nuclear magnetic resonance (NMR) spectra was carried out, respectively, by a NMR spectrometer (400 MHz, Bruker AVIII, Switzerland) in CDCl3 at ambient temperature. The measurements of the size and zeta potential of polymers were conducted by dynamic light scanning (DLS) applying a Zetasizer Nano ZS instrument (Malvern, UK). The morphology of the polymeric nanoparticles was captured by transmission electron microscope (TEM) performed on a Tecnai G2 Spirit microscope (120 kV) after phosphotungstic acid staining. The critical micelle concentration (CMC) of copolymers was determined by pyrene fluorescence spectroscopy. Inductively coupled plasma-mass spectrometry (ICP-MS, NexION 300X, PerkinElmer, USA) was used to measure boron content, and the DOX loading content (LC) was estimated by UV analysis at 480 nm of DOX absorbance as follows:
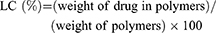
To evaluate the stability of polymers, the PBS solution (pH 7.4) of polymeric nanoparticles was analyzed by DLS at predetermined time points.
In Vitro Release Profile Of DOX
In vitro release rate of DOX from the polymers was determined in PBS solutions (pH 5.0, pH 7.4). Briefly, 2 mL of DOX-encapsulated polymer solution was introduced into a dialysis bag (MWCO: 3500), then placed into 18 mL of PBS solution at 37°C with steady shaking at 100 rpm/min. 1 mL of buffer outside the dialysis bag was gathered for M5 Multi-Mode Plate Reader (Molecular Devices) measurement and replenished equal volume of fresh buffer at different time points.
Cell Lines
The cancer cell lines A549, B16F10, C6, 4T1, Hela [Institute of Biochemistry and Cell Biology of Chinese Academy of Science (Shanghai, China)] were cultured in complete growth media (RPMI-1640 or DMEM medium supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin) at 37°C in a humid atmosphere maintained of 5% CO2.
Cell Viability Assay
A549 and B16F10 Cells were seeded in 96-well plates with a concentration of 3.0 × 103 cells/well, respectively. After 24 hrs hatch in cell culture incubator, different polymeric formations (n=6) were added into each well in various concentrations for 48 hrs incubation followed by Cell Counting Kit-8 (CCK8, Beyotime, Shanghai, China). And cell viability was calculated as percentages by compared with control (without polymers incubation). The toxicities of DOX and BSH were also assessed by the same procedure.
Hemolysis Assay
The hemolytic activities of mPEG-PCCL-B and iRGD-PEG-PCCL-B were evaluated by the issued protocol.19 Briefly, the erythrocytes were collected from the blood of New Zealand white rabbit by centrifuging at 2500 rpm for 10 mins, rinsed three times with PBS (pH 7.4) at 2500 rpm for 10 mins, and diluted with PBS to obtain a concentration of 1.5% (v/v). Polymers were dispersed in PBS and diluted into various concentrations of 0.3125, 0.625, 1.25, 2.5, 5, 10 mg/mL. PBS (pH 7.4) was introduced as negative control, while Triton X-100 (0.2%) as positive control. For each polymer solution, equal volume of 1.5% erythrocytes was appended. The resultant solutions were incubated at 37°C for 4 hrs and then centrifuged for 12 mins at 13,000 rpm. The supernatant was investigated by photometric analysis at 540 nm to assess the hemolytic percentage of polymers.
Cellular Uptake And Distribution
To determine the cellular uptake of DOX, A549 cells were seeded into glass bottom dishes (20 mm, Nest) at a cell density of 1.0 × 103 cells/dish, incubated for 24 hrs, washed by PBS and then coincubated for another 2 hrs, respectively, with free DOX, mPEG-PCCL-B/DOX, or iRGD-PEG-PCCL-B/DOX of an equivalent DOX dose of 0.5 μg/mL. After washing with cold PBS for three times, the cells were fixed in 4% paraformaldehyde, incubated with DAPI for 10 mins, and rinsed with PBS for three times. The DAPI and intracellular DOX were intuitively observed by laser scanning confocal microscope (LSCM, Zeiss LSM 800, Germany). The fluorescence channels for DOX were kept at the same laser intensity so as to quantify cellular uptake of DOX by an US National Institutes of Health-ImageJ software. For intracellular distribution of DOX, the cells were seeded into dishes, and incubated with free DOX (1 μg/mL) or iRGD-PEG-PCCL-B/DOX of the equivalent DOX dose for 2, 4, and 8 hrs. Samples were then fixed, stained with DAPI, and visualized by LSCM eventually.
The subcellular localization of polymers was primarily assessed by blue fluorescence molecular labeling of iRGD-PEG-PCCL with 2-butyl(4-aminohexyl)-6-(dimethylamino)-1H-benz[de]isoquinoline-1,3(2H)-dione (kindly provided by Dr. Xin Li, Zhejiang University, China). More procedure details can be found in our previous work.20 After incubated with blue labeling iRGD-PEG-PCCL for 2, 4, and 8 hrs, the cells were stained with Lyso-Tracker Red followed by a rinse with PBS. The distribution of iRGD-PEG-PCCL in A549 and B16F10 cells was observed under LSCM.
In Vitro Boron Incorporation Into Cells
To compare the difference in cellular boron uptake between BSH and boroned polymers, we conducted ICP-MS-based-experiments. Briefly, A549 cells were suspended in medium, seeded on 150 mm culture dishes and treated with BSH containing nutrient medium (20 μg/mL) of BSH, mPEG-PCCL-B, or iRGD-PEG-PCCL-B (n = 3). After 8 hrs, the cells were washed thrice with cold PBS, harvested by trypsin digestion, and collected by centrifugation. Cell pellets were later digested in 400 μL mixture of nitric acid and hydrogen peroxide (1:3, v/v) at 80°C for 24 hrs. Until the solution was transparent, ultra-pure water prepared by Milli-Q (Millipore, USA) was added to 5 mL before measuring the boron concentration using ICP-MS. The boron incorporation into B16F10 cells was processed for 4, 12, and 24 hrs following the same operation above.
Western Blot Analysis
To examine the integrin receptors and neuropilin-1 (NRP-1) expression, we collected cells (B16F10, C6, 4T1, Hela and A549) and obtained cellular proteins using T-PER Tissue Protein Extraction Reagent (Thermo Pierce). The total protein concentration was then quantified by a BCA protein assay kit (P0010, Beyotime Company). Western blot tests were processed in the standard fashion, and analyzed quantitatively protein expressions by the ImageJ software. The following antibodies were used in the procedure: integrin αv (ab179475, Abcam), integrin β3 (ab119992, Abcam), NRP-1 (ab81321, Abcam), integrin β5 (ab184312, Abcam), GAPDH (ab181602, Abcam), and goat anti-rabbit IgG (H+L) secondary antibody (31,210, Thermo Pierce).
Subcutaneous Tumor Xenografts
Female BALB/c nude mice (4–5 weeks) were purchased from Shanghai Slake Experimental Animal Co., Ltd. (Shanghai, China), and housed under standard conditions. Mice were injected with tumor cells by subcutaneous tissue near the right armpit. Tumor volumes (mm3) were evaluated with a caliper (length × width2/2). All relevant experiments were in accordance with the guidelines for the care and use of laboratory animals which were approved by the Animal Care and Use Committee of Zhejiang University.
Pharmacokinetics
Subcutaneous tumor xenografts were established by injected A549 cells with high viability (3.0 × 106 cells/100 μL PBS). When the tumor volume approached 150~200 mm3, 200 μL of a PBS solution of BSH at a boron concentration of 20 mg/kg body weight, equivalent dose of mPEG-PCCL-B or iRGD-PEG-PCCL-B with respect to the boron dose, was intravenously injected through tail vein (n = 3). To compare the biodistribution of different boron species, we collected blood, muscle, and tumor samples and recorded their weights at predetermined time points (6, 12, and 24 hrs). Samples were then treated with 65% nitric acid and 30% hydrogen peroxide (1:3, v/v) solution at 80°C for 48 hrs, followed by water bath with ultrasound for another 48 hrs. Finally, the solutions were diluted with ultra-pure water to measure the boron concentration with ICP-MS.
Biodistribution Of DOX-Encapsulated Polymers
For investigating the DOX distribution and tumor accumulation capacity of formations, A549 tumor-bearing BALB/c nude mice were then developed and randomly divided into three groups when tumors reached a size of 150~200 mm3, followed by tail vein injection with 200 μL of free DOX, mPEG-PCCL-B/DOX or iRGD-PEG-PCCL-B/DOX individually at an equivalent DOX dose of 2 mg/kg body weight. A Maestro Imaging System (Cambridge Research and Instrumentation, USA) was carried out to observe the biodistribution of the fluorescent polymers ex vivo at 24 hrs after injection. The exposure parameter of each group remained constant and images were preserved using a CCD camera.
In order to quantify the DOX concentration in targeted tissue, tumor tissues were weighed, suspended in 70% ethanol with 0.3 M HCl, vigorously homogenated, and centrifuged afterwards.21,22 Subsequently, supernates were shifted for M5 Multi-Mode Plate Reader (Molecular Devices) measurement.
Tumor Vascular Normalization
B16F10 tumor-bearing mice models were acquired by subcutaneous injections of B16F10 cells (3.0 × 105 cells/100 μL PBS) near the right armpit. At the seventh day after injection, mice were randomly allocated to receive daily intravenous injection of Endostar (100 μL, 15 mg/kg body weight) or the same volume of PBS solution for five days. A half portion of mice containing both Endostar and PBS administration were sacrificed to get tumor tissues fixed with 4% formaldehyde, then processed into paraffin sections. For vascular integrity assessment, the tumor sections were incubated with rabbit anti-CD34 antibody (1:40, Abcam) and mouse anti-α-SMA antibody (1:1000, Abcam) at 4°C overnight, subsequently with Alex 488-conjugated goat-anti-rabbit secondary antibody (1:500) and Alex 555-conjugated goat-anti-mouse secondary antibody (1:500). Cell nucleus were stained with DAPI, and the expressions of CD-34 and α-SMA were directly observed under LSCM. Besides, tumor sections were performed following CD34-PAS immunohistochemistry dual staining method23 to identify the endothelium-dependent vessels (EDV) and vascular mimicries (VM) on an upright fluorescence microscope (Olympus, Tokyo, Japan).
For investigating the real-time distribution of polymers after tumor vascular normalization, ICG, a near-infrared dye, was introduced into polymers through a solvent evaporation method.24 B16F10 tumor-bearing BALB/c nude mice were grouped evenly into two groups (there were both PBS and Endostar administration mice in each group), respectively, injected intravenously with 200 μL of free ICG, or iRGD-PEG-PCCL/ICG at an equivalent ICG dose of 2 mg/kg body weight. In vivo distribution of ICG at different time points (4, 9, 24, 48, and 72 hrs) after injection was assessed by Maestro imaging system. All mice were sacrificed to harvest tumors and main organs for fluorescence imaging analysis.
To assess the effect of boron accumulation in tumor using tumor vascular normalization, B16F10 tumor-bearing BALB/c nude mice following either PBS or Endostar administration were intravenously injected with 200 μL of iRGD-PEG-PCCL-B at an equivalent boron dose of 20 mg/kg body weight. Subsequently, we collected tumor samples and recorded their weights at 24 hrs. Tissue samples were then treated with 65% nitric acid and 30% hydrogen peroxide (1:3, v/v) solution at 80°C for 48 hrs, followed by water bath with ultrasound for another 48 hrs, and diluted with ultra-pure water to measure the boron concentration with ICP-MS.
Statistical Analysis
Quantitative data are expressed as mean ± standard deviation (SD). The statistical differences among groups were analyzed by the one-way ANONA test, and P<0.05 was considered statistically significant.
Results And Discussions
Synthesis Of Copolymers
To enhance the targeted delivery efficacy of boron into neoplasms, iRGD peptide-grafted polymeric nanoparticles comprising BSH and amphiphilic block copolymers were prepared in this work. The general sketch of synthetic routes of polymers is illustrated in Figure 2A–C. We first successfully synthesized monomer according to a modified documented method,18 which was confirmed by 1H NMR spectroscopy. The signals ranging from 7.5 to 8.0 ppm (n) attributed to the protons of the phenyl ring of monomer (Figure S1A-B). Ring-opening polymerization of monomer was then developed applying mPEG or Acetal-PEG as the initiator to obtain resultant diblock copolymer (mPEG-PBCL, Acetal-PEG-PBCL), and the benzyl groups of mPEG-PBCL (or Acetal-PEG-PBCL) were further removed in a reductive reaction system containing hydrogen. The chemical structures of different polymers were characterized using 1H NMR (Figure S1B), in which the peak of the phenyl ring (n) was dismissed in PCCL. The molecular weights of prepared copolymers were calculated to be 9160 g/mol (Acetal-PEG-PCCL) and 8185 g/mol (mPEG-PCCL) by comparing the peak intensities of PEG (f) or Acetal-PEG (b, e, f) to that of PCCL (k, n).
 |
Figure 2 Synthetic scheme for the preparation of (A) monomer, (B) mPEG-PCCL-B, and (C) iRGD-PEG-PCCL-B. |
Next, BSH was conjugated to the mPEG-PCCL (or Acetal-PEG-PCCL) by introducing linkers N-(2-aminoethyl) maleimide to connect the hydrosulfide groups of BSH and the free carboxyl groups on the PCCL chain. Ultimately, we estimated the boron content using ICP-MS approach to prove the formation of boroned polymers. Measurement of the concentration of intact iRGD after four days reaction by HPLC indicated an ideal conjugation efficiency of 95.76% for iRGD-PEG-PCCL, and 95.40% for iRGD-PEG-PCCL-B, respectively.
Assembly And Characterization Of Polymeric Nanoparticles
DOX was physically incorporated into iRGD-PEG-PCCL-B by solvent evaporation technique and the subsequent polymeric nanoparticles were acquired with 2.30% of DOX loading content, named as iRGD-PEG-PCCL-B/DOX. The investigations of characterization on iRGD-PEG-PCCL-B/DOX with respect to the particle size, zeta potential, boron content, and CMC are summarized in Table 1. To realize BNCT effect and reduce toxicity, FDA-approved 10BSH was protected in the hydrophobic core of polymers followed by desalinization, and 3.81% of boron content was evaluated by ICP-MS method. The morphology of iRGD-PEG-PCCL-B/DOX is monodisperse (PDI = 0.26) and spheroidal in water with an average diameter of 24.97 nm (Table 1, Figure 3A and B) for that particle size of ~100 nm is suitable for cultivated neoplasm accumulation of nanocarriers through EPR effect.8,25,26 The negative charges (−15.50 mV) of nanoparticles could protect them from influence of positive-charged materials in blood after intravenous injection. TEM image in Figure S2A also shows that the spherical appearance of mPEG-PCCL-B/DOX was similar to iRGD-PEG-PCCL-B/DOX, indicating no significant effect on the size with or without iRGD grafting. The hydrophobic interactions between conjugated BSH resulted in a lower CMC value (30.44 μg/mL) compared with iRGD-PEG-PCCL (43.47 μg/mL), and a more homogeneous appearance in comparison to iRGD-PEG-PCCL/DOX (Figure S2B). To further assess the stability of polymers, iRGD-PEG-PCCL-B/DOX was placed at near-physiological pH and characterized by particle size over time. As shown in Figure S3, there was no significant variation in size over 48 hrs, indicating that the prepared polymers were much stable at physiological pH. All the properties described illustrated that the nano-scaled drug delivery system with good stability was already established.
 |
Table 1 Properties Of iRGD-PEG-PCCL-B/DOX Polymers |
 |
Figure 3 Characteristics of iRGD-PEG-PCCL-B/DOX polymers. (A) Size distribution and (B) TEM image of the polymeric nanoparticles. Scale bar = 50 nm. |
In Vitro Release Profile Of DOX
The cumulative releases of DOX from iRGD-PEG-PCCL-B/DOX at pH 5.0 and pH 7.4 were 66.64 ± 3.58% and 15.66 ± 8.71%, respectively, after 72 hrs of incubation (Figure S4), suggesting much faster release of DOX at endolysosomal pH than that at physiological pH. These results showed that iRGD-PEG-PCCL-B/DOX kept relatively stable to prevent DOX from leaking at physiological pH, and possessed a controlled-release effect to enable the sustained release of DOX, especially during endocytosis.
In Vitro Cytotoxicity And Hemolysis Assay
The cytotoxicity of free BSH, mPEG-PCCL-B, iRGD-PEG-PCCL-B, free DOX, iRGD-PEG-PCCL-B/DOX, and blank nanoparticles (mPEG-PCCL, iRGD-PEG-PCCL) against A549 cells for 48 hrs was examined by CCK8 assay, and the results are featured in Figure 4A and B. It is known that PEG-PCL are approved by the FDA mainly due to the biodegradability, great biocompatibility of PEG and PCL. Likewise, the prepared blank nanoparticles (mPEG-PCCL and iRGD-PEG-PCCL) were non-toxic to A549 and B16F10 cells in the displayed concentrations (Figure 4A, Figure S5A). The biosafety of iRGD peptides was also proved. Moreover, BSH encapsulated nanoparticles (mPEG-PCCL-B and iRGD-PEG-PCCL-B) presented low toxicity as BSH. This is because the acute toxicity of BSH is very limited, as compared to other drugs in clinical practice.2
 |
Figure 4 Biocompatibility assessment of formations. (A, B) Evaluation of cell viability (CCK-8 assay) of A549 cell lines after 48 hrs of treatment with drugs in particle form or in solution, respectively. Values are expressed as mean ± SD (n = 6). (C) Hemolytic effect was estimated by incubating 1.5% (v/v) rabbit erythrocytes with various gradient concentrations of mPEG-PCCL-B or iRGD-PEG-PCCL-B for 4 hrs at 37°C. And, physical observation of the supernatant after mPEG-PCCL-B (D) or iRGD-PEG-PCCL-B (E) addition and centrifugation was shown. Extreme values (100% and 0) are positive control (0.2% Triton X-100, D–H and E–H) and negative control (pH 7.4 PBS, D–A and E–A). Samples B–G in (D) and (E) represented the gradient concentrations (0.16 ~ 5 mg/mL) of polymers. |
As for DOX-loaded nanoparticles, the cytotoxicity of iRGD-PEG-PCCL-B/DOX (IC50, 1.24 μM) against A549 cells was weaker than that of free DOX (IC50, 0.65 μM). It is highly plausible that nanoparticle essence and the dihydrogen bond between B-H (BSH) and N-H (DOX) could sustain release of DOX. On the other hand, the cellular entry of DOX was by passive diffusion through membranes instead of receptor-mediated internalization, that may be a benefit for intracellular accumulation during the long-term incubation. However, the cytotoxicity (Figure S5B) of iRGD-PEG-PCCL-B/DOX (IC50, 0.13 μM) against B16F10 cells was similar to free DOX (IC50, 0.11 μM). It is because the susceptibility of B16F10 to DOX may compromise the finite dissimilarity of cellular uptake and release property of polymers. B16F10 cells were bound to be suffering even at a low concentration of DOX. Importantly, polymeric nanoparticles can serve as a good targeted carrier of DOX for limiting systemic toxicity in vivo.
Figure 4C–E demonstrates the low hemolytic toxicity of the mPEG-PCCL-B and iRGD-PEG-PCCL-B. Taking PBS as the negative control, which was regarded as zero percent of hemolysis in this assay, the hemolytic percentage of iRGD-PEG-PCCL-B was about 1.80% at an utmost high concentration of polymers (5 mg/mL), while it was 3.31% for mPEG-PCCL-B. The negligible cytotoxicity and hemolysis of synthetic polymers make it possible for further application in vivo.
Cellular Internalization And Localization
Unlike cytotoxicity assessment, we were focusing on intracellular uptake and localization of DOX for shorter incubation time, to monitor the drug delivery activities of polymers in cells. A549 cells were cultured with free DOX, mPEG-PCCL-B/DOX, and iRGD-PEG-PCCL-B/DOX for 2 hrs followed by CLSM operation, thanks to the fluorescence of DOX. The results shown in Figure 5A directly screened apparent enhancement of DOX uptake in iRGD-PEG-PCCL-B/DOX group compared with mPEG-PCCL-B/DOX group, and significant difference was shown by quantitative analysis of fluorescence intensities (Figure 5B) using Image J software according to the confocal images. Though polymer micelles loading DOX shared indistinguishable uptake of DOX with free DOX, the reason for this is probably related to the passive diffusion and susceptibility of A549 cells to DOX in vitro.
 |
Figure 5 Visualization and graph-based quantification of intracellular DOX. (A) Confocal fluorescence images of A549 cells localization of DOX 2 hrs post-incubation at the equivalent DOX dose of 0.5 μg/mL. Blue fluorescence signifies positive staining of DAPI and DOX is shown in red. Scale bar = 60 μm. (B) Quantified intensities (average optical density) of DOX signal by ImageJ ver 1.50i. Values are expressed as mean ± SD. ## P < 0.01 vs mPEG-PCCL-B/DOX group. |
When it comes to the subcellular distribution of DOX, we found that DOX (red fluorescence) were limited at the nucleus (blue fluorescence) in A549 and B16F10 cells at 2 hrs post-incubation interval (Figure 6), which is of vital importance for DOX-induced DNA cross-linking.27 To further investigate the intracellular movement route of polymers, cells were incubated with blue labeling polymeric nanoparticles and stained lysosome after incubation. The samples observed under CLSM showed that there was less overlap between blue (polymers) with red dyes (lysosomes) in some space of cytosol (Figure 7). This implied that polymers may partly get away from lysosomes to shift elsewhere.28 Interestingly, we also noticed there were some blue dyes located in the central area of cells as marked by the white arrow in Figure 7 (enlarged graph shown in Figure S6), speculating that polymers may enter nucleus due to its minimal size of ~25 nm. Currently, the vital design principles involved in cellular nucleus-targeting nanotherapeutics include shape regulation, size control, and surface modification according to the literature.29 It is well known that the sufficient entry of 10B into cell is one of the key to successful BNCT, either sited in the cytoplasm or lysosome. Yet, 10B located in the nucleus is going to achieve a better therapeutic effect more than tenfold of that of the cytosol location in BNCT.2,30–33
 |
Figure 6 Time-lapse confocal images of A549 and B16F10 cells that had been incubated with the DOX solution or the iRGD-PEG-PCCL-B/DOX formulations (1 μg/mL equivalent DOX) for 2, 4, and 8 hrs. DAPI is shown in blue, and DOX in red. Scale bar = 20 μm. |
 |
Figure 7 Time-lapse confocal images of A549 and B16F10 cells that have been incubated with the 2-butyl(4-aminohexyl)-6-(dimethylamino)-1H-benz[de]isoquinoline-1,3(2H)-dione-labeled iRGD-PEG-PCCL polymers for 2, 4, and 8 hrs. Lyso-Tracker Red were used to mark lysosomes (red fluorescence), and labeled iRGD-PEG-PCCL fluorescence is blue. The white arrows show the locations of blue fluorescence in the central region of cells. Scale bar = 20 μm. |
Intracellular Boron Uptake Efficacy
To date, only two boron agents (BSH and BPA) are used for BNCT in clinic. However, both are lacking targeting ability to tumor cells, thereby limiting their applications. To determine the advance of our developed BSH-conjugated nanoparticles in comparison to clinically used BSH, the intracellular uptake of boron by A549 and B16F10 cells was assessed by ICP-MS. Figure S7 shows the time-dependent uptake of boron by B16F10 for BSH, mPEG-PCCL-B, and iRGD-PEG-PCCL-B. The uptake amounts of BSH-conjugated polymers (21.90 ng 10B/106 cells) significantly exceeded than that of intact BSH (12.48 ng 10B/106 cells) at 12 hrs post-incubation interval, which was because BSH is not easy to transduce into cells due to the structure of anionic boron cluster,6,34 while boroned polymers process a better retention and uptake characteristic. After another incubation of 12 hrs, cells administrated with iRGD-PEG-PCCL-B disclosed a higher concentration of boron uptake (45.14 ng 10B/106 cells) than that of mPEG-PCCL-B (34.00 ng 10B/106 cells). Surprisingly, iRGD-PEG-PCCL-B (209.83 ng 10B/106 cells) in A549 cells showed a remarkable enhancement of boron uptake in respect to that of BSH (33.88 ng 10B/106 cells) and mPEG-PCCL-B (148.28 ng 10B/106 cells) under a condition of incubation of 8 hrs (Figure 8). There are seldom documents reported such a better cellular uptake of 10B by organic materials. A brief overview was presented in Table S1.30,35–39 BSH was encapsulated into liposomes to form Liposome-ZZ-mAb or TF-PEG liposomes, while BNPs were linked with BSH by covalent bonds. As for BPN and AT101/LDL particles, boron agents were individually coated with micelles or LDL to conduct corresponding nanoparticles. The reason for the increased cellular uptake attributed to the endocytosis for polymers, especially the ligand-receptor-binding-based endocytosis for iRGD grafted polymers. Hence, iRGD-PEG-PCCL-B is a promising boron delivery agent for BNCT.
 |
Figure 8 Intracellular boron uptake of the BSH solution or the polymers containing BSH. A549 cells were incubated for 8 hrs in the presence of each boroned polymers or BSH at the dose of 20 μg/mL on a boron basis, then digested and their boron concentration were measured by ICP-MS. The data are expressed as mean ± SD (n = 3). ***P < 0.001 vs BSH group, # P < 0.05 vs mPEG-PCCL-B group. |
Expressions Of Integrins And NRP-1 Receptors
iRGD-based drug delivery system is of improved binding and internalization in tumor site in terms of the interaction between receptors (integrins and NRP-1) located on the cell surface and iRGDs.40 We performed Western blot analysis to verify the integrins and NRP-1 expressions of several cancer cell lines (B16F10, C6, 4T1, Hela, and A549). As Figure 9A indicates, all the tested cell lines beared with more or less expressions of αvβ3/β5 and NRP-1. The quantitative analysis of protein expressions compared with GAPDH conducted by Image J software revealed intuitively the superiority of A549 cell line among all the cell lines (Figure 9B). In contrast to A549, the condition for B16F10 was almost opposite except for integrin β5. That is what exactly accounts for the dominant boron uptake by A549 cells compared with B16F10 cells.
 |
Figure 9 Expressions and quantifications of integrin αv/β3/β5 and neuropilin-1 proteins in several cell lines (B16F10, C6, 4T1, Hela, and A549). (A) Representative Western blot diagram of the selected proteins in different cell lines. (B) Western blot-based quantifications of the chosen protein expressions in various cell lines. The results are expressed as mean ± SD (n = 3). |
Boron Biodistribution In Mice Bearing A549 Tumor Xenografts
In vivo experiments were administrated to evaluate the biodistribution of boron in blood and tumors employing ICP-MS approach, which is an important indicator for predicting the treatment efficiency of BNCT. We used a lower dose (20 mg/kg body weight) for vein injection in comparison to the literatures.30,35–39 The data showed the time-dependent boron concentration in blood, tumor, and muscle, indicates the time point of 6 hrs for highest boron accumulation as for BSH, while 24 hrs for highest boron accumulation for mPEG-PCCL-B and iRGD-PEG-PCCL-B (Figure 10A–C). What is more, iRGD-PEG-PCCL-B represented favorable tumor:normal tissue boron concentration ratios (tumor:blood = 14.11, tumor:muscle = 19.49) in A549 tumor-bearing mice after 24 hrs intravenous injection of formations (Figure 10D and E). The excellent performance of iRGD-PEG-PCCL-B was probably attributed to EPR effect and active targeting properties of iRGD. It is known that nanoparticles could prolong blood circulation and gather in tumor tissue as time goes because of EPR effect. In addition, active targeting peptides grafted nanomaterials would achieve more outstanding bioavailability in contrast to passive targeting nanomaterials. RGD-based nanocarriers have been used to delivery boron to tumor site, but penetrating the tumor parenchyma remains challenging.40 iRGD cyclic peptide, which is rarely introduced into BNCT, possessed excellent cell-penetrating activities to deliver a large variety of cargoes into tumor parenchyma, was selected for targeting motif in our work. The binding of RGD motif to the integrin in vivo is followed by the proteolytic cleavage and release of the CDPG sequence, also trimmed the C-terminal rule (CRGDK) motif, which integrates with the NRP-1 receptor and leads to eliciting of an endocytic transcytosis and trans-tissue transport path that facilitates boron delivery into tumor region. It was inferred from the results that iRGD-based nanoparticles possessed an extraordinary performance in drug delivery in vivo. Meanwhile, honestly speaking, it is better to carry out more investigations on the penetration of iRGD in the future study.
 |
Figure 10 Blood pharmacokinetics and tissue distribution of BSH and BSH-conjugated polymers. Boron concentrations (%ID/g of tissue) in (A) blood, (B) tumor, and (C) muscle tissue were evaluated on A549 subcutaneous tumor-bearing nude mice at 6, 12, and 24 hrs after intravenous administration of BSH or BSH-conjugated polymers (20 mg/kg on a boron basis). (D) Tumor to blood distribution ratio of boron after injection of BSH or BSH-conjugated polymers. (E) Tumor to muscle distribution ratio of boron after injection of BSH or BSH-conjugated polymers. The results are expressed as mean ± SD (n = 3). **P < 0.01, ***P < 0.001. |
Biodistribution Of DOX-Encapsulated Polymers
Thanks to the fluorescence Properties of DOX (Ex: 488 nm, Em: 590 nm), we developed an ex vivo fluorescence imaging study to quantify DOX concentration at tumor. At 24 hrs after injecting of formations, the tumors and organs were harvested and operated by fluorescence imaging. As for iRGD-PEG-PCCL-B/DOX, it not only disclosed the highest intensity of fluorescence in the tumor tissue, but also the lowest signal in the heart tissue (Figure 11A), suggesting iRGD-PEG-PCCL-B/DOX processed the high therapeutic efficacy, together with the minimal heart side effect compared with free DOX and mPEG-PCCL-B/DOX.
 |
Figure 11 Biodistribution of DOX at 24 hrs after intravenous administration of various formations (2 mg/kg equivalent DOX) in nude mice bearing A549 tumors. (A) Fluorescence images were obtained over the scarified organs. The signal intensities were denoted by the respective color bar. (B) DOX concentrations in tumor were quantified by M5 Multi-Mode Plate Reader. The data are expressed as mean ± SD (n = 3). *P < 0.05, ***P < 0.001. |
Meanwhile, Figure 11B demonstrates the highest concentration of DOX in tumor at 24 hrs after injection for iRGD-PEG-PCCL-b/DOX (5.05 ± 0.36% ID/g) compared with free DOX (2.47 ± 0.31% ID/g) and mPEG-PCCL-B/DOX (4.28 ± 0.19% ID/g) (Figure 11B). It once again proved that the iRGD-based polymers would improve the accumulation of DOX in the tumor for promising chemotherapy.
Tumor Vascular Normalization
As for fluorescence study in mice bearing B16F10 tumor xenografts, no signal was detected at the tumor. Unlike A549 tumor tissue, the watery appearance and fast-growing behavior were found in B16F10. The elevated interstitial fluid pressure (IFP) in B16F10 tumor keep drugs out of vascular wall probably accounted for that.41–43 We further administrated Endostar to promote tumor vessel normalization following procedure addressed before,44,45 expected to normalize the tumor microenvironment (improving vascular integrity, reducing IFP, and so on). The alternation of vasculatures of tumor was presented in Figure 12A and B. ICG biodistribution in vivo and ex vivo were shown in Figures S8A–D and Figures S9A–D. Similar to other studies, improvement of vascular integrity and decrease of VM were found after Endostar injection. But there was still no signal in tumor tissue probably because of the remaining evaluated IFP. From what harvested, we noticed the watery tumor tissues. Or the quite weak fluorescence in tumor site owing to the slight modification of ICG distribution was absorbed by melanin.
 |
Figure 12 The effect on the tumor following either PBS or Endostar administration in mice bearing B16F10 tumors. (A) Immunohistochemistry of tumor tissues. The vascular mimicries (top arrow) are PAS+ and show purple, while the endothelium-dependent vessels (bottom arrow) are CD34+ and show brown. Original magnification: 20X and 40X. (B) Immunofluorescence staining for CD34 and α-SMA in tumor tissues. The tumor tissues were also incubated with DAPI to show blue fluorescence. The degree of overlap of CD34 and α-SMA indicates vascular maturity. Scale bar = 50 μm. (C) Boron concentrations in tumor were evaluated at 24 hrs after intravenous administration of iRGD-PEG-PCCL-B (20 mg/kg on a boron basis). The data are expressed as mean ± SD (n = 3). ***P < 0.001. |
Furthermore, in vivo distribution study of iRGD-PEG-PCCL-B was done to estimate the effect on boron accumulation in tumor following Endostar injection. As seen in Figure 12C, improvement of boron concentration was found after Endostar treatment in comparison to PBS control treatment. However, more studies focusing on BNCT combined with tumor vascular normalization are required afterwards so as to achieve a better boron accumulation in tumor.
Conclusions
In this contribution, we have mainly engineered a polymer-based tumor-targeting boron delivery system for BNCT, and combined with chemotherapy by physically incorporated with DOX into the core of the biodegradable amphiphilic polymers. Compared with many inorganic materials studies in the related field, iRGD-PEG-PCCL-B meet the biocompatible criteria for clinical application. The relatively stable polymeric nanoparticles, with an appropriate size of 24.97 nm, could transport DOX into nucleus, and BSH into cytosol correspondingly. Owing to the multifunctionalized iRGD peptide, the cellular uptake of drugs in integrins/NRP-1 overexpressed cells was dramatically increased to 209.83 ng 10B/106 cells. Moreover, the developed polymers processed obvious prolonged blood circulation and enhanced tumor accumulation of drugs in A549 tumor-bearing mice, which exhibited a great potential to be applied into combined BNCT with chemotherapy. Future studies will pay more attention to the combined DOX-BNCT management against a variety of solid tumor in the tumor-bearing mice model, and the specific contribution of DOX for BNCT. What is more, the results of tumor vascular normalization assay demonstrated that Endostar could promote the normalization of tumor vessels on B16F10 tumor tissues to some extent, and promote delivery efficiency of the prepared polymers. More works are required to lighten the dark box of combined tumor vascular normalization and BNCT in refractory cancer.
Acknowledgment
This study was funded by the National Natural Science Foundation of China (No. 81572952, No. 81673022, and No.81373346), and supported by National Key R&D Program of China (No.2017YFE0102200).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Barth RF, Zhang Z, Liu T. A realistic appraisal of boron neutron capture therapy as a cancer treatment modality. Cancer Commun (Lond). 2018;38(1):36. doi:10.1186/s40880-018-0280-5
2. Barth RF, Mi P, Yang W. Boron delivery agents for neutron capture therapy of cancer. Cancer Commun (Lond). 2018;38(1):35. doi:10.1186/s40880-018-0299-7
3. Barth RF, Soloway AH, Fairchild RG, Brugger RM. Boron neutron capture therapy for cancer. Realities and prospects. Cancer. 1992;70(12):2995–3007. doi:10.1002/1097-0142(19921215)70:12<2995::aid-cncr2820701243>3.0.co;2-#
4. Kreiner AJ, Baldo M, Bergueiro JR, et al. Accelerator-based BNCT. Appl Radiat Isot. 2014;88:185–189. doi:10.1016/j.apradiso.2013.11.064
5. Kreiner AJ, Bergueiro J, Cartelli D, et al. Present status of accelerator-based BNCT. Rep Pract Oncol Radiother. 2016;21(2):95–101. doi:10.1016/j.rpor.2014.11.004
6. Michiue H, Sakurai Y, Kondo N, et al. The acceleration of boron neutron capture therapy using multi-linked mercaptoundecahydrododecaborate (BSH) fused cell-penetrating peptide. Biomaterials. 2014;35(10):3396–3405. doi:10.1016/j.biomaterials.2013.12.055
7. Wang J, Wu W, Jiang X. Nanoscaled boron-containing delivery systems and therapeutic agents for cancer treatment. Nanomedicine (Lond). 2015;10(7):1149–1163. doi:10.2217/nnm.14.213
8. Golombek SK, May JN, Theek B, et al. Tumor targeting via EPR: strategies to enhance patient responses. Adv Drug Deliv Rev. 2018;130:17–38. doi:10.1016/j.addr.2018.07.007
9. Hoffman AS. The origins and evolution of “controlled” drug delivery systems. J Control Release. 2008;132(3):153–163. doi:10.1016/j.jconrel.2008.08.012
10. Meo CD, Panza L, Capitani D, et al. Hyaluronan as carrier of carboranes for tumor targeting in boron neutron capture therapy. Biomacromolecules. 2007;8(2):552–559. doi:10.1021/bm0607426
11. Liko F, Hindre F, Fernandez-Megia E. Dendrimers as innovative radiopharmaceuticals in cancer radionanotherapy. Biomacromolecules. 2016;17(10):3103–3114. doi:10.1021/acs.biomac.6b00929
12. Danhier F, Feron O, Preat V. To exploit the tumor microenvironment: passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J Control Release. 2010;148(2):135–146. doi:10.1016/j.jconrel.2010.08.027
13. Sugahara KN, Teesalu T, Karmali PP, et al. Coadministration of a tumor-penetrating peptide enhances the efficacy of cancer drugs. Science. 2010;328(5981):1031–1035. doi:10.1126/science.1183057
14. Curran WJ
15. Lawrence TS, Haffty BG, Harris JR. Milestones in the use of combined-modality radiation therapy and chemotherapy. J Clin Oncol. 2014;32(12):1173–1179. doi:10.1200/JCO.2014.55.2281
16. Xu J, Zhang BC, Li XL, et al. Chemosensitization and radiosensitization of a lung cancer cell line A549 induced by a composite polymer micelle. Discov Med. 2016;22(119):7–17.
17. Xu W, Han M, Diao Y, et al. Doxorubicin encapsulated in micelles enhances radiosensitivity in doxorubicin-resistant tumor cells. Discov Med. 2014;18(99):169–177.
18. Xiong XB, Mahmud A, Uludag H, Lavasanifar A. Multifunctional polymeric micelles for enhanced intracellular delivery of doxorubicin to metastatic cancer cells. Pharm Res. 2008;25(11):2555–2566. doi:10.1007/s11095-008-9673-5
19. Qi R, Gao Y, Tang Y, et al. PEG-conjugated PAMAM dendrimers mediate efficient intramuscular gene expression. Aaps J. 2009;11(3):395–405. doi:10.1208/s12248-009-9116-1
20. Liu HN, Guo NN, Wang TT, et al. Mitochondrial targeted doxorubicin-triphenylphosphonium delivered by hyaluronic acid modified and pH responsive nanocarriers to breast tumor: in vitro and in vivo studies. Mol Pharm. 2018;15(3):882–891. doi:10.1021/acs.molpharmaceut.7b00793
21. Han HD, Lee A, Hwang T, et al. Enhanced circulation time and antitumor activity of doxorubicin by comblike polymer-incorporated liposomes. J Control Release. 2007;120(3):161–168. doi:10.1016/j.jconrel.2007.03.020
22. Xiong H, Zhou D, Qi Y, et al. Doxorubicin-loaded carborane-conjugated polymeric nanoparticles as delivery system for combination cancer therapy. Biomacromolecules. 2015;16(12):3980–3988. doi:10.1021/acs.biomac.5b01311
23. Xu Y, Li Q, Li XY, Yang QY, Xu WW, Liu GL. Short-term anti-vascular endothelial growth factor treatment elicits vasculogenic mimicry formation of tumors to accelerate metastasis. J Exp Clin Cancer Res. 2012;31:16. doi:10.1186/1756-9966-31-95
24. Han M, Huang-Fu MY, Guo WW, et al. MMP-2-sensitive HA end-conjugated poly(amidoamine) dendrimers via click reaction to enhance drug penetration into solid tumor. ACS Appl Mater Interfaces. 2017;9(49):42459–42470. doi:10.1021/acsami.7b10098
25. Radomska A, Leszczyszyn J, Radomski MW, Nanopharmacology T. Nanotoxicology of nanomaterials: new opportunities and challenges. Adv Clin Exp Med. 2016;25(1):151–162. doi:10.17219/acem/60879
26. Gidwani B, Vyas A. The potentials of nanotechnology-based drug delivery system for treatment of ovarian cancer. Artif Cells Nanomed Biotechnol. 2015;43(4):291–297. doi:10.3109/21691401.2013.853179
27. Zaleskis G, Berleth E, Verstovsek S, Ehrke MJ, Mihich E. Doxorubicin-induced DNA degradation in murine thymocytes. Mol Pharmacol. 1994;46(5):901–908.
28. Xiong XB, Lavasanifar A. Traceable multifunctional micellar nanocarriers for cancer-targeted co-delivery of MDR-1 siRNA and doxorubicin. ACS Nano. 2011;5(6):5202–5213. doi:10.1021/nn2013707
29. Pan L, Liu J, Shi J. Cancer cell nucleus-targeting nanocomposites for advanced tumor therapeutics. Chem Soc Rev. 2018;47(18):6930–6946. doi:10.1039/c8cs00081f
30. Shi Y, Li J, Zhang Z, et al. Tracing boron with fluorescence and PET imaging of boronated porphyrin nanocomplex for imaging guided boron neutron capture therapy. ACS Appl Mater Interfaces. 2018;10(50):43387–43395. doi:10.1021/acsami.8b14682
31. Okumura K, Kinashi Y, Kubota Y, et al. Relative biological effects of neutron mixed-beam irradiation for boron neutron capture therapy on cell survival and DNA double-strand breaks in cultured mammalian cells. J Radiat Res. 2013;54(1):70–75. doi:10.1093/jrr/rrs079
32. Hartman T, Carlsson J. Radiation dose heterogeneity in receptor and antigen mediated boron neutron capture therapy. Radiother Oncol. 1994;31(1):61–75. doi:10.1016/0167-8140(94)90414-6
33. Barth RF, Coderre JA, Vicente MG, Blue TE. Boron neutron capture therapy of cancer: current status and future prospects. Clin Cancer Res. 2005;11(11):3987–4002. doi:10.1158/1078-0432.CCR-05-0035
34. Soloway AH, Hatanaka H, Davis MA. Penetration of brain and brain tumor. VII. Tumor-binding sulfhydryl boron compounds. J Med Chem. 1967;10(4):714–717. doi:10.1021/jm00316a042
35. Wada Y, Hirose K, Harada T, et al. Impact of oxygen status on 10B-BPA uptake into human glioblastoma cells, referring to significance in boron neutron capture therapy. J Radiat Res. 2018;59(2):122–128. doi:10.1093/jrr/rrx080
36. Alberti D, Deagostino A, Toppino A, et al. An innovative therapeutic approach for malignant mesothelioma treatment based on the use of Gd/boron multimodal probes for MRI guided BNCT. J Control Release. 2018;280:31–38. doi:10.1016/j.jconrel.2018.04.043
37. Gao Z, Horiguchi Y, Nakai K, et al. Use of boron cluster-containing redox nanoparticles with ROS scavenging ability in boron neutron capture therapy to achieve high therapeutic efficiency and low adverse effects. Biomaterials. 2016;104:201–212. doi:10.1016/j.biomaterials.2016.06.046
38. Feng B, Tomizawa K, Michiue H, et al. Delivery of sodium borocaptate to glioma cells using immunoliposome conjugated with anti-EGFR antibodies by ZZ-His. Biomaterials. 2009;30(9):1746–1755. doi:10.1016/j.biomaterials.2008.12.010
39. Miyata S, Kawabata S, Hiramatsu R, et al. Computed tomography imaging of transferrin targeting liposomes encapsulating both boron and iodine contrast agents by convection-enhanced delivery to F98 rat glioma for boron neutron capture therapy. Neurosurgery. 2011;68(5):1380–1387. discussion 1387. doi:10.1227/NEU.0b013e31820b52aa
40. Nieberler M, Reuning U, Reichart F, et al. Exploring the role of RGD-recognizing integrins in cancer. Cancers (Basel). 2017;9(9):116. doi:10.3390/cancers9090116
41. Burazin A, Drapaca CS, Tenti G, Sivaloganathan S. A poroelasticity theory approach to study the mechanisms leading to elevated interstitial fluid pressure in solid tumours. Bull Math Biol. 2018;80(5):1172–1194. doi:10.1007/s11538-017-0383-1
42. Seymour LW, Ulbrich K, Steyger PS, et al. Tumour tropism and anti-cancer efficacy of polymer-based doxorubicin prodrugs in the treatment of subcutaneous murine B16F10 melanoma. Br J Cancer. 1994;70(4):636–641. doi:10.1038/bjc.1994.363
43. Walker-Samuel S, Roberts TA, Ramasawmy R, et al. Investigating low-velocity fluid flow in tumors with convection-MRI. Cancer Res. 2018;78(7):1859–1872. doi:10.1158/0008-5472.CAN-17-1546
44. Ma CH, Jiang R, Li JD, Wang B, Sun LW, Lv Y. Experimental study of endostar injection concomitant with cryoablation on lung adenocarcinoma A549 xenografts. Asian Pac J Cancer Prev. 2014;14(11):6697–6701. doi:10.7314/apjcp.2013.14.11.6697
45. Xu Q, Gu J, Lv Y, et al. Angiogenesis for tumor vascular normalization of endostar on hepatoma 22 tumor-bearing mice is involved in the immune response. Oncol Lett. 2018;15(3):3437–3446. doi:10.3892/ol.2018.7734
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.