Back to Journals » Drug Design, Development and Therapy » Volume 19
Relative Bioavailability of Inhaled Fluticasone Propionate and Salmeterol – is Population Pharmacokinetic Modelling a Relevant Alternative to a Non-Compartmental Approach?
Authors Rosenborg J ![]() , Bäckman P
, Bäckman P ![]() , Bengtsson T, Haughie S
, Bengtsson T, Haughie S
Received 20 August 2024
Accepted for publication 19 July 2025
Published 27 October 2025 Volume 2025:19 Pages 9653—9670
DOI https://doi.org/10.2147/DDDT.S480189
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Yan Zhu
Johan Rosenborg,1 Per Bäckman,2 Thomas Bengtsson,1 Scott Haughie3
1StatMind AB, Lund, Sweden; 2Emmace Consulting AB, Lund, Sweden; 3Viatris, Mylan Pharma UK Ltd, Sandwich, UK
Correspondence: Johan Rosenborg, Email [email protected] Per Bäckman, Email [email protected]
Background: Conventional statistical analysis of clinical data based on standard methods suggests that fixed combinations of fluticasone propionate (FP) and salmeterol (SALM) inhaled via Wixela® Inhub® dry powder (test) are therapeutically and pharmacokinetically equivalent to Advair® Diskus® (reference). Assessment of bioequivalence, test/reference, based on empirical population pharmacokinetic modelling was compared with published outcomes based on a non-compartmental analysis (NCA) approach.
Methods: Three inhalations of FP/SALM were administered to healthy subjects with 7-day washouts via test and reference and delivered doses of 100/50, 250/50, or 500/50 μg (total matched doses of 300/150, 750/150, or 1500/150 μg) in studies 1, 2, and 3, respectively. Empirically derived pharmacokinetic structural models, considering random effects of subject (FP, SALM) or study (SALM), emulating linear first-order input, disposition and elimination were fitted to plasma concentrations with a frequentist approach, by study/dose of FP, and across studies of SALM, using product as categorical covariate. Bioequivalence was assessed using individually predicted relative systemic extent of bioavailability (F4_rel) and ratio of individually predicted peak concentrations (Cmax).
Results: The models adequately described plasma concentration versus time data. Descriptive statistics and bioequivalence assessments (90% confidence interval generally within 0.80– 1.25) reflected NCA-derived outcomes.
Conclusion: Model- and NCA-based approaches to assess bioequivalence were consistent. However, a frequentist parametric approach that fully utilizes the population analysis potential and mechanistically predicts regional pulmonary deposition of FP and SALM would require a demographically more heterogenous data set and analysis including additional outcomes of parenteral and oral administration to discriminate presystemic from systemic disposition.
Keywords: fluticasone propionate, salmeterol, inhalation, pharmacokinetics
Introduction
The Wixela Inhub dry powder inhaler, the generic equivalent to Advair Diskus for the treatment of asthma and chronic obstructive pulmonary disease, is available at the nominal doses of the glucocorticoid fluticasone propionate (FP) 100 µg, 250 µg or 500 µg combined with 50 µg of the long-acting beta2-agonist salmeterol base (SALM) per inhalation. A clinical study of the Wixela Inhub dry powder inhaler, with assessment of bronchodilation, 12-h area under the curve of forced expiratory volume during the first second (FEV1) on day 1 and the pre-dose value on day 29 during twice-daily inhalation of FP [100 µg] and SALM [50 µg] demonstrated therapeutic equivalence to Advair Diskus.1 Non-compartmental analysis (NCA) of peak plasma concentration (Cmax) and area under the plasma concentration vs time curve (AUC) of FP and SALM after single-dose inhalation of the combination in three separate studies [FP 3 × 100, 3 × 250, or 3 × 500 µg with SALM 3 × 50 µg] demonstrated pharmacokinetic bioequivalence between the Wixela Inhub dry powder inhaler and Advair Diskus.2 Comparisons across the three studies suggested consistently between products, dose proportional increase in AUC but less than dose proportional increase in peak plasma concentration of FP.
An interdisciplinary group led by Günther Hochhaus explored and compared non-parametric and parametric pharmacokinetic methods of analysis considering product characteristics as a means to describe regional pulmonary deposition, particularly the impact of variable particle size distribution on the local pulmonary effect potential.3,4 This parametric approach is supported by fixed systemic three-compartment disposition pharmacokinetic (PK) model parameters and parameters describing non-absorptive mucociliary clearance from conducting airways.5–7 The variable composition of lactose fine particles was employed to control the particle size distribution, expressed as the median mass aerodynamic diameter (MMAD). The total lung dose, assessed in vitro using three independent anatomical mouth throats, showed an inverse correlation with MMAD; the larger the MMAD, the more central deposition and, because of mucociliary clearance, the lower the fraction absorbed.3 The outcome of model-based analysis suggested that i) a biphasic absorption model was superior to a monophasic model for describing absorption, and ii) the total and regional extent of absorption was inversely correlated with MMAD.4
We present an alternative analysis of previously generated results (please note that any reference to data based on NCA has been retrieved from Haughie et al (2019)2). The objective was to compare the NCA with a population pharmacokinetic (PopPK) approach for testing bioequivalence. Only properties which were considered to impact NCA and PopPK differently would impact the conclusions drawn. If data had been generated for a study evaluating BE using PopPK directly, then there may have been other choices made to benefit from the PopPK properties. However, to compare the methods, the standard choices for an NCA approach should be used. The purpose was not to disqualify one method compared to another, but to add a data driven, empirical model-based analysis8 as a means to derive individual post-hoc parameter estimates reflecting relative extent (F4_rel), and a surrogate marker for relative rate (ratio of predicted peak concentrations, test/reference) of bioavailability, thus representing NCA-compatible outcome variables for testing bioequivalence (Figure 1). The relevance of empirically model-based parameter estimates (apparent clearance and relative extent of bioavailability) and mean individual post-hoc predictions (terminal half-life and peak plasma concentration) are discussed based on comparisons with literature data.
 |
Figure 1 Empirical pharmacokinetic structural models fitted to available pharmacokinetic data of FP and SALM, implying monophasic absorption and three-compartment systemic disposition. K41 and F4_rel, apparent rate and relative extent of absorption; CL/F and V1/F, apparent elimination clearance and central volume of distribution; Q2/F and Q3/F, apparent distribution clearances; V2/F and V3/F, apparent peripheral volumes of distribution; ETA1-8 indicates between subject variability, ETA9 between study variability, TREA constitutes a categorical treatment factor on k41 and F4_rel. |
A possible flaw of the iterative least square curve fitting method is the dependence on treatment differences represented by the empirically rationalized parametrization. Any potential difference must be accounted for, at least in the initial model attempts. An additional flaw is the time-consuming work to reach convergence. NCA-based test of bioequivalence addresses differences between devices/formulations independent of how they emerge and so does not rely upon compartmental assumptions; except for estimation of half life. In addition, the analysis is less complex than compartmental methods, but is dependent on more frequent blood sampling to give accurate results, in this case both with respect to the integrated estimate of exposure extent (AUC) and proper capture of the peak concentration. Moreover, results based on NCA cannot be used quantitatively for simulation taking into account uncertainty factors in parameter estimation at individual, group or bioanalysis level.
Materials and Methods
This analysis was based on plasma concentrations in three separate two-way crossover studies of homogenously composed subject panels in the USA, each recruiting 66 healthy subjects, black and white, mean BMI ≈ 26, of which 61 to 65 completed both treatment periods; study 1: 29 female/37 male; mean age 33.8 years; study 2: 36 female/30 male; mean age 37.7 years; study 3: 42 female/24 male; mean age 35.7 years.2 Study protocols and patient informed consents were subjected to ethical review and approval according to guidelines outlined in the Declaration of Helsinki. The institutional review board processes were centrally administered via IntegReview IRB, an organisation accredited by the Association for the Accreditation of Human Research Protection Programs that is affiliated to Advarra (https://www.advarra.com/review-services/institutional-review-board/?campaign=integreview). Three inhalations of either the test product (test), Wixela Inhub, or the reference product (reference), Advair Diskus were performed on two separate occasions with a 7-day washout between inhalations. Nominal doses of FP were 3 × 100 µg (Study 1, 300 µg), 3 × 250 µg (Study 2, 750 µg), and 3 × 500 µg (Study 3, 1500 µg). The nominal dose of SALM was 3 × 50 µg in Studies 1–3 (150 µg). The aerodynamic particle size distribution (APSD) of the formulations was equivalent at all strengths (Viatris Mylan Pharma, data on file; supportive data focusing on MMAD in Advair Diskus9). The lower limit of quantification (LLOQ) was 1 ng/L. Gastrointestinal absorption was not blocked. The original sample size calculation was based on the within-subject variability of salmeterol peak plasma concentration (Cmax).2
Training in inhalation technique was provided to all participants, prior to any drug intake. In short, subjects practiced “deep and forceful” inhalation before the first administration with the aim of optimizing drug delivery. Blood sampling at study visits was planned to take place before drug administration and at 2, 5, 10, 15, 20, 30, and 45 min, and at 1, 1.5, 2, 3, 4, 6, 8, 12, 24, 36, and 48 hours post-dose. Individual sampling times were used for the analysis.
The empirical parametric population PK analysis approach should primarily be regarded as an alternative to that based on the individual observed rate (Cmax) and calculated extent (AUC) of absorption, as applied by Haughie et al and Hochhaus et al2,3 Empirical in this context means that no mechanistic assumptions were made regarding lung deposition, either in terms of the magnitude or regional distribution of the systemically absorbed dose fraction. In order to make the comparison between outputs of model- and NCA-based analysis relevant, conditions must be similar (cf. Introduction). The frequentist empirical model as well as the previous NCA approach to data meant that we let the experimental plasma concentrations speak for themselves with a minimal number of assumptions, possibly a relevant starting point to make model analysis an apt alternative for bioequivalence tests of systemic exposure. The simplified starting points for the analysis technically meant i) that the inhaled nominal dose was assumed to be quantitatively and instantaneously deposited in the lungs, and henceforth absorbed into the systemic circulation. In the absence of sharply monitored duration of the inhalation procedure, we argue that it is reasonable to disregard the short delay that the administration procedure entailed before the dose – once it left the inhaler – almost instantaneously was available for dissolution and absorption in the lungs. The fact that the nominal dose is not quantitatively deposited in the lungs after inhalation was implicitly account for by – in the context – an unknown absolute bioavailability factor <1; ii) the assessment of potential product differences focused parametrically on mono- or biphasic input rates, and the relative extent of bioavailability, test vs reference product (F4_rel) using product as a categorical covariate (cf. Figure 1), and iii) that systemic disposition was product independent and could be described using a three-compartment model.4,10 The applied parametric approach implies that the body is presumed to consist of interconnected compartments; in the study first order transfer was presumed to take place i) from theoretical deposition site(s) to the central blood compartment, ii) distribution from the central blood compartment to and redistribution from two peripheral compartments back to the central compartment from which finally drug was eliminated. Basic elements of our strategy to consider model robustness: i) Adequate and fit for purpose assignment of one or two deposition sites to reflect mono- or biphasic absorption and apparent clearance (CL/F) were prerequisites for the estimates of Cmax by product, and F4_rel between test and reference, the primary outcome variables for assessment of bioequivalence. ii) Adequate description of the concentration profile, its magnitude, change over time, and ultimate rate of elimination (kel), requires balance in the model between, on the one hand, predicted distribution and elimination clearance (CL/F, Q2/F, and Q3/F) and, on the other hand, central and peripheral volumes of distribution (V1/F, V2/F, and V3/F). Therefore, post-hoc estimation of T½ (= ln(2)/kel), the value of which depends on all these primary model disposition parameters, was used as means to check that the fates of FP and SALM, once they have reached the systemic circulation, were adequately handled by the empirically derived model parameters for fixed and random effects. Reasonable initial estimates, especially on clearance, taking into account expected absolute systemic bioavailability after inhalation, were taken from the literature (eg Haughie et al, 2019)2 and were an important element in order to get reasonable results. Sanity checks were performed continuously based on preliminary population estimates, starting with reduced data sets and ending with the final results from fits to all data tabulated in the manuscript.
Values below the LLOQ (1ng/L) were considered. Note that the lack of a parenteral reference treatment and information on absolute bioavailability means that estimated apparent clearances and apparent volumes of distribution have a systematic bias towards higher values compared to the PK analysis outcome of an intravenous administration. The between-subject variability (IIV) of elimination and inter compartmental clearances and volumes of distribution parameters were considered for FP and SALM, and the between study variability (ISV) of relative bioavailability for SALM. Shrinkage in between subject variability is calculated as 100 x(1-SD(ETA)/ω)%. Apart from the categorical effect of product, neither demographic nor other covariates were considered in the evaluation of NCA-based results2 and therefore not in this alternative model analysis either, partly due to the crossover design (each subject is used as their own control) and partly to the homogeneously composed panels of subjects. This is the context in which the alternative proposed method was developed and is also assumed to be the future context where the method could be applied. Shrinkage approaching 0% suggests very informative data, whereas a value >30% suggests the opposite whereby the individual parameter estimates “shrink” to the population parameter estimate.11 The residual error was considered proportional or additive + proportional, the standard deviations of which were coded as fixed parameters and henceforth denoted as CP_ADD (additive) and CP_PROP (proportional).
The empirical linear models were fitted to the data of all subjects simultaneously, if possible across studies, considering the product differences, as described above. The fit was optimized using a non-linear mixed-effect model approach as implemented in NONMEM, version 7.5.0, using the subroutine ADVAN5 (Icon development Solutions, Hannover, MD, USA) and the Pearl Speaks NONMEM (PsN) run tool “Execute”.12 Pharmacokinetic parameters and associated random between-subject variability (denoted ETA1-ETA8 in Figure 1) or random between study variability (denoted ETA9 in Figure 1) were assumed to be normally distributed on the log scale (except for pharmacokinetic parameters without consideration of random effects), means of the latter ideally equal to zero, and variance ω2. Model-based analyses were performed, and the impact of data below the lower limit of quantitation was considered as outlined by Bauer (2019), implying Markov Chain Monte Carlo stochastic approximation expectation maximization (SAEM) followed by Monte Carlo importance sampling (IMP) combined with second-order conditional estimation with interaction,13 jointly denoted SAEM_I-IMP*_I in the NONMEM output. Alternative approaches suggested by the same author were less appropriate for this exercise. Possible trends in random effects, that is, deviations from the expected zero mean of individual random effects on typical values of pharmacokinetic parameters, were explored using box plots.
The NONMEM output was further processed in R 4.2.3,14 using the package ggplot2 alone or with its application xpose.15 The model assessment criteria are listed in Table 1. Final model optimization particularly involved fine-tuning of initial parameter estimates, and residual models adapted for exponentiated or logged data, thereby producing alternatives not apt for discrimination using the OFV-derive Akaike’s Information Criterion (AIC). Mechanistic accuracy, except for what was necessary for bioequivalence testing, was not considered within the scope nor realistic due to long run times; neither boot strapping nor log likelihood profiling was considered. Central NONMEM-code elements of final models, Model 1 (FP 3 × 100µg), Model 2 (FP 3 × 250µg), Model 3 (FP 3 × 500µg), and Model 4 (SALM 3 × 50µg, across studies),13 R-code for individual post-hoc estimation of terminal half-life,16 and output of R-based basic goodness-of-fit plots are supplied as a supplement. The numerical results are presented using four valid digits. Pharmacokinetic parameters were presented as typical mean estimates, and the relative standard error of the mean was calculated as the square root of the variance of the parameter estimate divided by the typical parameter value. Random effects are presented as the standard deviation from an associated typical parameter value, that is, the square root of the estimated variance. The relative standard error (RSE) is calculated as the standard error of the estimated variance (SE) divided by two times the variance estimate.15
 |
Table 1 Model Assessment Criteria |
The terminal half-life, T½, was calculated based on individual estimates of systemic disposition micro constants,16 under the assumption that absorption was not rate-limiting for terminal elimination. The Cmax was identified based on individually predicted peak concentrations within the framework of actual discrete individual sampling times. Post-hoc predictions of T½ and Cmax were described using the mean ± standard deviation as the basis for outcome comparison with non-compartmental analysis.
Results
The analysis dataset comprised 7184 observations of FP and 7186 observations of SALM observations. All administrations were treated as single administrations with no carryover effect on the PK of either substance and were analyzed using structural models, as indicated in Figure 1. Four subjects had low but measurable pre-dose concentrations before administration of FP 3 × 500 µg, one in the first and three in the second period; these values were disregarded and set to zero in the analysis on the grounds of likely random bioanalytical error combined with the fact that >10 half-lives of FP had passed during the minimum of a 7-day wash-out period between first and second dose event.
Population Analysis
The empirical structural models fit the FP and SALM data well, both at the population and individual levels, apparently without bias, ie the median individual ln(deviation) from the population estimate was generally close to zero (cf. below).
Alternative absorption (biphasic first order, zero order, bolus, or combinations) and distribution models were exhaustively explored, with or without censored data, to exponentiated or log transformed data, using different minimization routines. Provisional linear models, fitted to all data of inhaled FP across studies/doses, considering biphasic- vs monophasic absorption (full vs a reduced model), indicated that systemic absorption of inhaled FP might be better explained by a biphasic rather than a monophasic model (AICfull model - AICreduced model = -75.614). However, the estimated absorption rate constants differed marginally with the full model; moreover, relative extent of bioavailability was not estimated and the simultaneous fit across studies revealed indistinguishable pre- and post absorption nonlinear pharmacokinetic elements of FP (cf. below). Hence, subsequent analysis was restricted to monophasic absorption and across studies only for SALM (only one dose across studies, 3 × 50 µg), in order to avoid unidentifiable Michaeli Menten kinetics of FP.
An alternative strategy to analyze bioequivalence of FP was explored; thereby, the categorical effects of treatment on CL/F and V1/F were estimated in the population analysis instead of that on the population PK parameter F4_rel (cf. Figure 1). With the alternative, relative extent of bioavailability between test and reference product was calculated as the inverse ratio between post-hoc estimates of treatment-differentiated apparent elimination clearance (relative rate of bioavailability was equally calculated with both options based on post-hoc estimates of Cmax). Good agreement between the two modeling approaches can be regarded as a sensitivity test of robustness with respect to the basic pharmacokinetic model, jointly applied for both FP and SALM (linear first order absorption and disposition). However, considering our particular objective, to compare the empirical model-based approach with conventional NCA, this should mean that parametrically post-hoc derived outcome variables match those used in NCA, ie F4_rel and Cmax.
Outcome details of the above alternative analyses, monophasic vs biphasic absorption and alternative parametrization to assess relative extent of absorption, including our reasoning around AIC as validation tool in these respects are provided as Supplement.
The chosen final generic linear model was a fair trade-off between simplicity and being fit for purpose. However, assessment of between subject variability indicated shrinkage particularly regarding apparent distribution clearance (Q2/F and Q3/F) of FP. The default action would be to reduce the complexity of the model, in this case to go for a two-instead of a three-compartment model. Considering i) visual inspection of log transformed mean plasma concentration vs time profiles,2 ii) outcome of previous model analysis,10 and iii) good precision in the population estimates of Q2/F and Q3/F of FP it was decided to retain a three-rather than a two-compartment systemic disposition model for FP, however disregarding between subject variability of apparent distribution clearances of FP owing to shrinkage.
Visual investigation of random effects using boxplots did not suggest systematic deviations from the typical values of pharmacokinetic parameters (Figures 2 and 3), although it was remarkably skewed for one of the two apparent peripheral volumes of distribution, V2/F, after FP 750 and 1500 µg (cf. Figure 2). The predicted plasma concentrations based on apparent population pharmacokinetic parameter estimates accurately described the plasma concentrations of FP and SALM in a typical subject with respect to magnitude and variation over time (Figure 4). The results obtained with these models also indicate that normalized prediction distribution errors (NPDE), guided by the information on measurements <LLOQ, “fill out” the lower regions in a concentration scatter plot vs population prediction variable (PREDV) (Figures 5). Visual predictive checks, 95% confidence interval percentiles of observations, and 95% confidence interval percentiles of 100 simulations are shown in Figures 6 and 7. Table 2 presents the typical mean values of the population pharmacokinetic parameters and the relative standard errors. Precision in estimates was generally good with the exception of one apparent peripheral volume of distribution for FP, V2/F, and the jointly estimated random effect of study on apparent elimination and inter compartmental clearances of SALM. Central parametric determinants of apparent rate and relative extent of absorption (deviation from unity), apparent clearance, and central volume of distribution (k41, F4_rel, CL/F, and V1/F) showed minor differences between test and reference products at equivalent strengths. The applied mono-phasic input parameter (k41) indicated a substantially faster absorption of SALM than FP. Outcomes of the separate analysis of FP by study/dose indicated similar apparent elimination clearance of typically less than 500 L/h across FP doses of 300 µg and 1500 µg, respectively, whereas the value after FP 750 µg was approximately 25% higher. Notably, across study/doses, the central apparent volume of the FP distribution, V1/F, tended to be directly correlated, whereas the empirically derived apparent rate of mono-phasic absorption, k14, tended to be inversely correlated with the dose – NB the primary reason why the generic linear model was not fitted to FP across studies/doses! The population estimates of F4_rel were close to unity for the dose of FP and across the SALM studies.
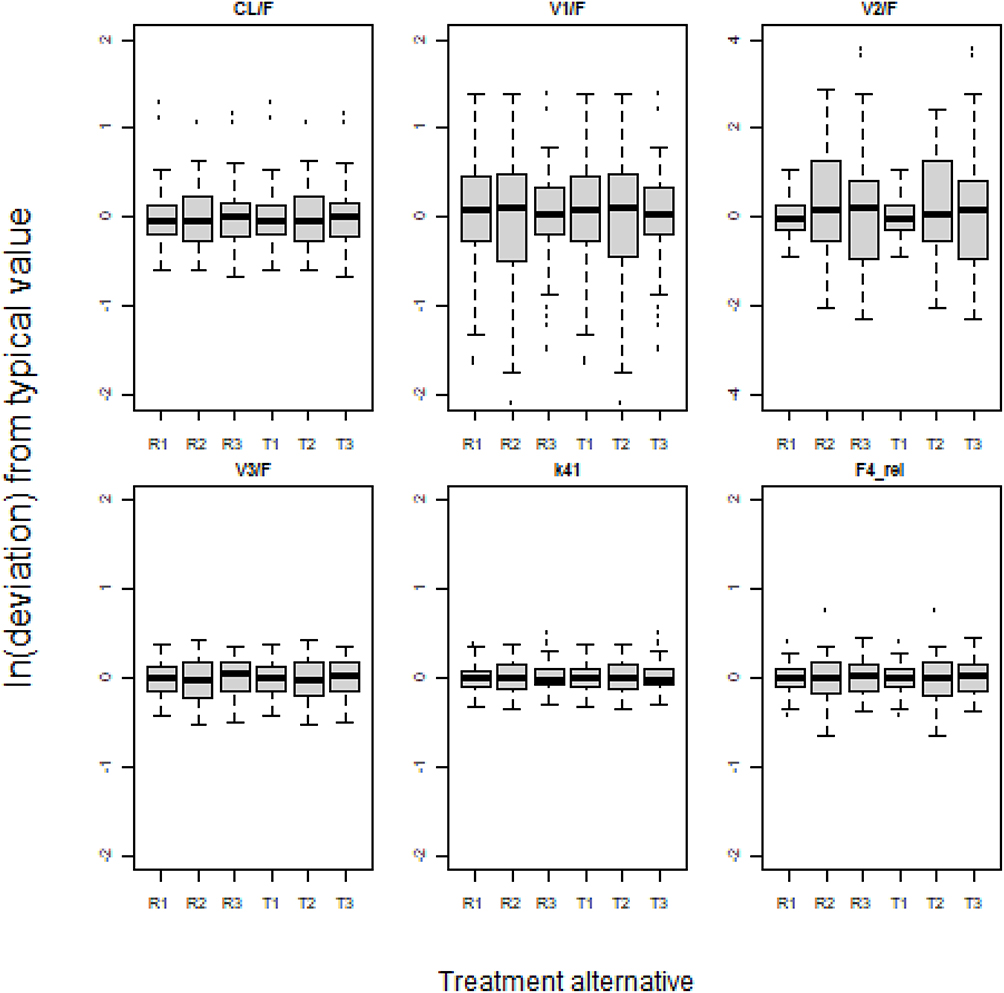 |
Figure 2 Between subject variability of empirically derived pharmacokinetic FP parameters considering categorical effects of treatment alternative on relative bioavailability and rate of absorption. R1: Reference product (FP 300µg); R2: Reference product (FP 750µg); R3: Reference product (FP 1500µg); T1: Test product (FP 300µg); T2: Test product (FP 750µg); T3: Test product (FP 1500µg). |
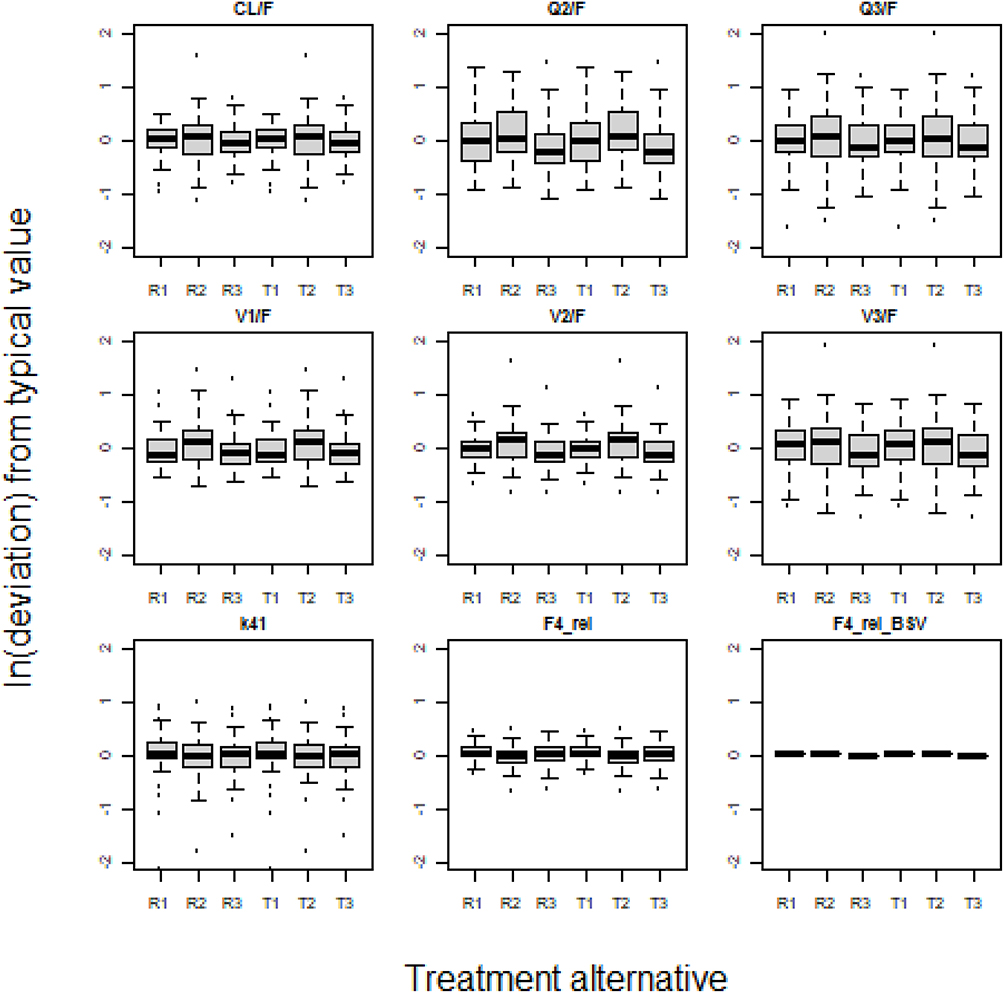 |
Figure 3 Between subject and between study variability of empirically derived pharmacokinetic SALM parameters considering categorical effects of treatment alternative on relative bioavailability, rate of absorption, and study. R1-3: Reference product (SALM 150µg) in studies 1–3; T1-3: Test product (SALM 150µg) in studies 1–3. |
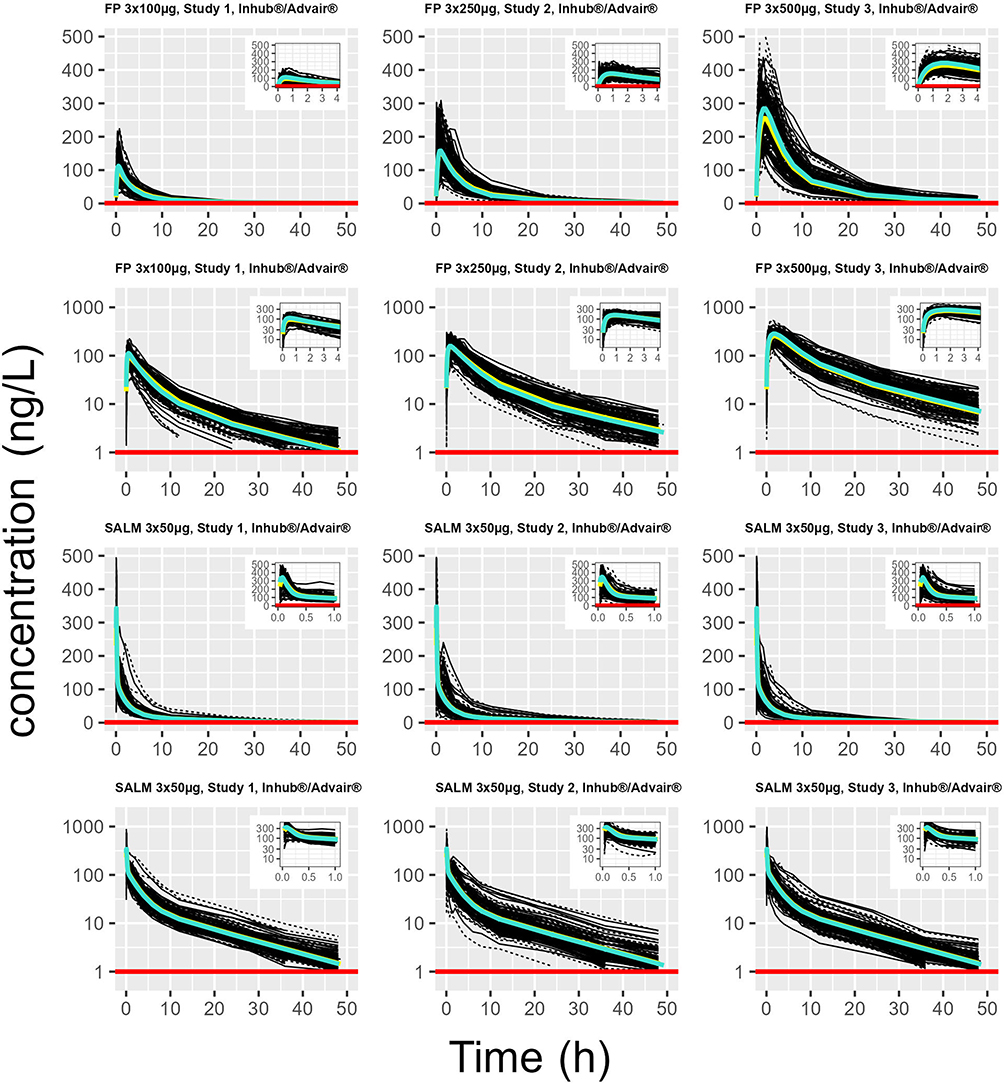 |
Figure 4 Empirical population pharmacokinetic structural model, implying apparent monophasic absorption and three-compartment systemic disposition, fitted to measured plasma concentrations of FP by study and dose (Models 1–3, from the top rows 1–2), and simultaneously across studies to plasma concentrations of SALM (Model 4, from the top rows 3–4). Each spaghetti plot illustrates, on log ordinate or exponentiated scale serial measured individual plasma concentrations (thin full black lines: test product; thin black dotted lines: reference product), and predictions in the typical subject (thick yellow solid line: test product; thick turquoise solid line: reference product), and LLOQ (red solid line). |
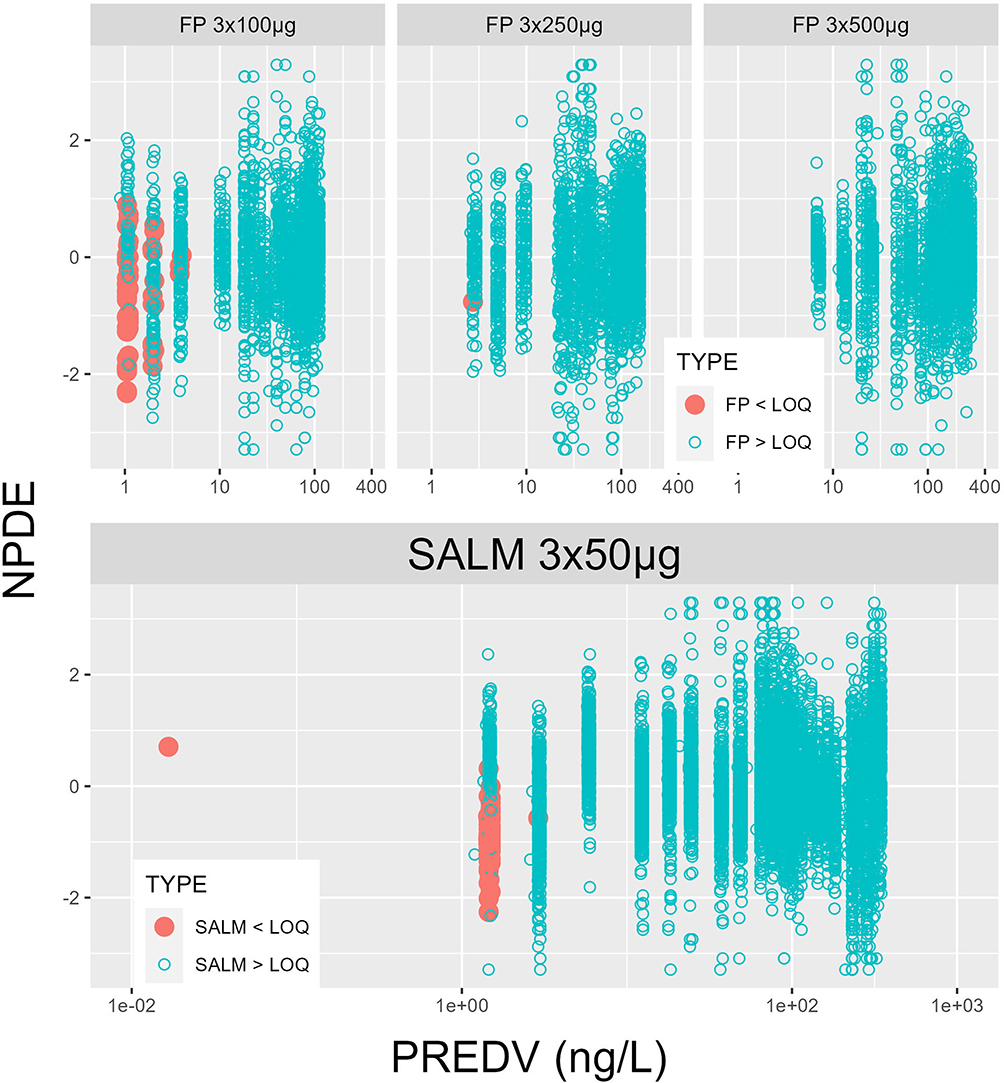 |
Figure 5 Normalized prediction distribution error (NPDE) plotted against population prediction variable (PREDV) with final model fits, FP by study and dose (Models 1–3, row 1), SALM across studies 1–3 (Model 4, row 2; the far left red dot represents an outlier, a sample of SALM below LLOQ in study 3, taken at 126 h instead of 48 h post-dose). Number of values below lower limit of quantitation (LLOQ) as from 24 h after the dose onwards by study/dose. FP, 3 × 100µg: 54 samples; FP, 3 × 250µg: 1 sample and SALM, studies 1–3: 55 samples. |
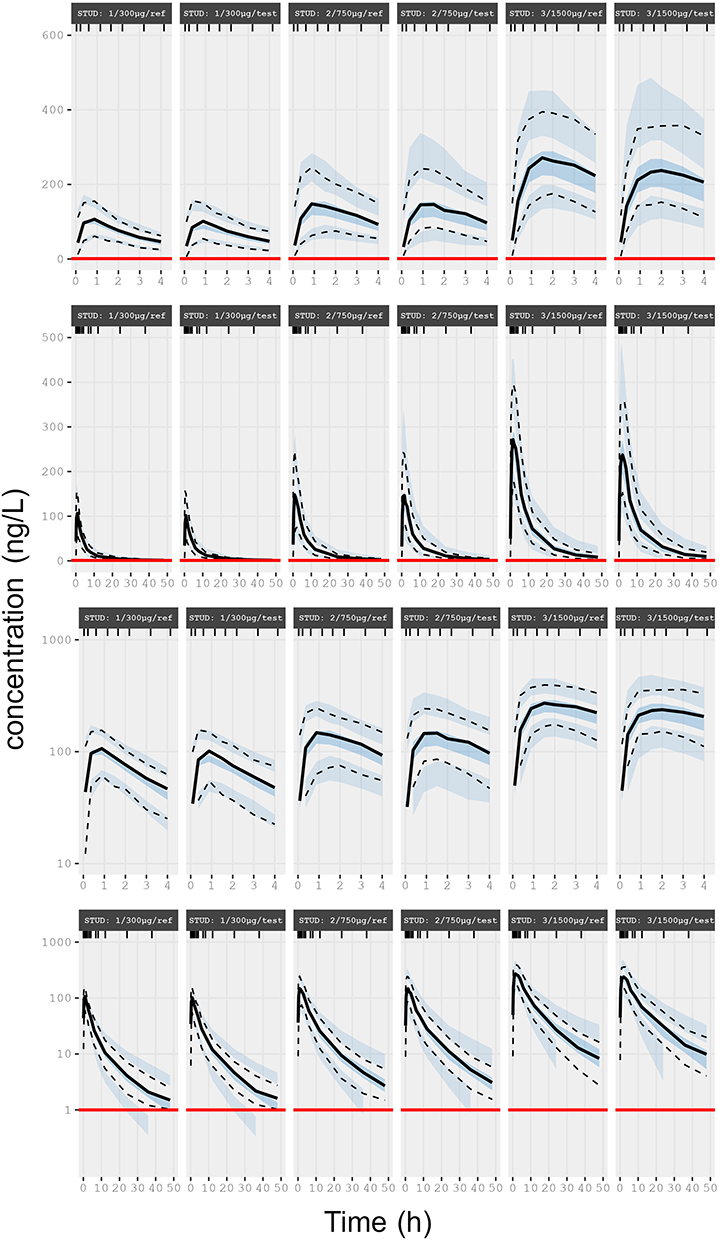 |
Figure 6 Final models, visual predictive checks of FP by dose (Models 1–3 by treatment alternative). Each plot illustrates, on exponentiated (rows 1–2) and log ordinate (rows 3–4) scale, median, 2.5th and 97.5th percentiles of observations (solid and dotted black lines), descriptive statistics of predictive checks (medians, 2.5th and 97.5th percentiles of 100 simulations, represented by coloured fields), and LLOQ (red solid lines). |
 |
Figure 7 Final model, visual predictive checks of SALM by study (Model 4 by treatment alternative). Each plot illustrates, on exponentiated (rows 1–2) and log ordinate (rows 3–4) scale, median, 2.5th and 97.5th percentiles of observations (solid and dotted black lines), descriptive statistics of predictive checks (medians, 2.5th and 97.5th percentiles of 100 simulations, represented by coloured fields), and LLOQ (red solid lines). |
 |
Table 2 Typical Empirical Population Pharmacokinetic Parameter Estimates (Relative Standard Error) Obtained with Final Models Fitted Simultaneously to Data by Study/Dose of FP (Models 1–3) and Across Studies of SALM (Model 4), Considering Relative Bioavailability Between Products and Accounting for Values Below Lower Limit of Quantification |
Analysis of Individual Post-Hoc Estimates
Individual post-hoc estimates of the terminal half-life and peak plasma concentration of the test vs reference product is described in Table 3. The mean terminal half-lives of FP and SALM were approximately 14 h and 12 h, respectively. Thus, the fractional rate of elimination of both substances (ln(2)/T½) was substantially lower than the apparent fractional rate of absorption (k14, cf. Table 2), suggesting that absorption was not a rate-limiting factor during the terminal elimination phases of FP and SALM. Despite the lower dose, the mean Cmax of SALM was generally higher than that after all three doses of FP, which is consistent with what would be expected, considering the differences in the absorption rate and central pharmacokinetic determinants of systemic exposure (CL/F and V1/F).
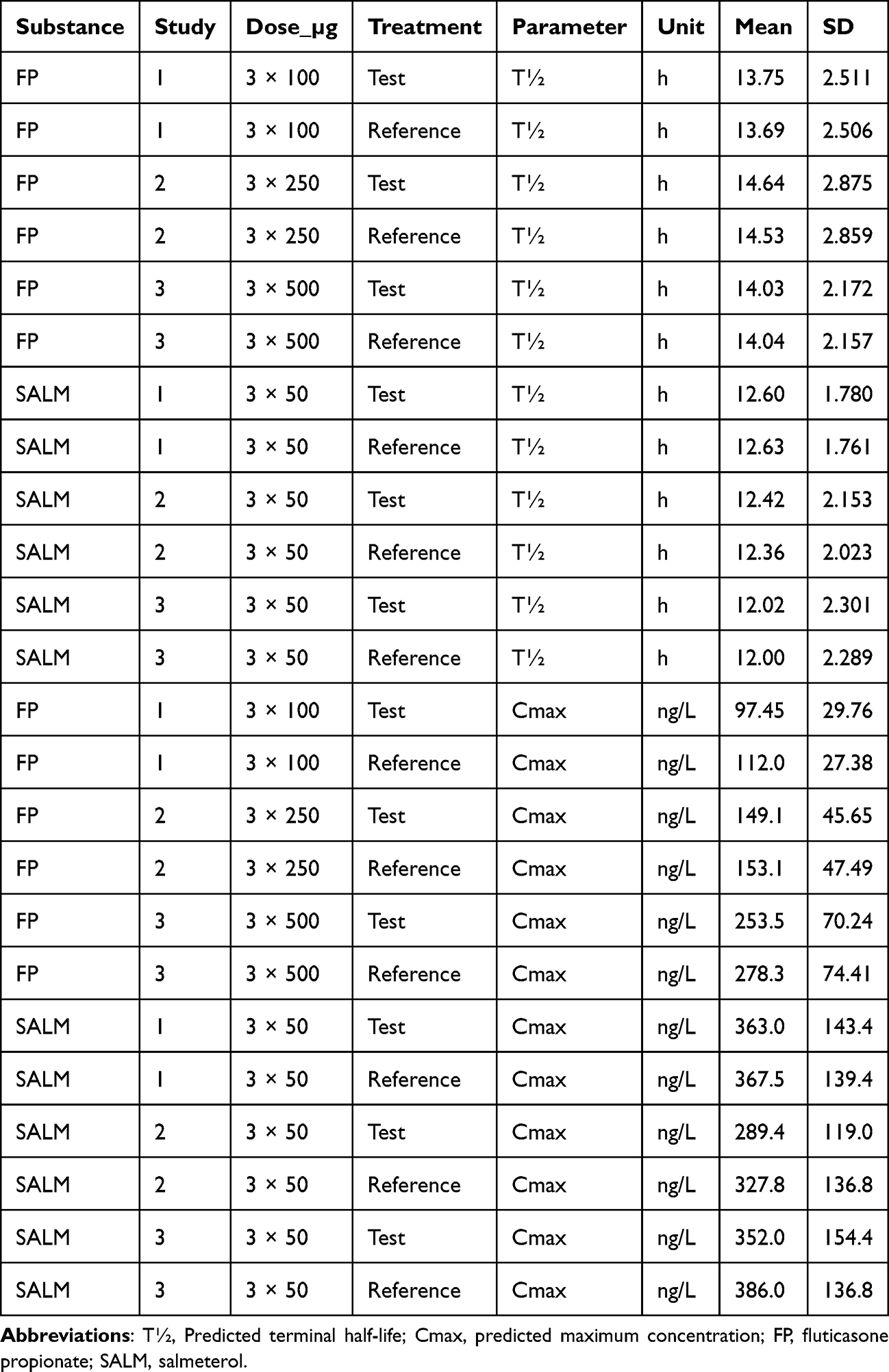 |
Table 3 Descriptive Statistics of Post-Hoc Individual Estimates of Terminal Half-Life and – Within the Framework of Sampling Occasions – Individually Predicted Plasma Concentration with a Monophasic Absorption and Three-Compartment Systemic Disposition Model Fitted Separately by Study/Dose to Plasma Concentration of Fluticasone Propionate and Simultaneously Across Studies to Plasma Concentration of Salmeterol |
Bioequivalence, Empirical Model- vs NCA-Based Assessment
Analysis of model based individual post-hoc predicted relative extent (F4_rel) and the surrogate rate marker of relative rate (ratio of predicted Cmax, denoted Cmax_ratio) of bioavailability, matched well previous analysis of NCA-based outcome variable2 (Table 4).
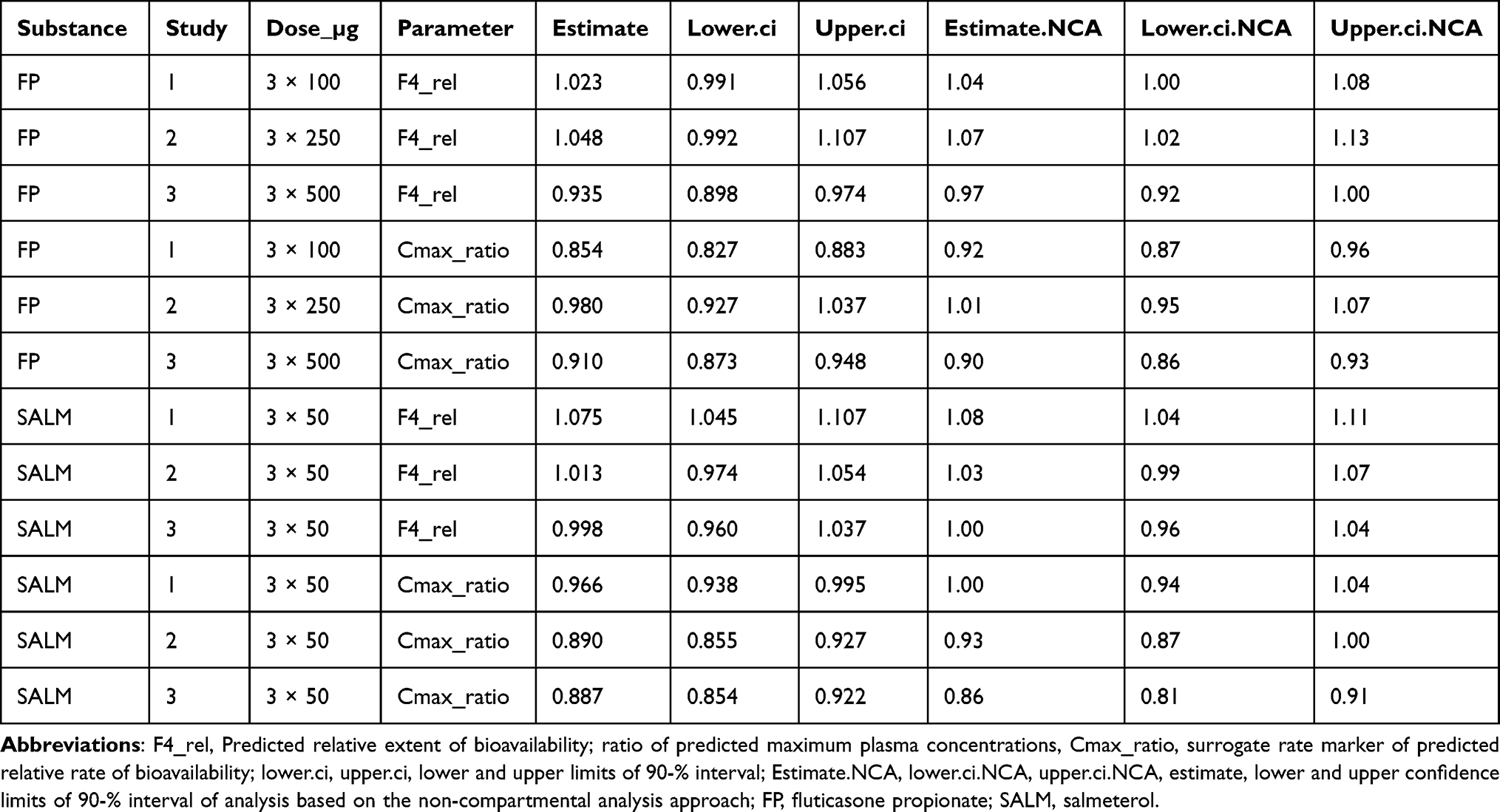 |
Table 4 Bioequivalence Test, Analysis of Model Based Individual Post-Hoc Predicted Relative Extent and a Surrogate Rate Marker of Bioavailability in Comparison with the Previous NCA-Based Approach (Cf. Text) |
Discussion
Inference and Relevance of Parameter Estimates
This study describes the empirical derivation of pharmacokinetic parameters for an inhaled combination of FP and SALM using a model-based approach. This outcome could complement the NCA-based assessment of relative systemic bioavailability.17
Thorsson et al (2001) estimated clearance (CL) of intravenously administered FP at 1308 mL/min, ie, 78 L/h, and systemic bioavailability of FP after inhalation via Diskus at 13%.5 The apparent mean FP clearance, CL/F (nominal dose/mean AUC), based on NCA analysis of the present dataset set was 493–606 L/h after inhalation via the reference product Advair Diskus, depending on study/dose (mean value across study/dose: 538 L/h),2 an indication of an absolute bioavailability similar to that presented by Thorsson et al (2001),5 just under 15% (78.48/538).2 The corresponding NCA-based mean CL/F for the test product Wixela Inhub was 470–578 L/h depending on the study/dose (mean across study/dose: 525 L/h).2 A typical model-based joint estimate of the apparent FP clearance, 498.1–618.4 L/h, respectively, reasonably confirmed the NCA (Table 2). NCA-based analysis of FP, test vs reference product, estimated the relative extent of bioavailability at 0.97–1.04 with respect to AUC, depending on the study/dose of FP, with all 90% confidence intervals within 0.80–1.252. Analyses of model-based post-hoc estimates of F4_rel and Cmax_ratio by dose (Table 4) were compatible with NCA-based bioequivalence tests of AUC with the same data set.2
Mean NCA-based estimates of FP Cmax and terminal T½ ranged 119–291 ng/L and 10.0–10.6 h with the reference and 110–262 ng/L and 10.2–12.2 h with the test product, respectively, depending on study/dose.2 Post-hoc, model-based means of individually predicted Cmax, 112.0–278.3 ng/L and 97.45–253.5 ng/L with reference and test product, respectively, depending on study/dose, confirmed the NCA-analysis. Post-hoc, model-based mean individually predicted T½, 13.69–14.53 h with the reference and 13.75–14.64 h with the test product, respectively, depending on study/dose (Table 3), were marginally longer than suggested by the NCA-analysis, although in line with what was presented by Allen et al (2012) for inhaled FP.6
The apparent mean SALM clearance, CL/F (nominal dose/mean AUC), based on NCA analysis of the present dataset, was 212–222 L/h (mean value across study/dose: 217 L/h) after inhalation via the reference product Advair Diskus, and 206–214 L/h (mean value across study/dose: 209 L/h) after inhalation via the test product Wixela Inhub, depending on study.2 Typical model-based estimates of apparent SALM CL/F across studies, 221.0 L/h, confirmed NCA (Table 2). NCA-based analysis of SALM estimated the relative extent of bioavailability at 1.00–1.08 with respect to AUC, depending on the study, with all 90% confidence intervals within 0.80–1.252. Analyses of model-based post-hoc estimates of F4_rel and Cmax_ratio by dose (Table 4) were compatible with NCA-based bioequivalence tests of AUC with the same data set.2
Mean NCA-based estimates of Cmax and terminal half-life of SALM, derived from an NCA of the present data set, were 353–418 ng/L and 11.56–12.21 h with reference, and 320–385 ng/L, and 11.21–11.87 h with test product, respectively, depending on product and study.2 Post-hoc, model-based means of individually predicted Cmax and T½, 327.8–386.0 ng/L and 12.00–12.63 h with the reference, and 289.4 –363.0 ng/L and 12.02–12.60 h with the test product, respectively, confirmed the NCA-analysis (Table 3).
We acknowledge that, despite homogeneous composition of the studied panels, the lack of considered possible impact of demographic variability will constitute major limitations on generalizability. We also acknowledge that instantaneous pulmonary deposition is a mechanistic simplification, albeit a necessary one due to limited information about the administration procedure itself. The fit to the data is based on a generic empirical model with the primary aim to describe the plasma concentration of the FP and SALM curves, respectively, over time as correct as possible, notwithstanding detailed physiologically mechanistic relevance of individual parameters. Several alternatives to the robust model we ultimately settled on have been tested, but we believe that information is lacking to further enable adequate discrimination between regional deposition, dissolution of powder, absorption into and – except for elimination – ultimate systemic disposition.
Advantages and Limitations of Population Pharmacokinetic Modelling
The overall benefit compared to conventional NCA is that the population model-based approach enables a continuous description of the rise and decline of plasma concentrations over time using a limited number of parameters while simultaneously considering uncertainties at the group level, random effects on PK parameters, and residual error. The parametric description facilitates interpolation. However, to best utilize the potential of population analysis to extrapolate to alternative clinical situations, even within the scope of the basic conditions on which the present model-based analysis was based, would require a demographically more heterogeneous dataset, characterized by balanced but greater variability between subjects to enable adequate assessments of covariate effects (eg, ethnic origin, sex, age, or body weight) on outcome variable. The aim to explore alternative tests of bioequivalence using the rich dataset, with all elements in the original study designs adapted for NCA, was in fact suboptimal for stand-alone model fitting. Our particular modeling approach – and alternative to NCA – meant that a generic and simplistic pharmacokinetic structural model was established early on; sensitivity analysis within that narrow scope by fixing critical parameters, for example the empirically simplified first order absorption process, was not done. However, the setup allowed for sensitivity-tests of the models’ abilities to adequately match NCA-based estimations of rate and extent of absorption (Cmax and CL/F), the terminal elimination constant λ (and so T½=ln(2)/λ), and necessary elements for successful assessment of the covariate product effect on relative bioavailability.
The aerodynamic particle size distribution (APSD) of the formulations explored in this study was equivalent for all FP strengths and products. An expected outcome would be the dose-independent regional deposition of FP and loss of the pulmonary deposited drug via mucociliary clearance.4 The fraction of the deposited dose subject to non-absorptive mucociliary clearance could be affected by dose but this should have been observed as a dose-dependent variation in CL/F across strengths in the model analyses (as well as dose-disproportional AUC in NCA), but that was not the case. The empirically derived monophasic absorption rate after FP 3 × 100 µg was between the pulmonary biphasic rapid peripheral and slow central absorption after FP 500 µg, as presented by Drescher et al (2023)4 but was slower after FP 3 × 250 µg and 3 × 500 µg with both test and reference products. We hypothesize that the less-than-dose-proportional Cmax was causally the consequence of a negative correlation to dose strength; the higher the strength, the longer the dissolution time, resulting in a wider and thus the dose disproportional plasma concentration peak of FP.9,18 An approximately 10% underestimation of the mean predicted (Table 2) versus the observed Cmax2 was the necessary trade-off to apply a monophasic rather than a multiphasic absorption model. However, difficulties in distinguishing dissolution, absorption, and systemic disposition in the present context (cf. above) implied that the impact of FP dose on these indistinguishable elements manifested across studies, both in an expected effect on k41 and unexpected effects on systemic disposition parameters when fitting the empirically derived model (Table 2). It is possible that fixed systemic disposition parameters ad modum Hochhaus et al4 would have yielded model outcomes that more distinctively linked product attributes to regional pulmonary absorption. However, the assumption that the systemic disposition of FP and SALM is identical in all three studies is not supported by the independent intravenous PK data.
Despite these limitations, an empirical population model analysis could be a useful complement to NCA when exploring the fate of inhaled FP/SALM formulations and similar medicines. The prediction of peak concentrations in a typical subject reflected previous results based on NCA analysis and indicated the relevance of the approach and its potential. Exploration of alternative parenteral and oral routes of administration and wider biopharmaceutical attribute ranges, such as particle size distribution, would facilitate differentiation of the presystemic and systemic fate of inhaled dry powder drug formulations, particularly a mechanistically relevant pharmacokinetic description of regional deposition.3,4 Moreover, while the bioequivalent test and reference products formed the basis for this model analysis, further work could explore the sensitivity of the method to detect unknown “known” differences, for example, by applying it to study data where the products were not bioequivalent.
Conclusion
Model- and NCA-based approaches to assess bioequivalence were consistent. However, a frequentist parametric approach that fully utilizes the population analysis potential and mechanistically predicts regional pulmonary deposition of FP and SALM would require a demographically more heterogenous data set and analysis including additional outcomes of parenteral and oral administration to discriminate presystemic from systemic disposition.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
The study was funded by Mylan Pharma UK Ltd, a Viatris Company.
Disclosure
Johan Rosenborg has shares in Viatris Inc as a result of the spin-off by Pfizer Inc. of its global primarily off-patent medicines business and is an employee of StatMind AB that receives consultancy fees for the work with the manuscript. Thomas Bengtsson is a co-owner of StatMind AB that receives consultancy fees for the work with the manuscript.; reports personal fees from Mylan Pharma UK Ltd, during the conduct of the study. Scott Haughie is an employee of Viatris, Mylan Pharma UK Ltd. Dr Per Bäckman reports personal fees from Viatris, during the conduct of the study; personal fees from Viatris, outside the submitted work; and PB is a senior adviser to the pharmaceutical industry and have a such received remuneration from several entities, including but not limited to Viatris Pharma.
References
1. Ng D, Kerwin EM, White MV, et al. Clinical bioequivalence of Wixela Inhub and Advair Diskus in adults with asthma. J Aerosol Med Pulmonary Drug Delivery. 2020;33(2):99–107. doi:10.1089/jamp.2019.1547
2. Haughie S, Allan R, Wood N, et al. Equivalent systemic exposure to fluticasone propionate/salmeterol following single inhaled doses from Advair Diskus and Wixela Inhub: results of three pharmacokinetic bioequivalence studies. J Aerosol Med Pulmonary Drug Delivery. 2020;33(1):34–42. doi:10.1089/jamp.2019.1537
3. Hochhaus G, Chen M-J, Kurumaddali A, et al. Can pharmacokinetic studies assess the pulmonary fate of dry powder inhaler formulations of fluticasone propionate? AAPS J. 2021;23(3):1–14. doi:10.1208/s12248-021-00569-x
4. Drescher SK, Jiao Y, Chen M-J, et al. Central and peripheral lung deposition of fluticasone propionate dry powder inhaler formulations in humans characterized by population pharmacokinetics. Pharm Res. 2023;40(5): 1177–1191. doi:10.1007/s11095-023-03472-6
5. Thorsson L, Edsbäcker S, Källén A, et al. Pharmacokinetics and systemic activity of fluticasone via Diskus ® and pMDI, and of budesonide via Turbuhaler ®. Br J Clin Pharmacol. 2001;52(5):529–538. doi:10.1046/j.0306-5251.2001.01493.x
6. Allen A, Bareille PJ, Rousell VM. Fluticasone furoate, a novel inhaled corticosteroid, demonstrates prolonged lung absorption kinetics in man compared with inhaled fluticasone propionate. Clin Pharmacokinet. 2013;52(1):37–42. doi:10.1007/s40262-012-0021-x
7. Hofmann W, Asgharian B. The effect of lung structure on mucociliary clearance and particle retention in human and rat lungs. Toxicol Sci. 2003;73(2):448–456. doi:10.1093/toxsci/kfg075
8. Borghardt JM, Weber B, Staab A, et al. Pharmacometric models for characterizing the pharmacokinetics of orally inhaled drugs. AAPS J. 2015;17(4):853–870. doi:10.1208/s12248-015-9760-6
9. Bäckman P, Olsson B. Pulmonary drug dissolution, regional retention & systemic absorption: understanding their interactions through mechanistic modeling. In: Byron PR, editor. Respiratory Drug Delivery. River Grove, Illinois: Davis Healthcare International Publishing; 2020:113–122.
10. Thorsson L, Dahlström K, Edsbäcker S, et al. Pharmacokinetics and systemic effects of inhaled fluticasone propionate in healthy subjects. Br J Clin Pharmacol. 1997;43(2):155–161. doi:10.1046/j.1365-2125.1997.d01-1425.x
11. Savic RM, Karlsson MO. Importance of shrinkage in empirical Bayes estimates for diagnostics: problems and solutions. AAPS J. 2009;11:558–569. doi:10.1208/s12248-009-9133-0
12. Lindbom L, Pihlgren P, Jonsson N. PsN-toolkit—a collection of computer intensive statistical methods for non-linear mixed effect modeling using NONMEM. Comput Methods Programs Biomed. 2005;79(3):241–257. doi:10.1016/j.cmpb.2005.04.005
13. Bauer RJ. NONMEM tutorial part II: estimation methods and advanced examples. CPT. 2019;8(8):538–556.
14. R Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2019.
15. Guiastrennec B, Hooker, AC, Ueckert, S, Karlsson, MO. xpose: Diagnostics for Pharmacometric Models 2025. Available from: https://uupharmacometrics.github.io/xpose/.
16. Upton RN. Calculating the hybrid (macro) rate constants of a three-compartment mamillary pharmacokinetic model from known micro-rate constants. J Pharmacol Toxicol Meth. 2004;49(1):65–68. doi:10.1016/j.vascn.2003.09.001
17. Hochhaus G, Chen M, Shur J, et al. Unraveling the pulmonary fate of fluticasone and friends: meeting the physiologic and pharmacokinetic challenges. Respir Drug Deliv. 2020;2020:139–146.
18. Price R, Shur J, Ganley W, et al. Development of an aerosol dose collection apparatus for in vitro dissolution measurements of orally inhaled drug products. AAPS J. 2020;22(2):1–9. doi:10.1208/s12248-020-0422-y
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.