Back to Journals » International Medical Case Reports Journal » Volume 8
Relapsing polychondritis with different types of ocular inflammations
Authors Furuya N, Oshitari T, Yotsukura J, Baba T, Yamamoto S
Received 29 May 2015
Accepted for publication 27 July 2015
Published 18 September 2015 Volume 2015:8 Pages 193—197
DOI https://doi.org/10.2147/IMCRJ.S89507
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Nana Furuya, Toshiyuki Oshitari, Jiro Yotsukura, Takayuki Baba, Shuichi Yamamoto
Department of Ophthalmology and Visual Science, Chiba University, Graduate School of Medicine, Chuo-ku, Chiba, Japan
Abstract: We were presented with two cases of relapsing polychondritis (RP) associated with different types of ocular inflammation. The first case was a 35-year-old man who had bilateral hyperemic conjunctiva and ocular pain, and was referred to Chiba University Hospital with a diagnosis of episcleritis refractory. He was treated with dexamethasone eye drops. He developed tinnitus, deafness in both ears, and left auriculitis. A left auricular biopsy showed an infiltration of lymphocytes surrounding the cartilage. He was diagnosed with RP and treated with 30 mg/day oral prednisolone. After tapering the prednisolone, the scleritis in both eyes improved. The second case was a 71-year-old man who was deaf in both ears and had bilateral scleritis. At the first visit to our hospital, his left eyelid and right auricula were reddish and swollen, and he reported some pain. He was treated with intravenous antibiotics, and the left orbital cellulitis quickly improved. However, he developed right scleritis and left gonitis. Magnetic resonance imaging showed bilateral posterior scleritis and right auricular perichondritis. Auricular biopsy showed an infiltration of lymphocytes into the periauricular tissue. He was diagnosed with RP, and 40 mg/day oral prednisolone was given and his symptoms improved. Although RP is rare, it is a life-threatening disease. Thus, ophthalmologists should consider RP in patients with both ocular and auricular inflammation.
Keywords: relapsing polychondritis, scleritis, oral prednisolone, ocular cellulitis
Introduction
Relapsing polychondritis (RP) is a rare but life-threatening disease, which affects caltilaginous and proteoglycan-rich structures such as ears, nose, respiratory tract, and sclera.1 Ocular manifestations are present in 50%–60% of the cases of RP.1 However, there are a variety of inflammatory symptoms and little specificity is present that can help in diagnosing RP.
We report our findings in two patients with RP and with different types of ocular inflammation. We had local ethical permission to access and publish the patients’ data.
Case reports
Case 1
A 35-year-old man who had bilateral hyperemic conjunctiva and ocular pain visited a private clinic in December 2007. He was diagnosed with episcleritis, and steroids and antibiotic eye drops were prescribed, but the signs and symptoms did not improve. In January 2008, he began a treatment of 20 mg/day oral prednisolone and was referred to the Chiba University Hospital in February 2008.
At his first visit, his visual acuity was 1.2 OU, and the intraocular pressure was 20 mmHg OU. Slit-lamp examinations showed hyperemic episcleral vessels in both eyes but no inflammatory signs in the anterior chamber (Figure 1). Fundus examinations showed no abnormal findings. The blood tests showed an elevation of white blood cells count (16,300/L), but C-reactive protein (CRP) was 0.1 mg/dL. The dosage of 15 mg/day oral prednisolone was continued. During the tapering of the oral prednisolone, he developed tinnitus, vertigo, deafness, and left auricular deformation. Positron emission tomography identified a mild accumulation of 18F-fluorodeoxyglucose in the left auricle. In January 2009, an auricular biopsy was performed, and an infiltration of inflammatory cells was detected in and around the cartilagenous tissues.
 | Figure 1 Appearances of Case 1 who was diagnosed with episcleritis. |
In February 2009, visual field defects were detected bilaterally, namely, right enlargement of Mariotte blind spot and left mild central scotoma. Fundus examination showed mild hyperemic disks bilaterally (Figure 2). Fluorescein angiography revealed hyperfluorescence of both optic disks but vasculitis was not found (Figure 2). Magnetic resonance imaging findings were negative, thus eliminating optic neuritis.
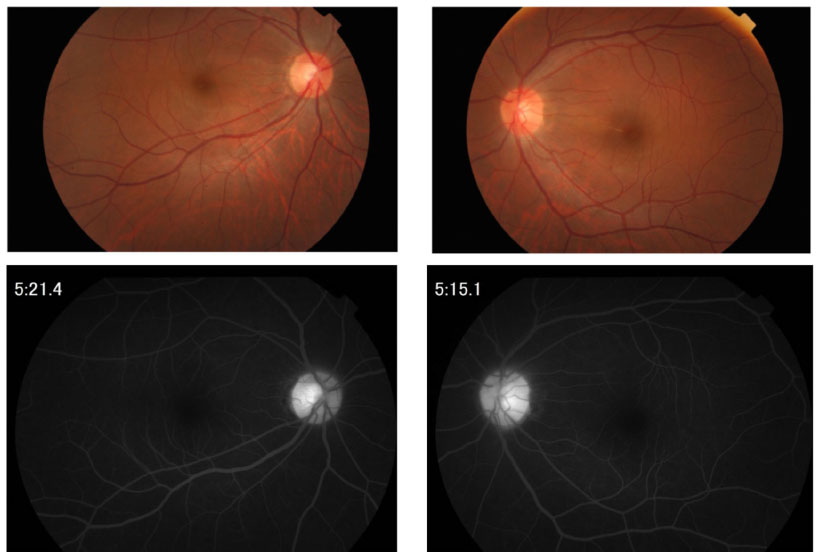 | Figure 2 Fundus photograph (upper) and fluorescein angiogram (lower) at the time of relapsing scleritis in Case 1. |
The presence of chondritis of the auricles, ocular inflammation (scleritis), and neurosensory hearing loss combined with the histologic findings led to the final diagnosis of RP. Systemic examinations showed no abnormal signs in the respiratory tract, heart, and kidney. The oral prednisolone was increased to 30 mg/day beginning in February 2009, and after a slow tapering, the oral prednisolone treatment was completed in July 2012. The scleritis was improved, and the visual acuities were 1.2 OU at the last visit in September 2014.
Case 2
A 71-year-old man had age-related macular degeneration and had been treated by an intravitreal injection of antivascular endothelial growth factor antibody in another hospital. Three months later (November 2013), he noticed that he was deaf in both ears. At the same time, he developed bilateral scleritis, which was improved by topical steroids. On January 2014, he had a reddish swelling on the left eyelid and was referred to the Chiba University Hospital. He had undergone cataract surgeries on both eyes in 2011 and Nd-YAG laser in 2012.
At the first visit, his visual acuities were 1.0 (OD) and 0.03 (OS). The intraocular pressures were 7 mmHg on the right and 17 mmHg on the left. The left eyelid was reddish and swollen with pain (Figure 3). The left conjunctiva was hyperemic, but no inflammation was found in both anterior chambers (Figure 3). Fundus examinations found late-phase age-related macular degeneration in the left eye. The right auricle was reddish and swollen (Figure 3).
 | Figure 3 Appearance of Case 2 at the first visit. |
Computed tomography showed no sinusitis, but the fat density on the upper surface of the eye was increased (Figure 4). Blood tests revealed an elevation of white blood cells (11,100/μL), the erythrocyte sedimentation rate (69 mm/h), and the CRP to 8.7 mg/dL. He was diagnosed with left orbital cellulitis and treated with intravenous antibiotics. The left orbital cellulitis rapidly improved but he developed right scleritis and left gonitis (Figure 5). A second blood test showed an elevation of erythrocyte sedimentation rate and CRP and positive antinuclear antibodies. The anti-type II collagen antibody was negative. Topical antibiotics and steroids were started. Magnetic resonance imaging showed bilateral posterior scleritis and right auricular perichondritis (Figure 5). Because he had auricular perichondritis and cochleovestibular disorders on January 30, 2014, an auricular biopsy was performed. An infiltration of lymphocytes into the periauricular tissue was found histologically (Figure 6).
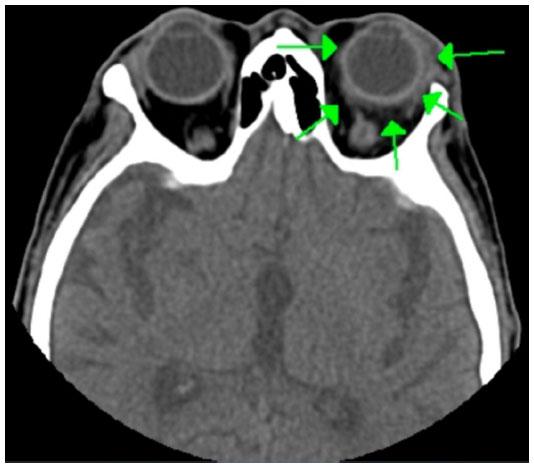 | Figure 4 Computed tomographic image showing the presence of orbital cellulitis. |
 | Figure 5 Ocular findings after scleritis. |
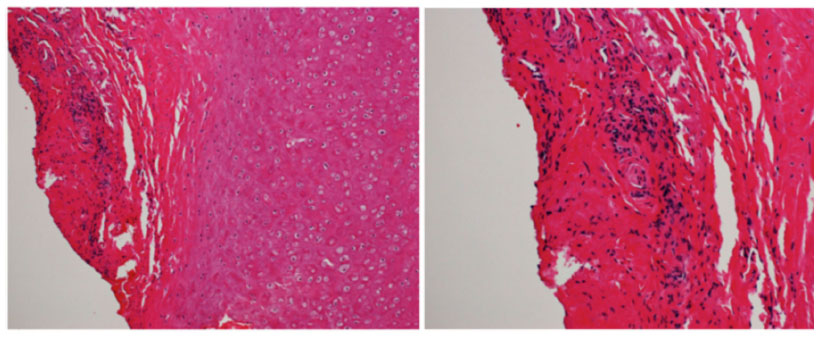 | Figure 6 Histologic findings of auricular biopsy specimen. |
From these findings, the patient was diagnosed with RP. Systemic examinations revealed no abnormalities of the respiratory tract, heart, and kidney, but he had the myelodysplastic syndrome. Blood test showed that anti-type II collagen antibody was negative. Approximately 40 mg/day oral prednisolone was given by a consulting rheumatologist, and both ocular and auricular inflammation improved. He was continued with 10 mg/day oral prednisolone, and his visual acuity at the last examination in July 2014 remained unchanged.
Discussion
McAdam et al were the first to propose the diagnostic criteria for RP in 1976.1 This criteria based on the clinical signs and symptoms include 1) bilateral auricular chondritis, 2) nonerosive inflammatory polyarthritis, 3) nasal chondritis, 4) ocular inflammation, 5) respiratory tract chondritis, and 6) chochlear and/or vestibular damage, resulting in neurosensory hearing loss, tinnitus, and/or vertigo. Damiani and Levine updated the criteria by adding the histopathologic criteria,2 which included 1) the presence of any three of the McAdam’s criteria, 2) at least one of the McAdam’s criteria confirmed by biopsy, and 3) the presence of chondritis in two or more separate anatomic locations with positive response to steroids or dapsone. Our cases met one or both criteria and both cases were diagnosed with typical RP.
The etiology of RP is still undetermined, but 30% of patients at the acute phase have increased type II collagen autoantibodies in their sera. The effectiveness of steroid therapy indicates that RP is probably an idiopathic autoimmune disease.
Because the ocular findings can be the initial findings of RP, ophthalmologists should know the major ocular findings of this disease. Isaak et al reported that the most common ocular finding is episcleritis (39%) and the second is scleritis (14%).3 Other signs are iritis (9%), retinopathy (9%), muscle paresis (5%), and optic neuritis (5%). Although Case 1 had the typical ocular manifestations, it took >1 year to make the final diagnosis probably because of the wide variety of clinical manifestations. Case 2 had ocular cellulitis, and antibiotics were effective, even though it is rare that ocular cellulitis accompanies RP. However, other clinical symptoms are typical and only 3 months were required to make the final diagnosis in this patient.
A recent case report reported that multiple myeloma developed 8 years after the diagnosis of RP.4 Case 2 also had the myelodysplastic syndrome. Thus, a long-term follow-up is required to monitor the development of hematopoietic malignancies.4
Although the standard treatment of RP is through the use of steroids, refractory cases require immunosuppressive therapies, eg, cyclosporine,5 cyclophosphamide,6 and methotrexate.7 Fortunately, in our cases, oral prednisolone was effective with successful tapering without any side effects.
RP is a life-threatening disease, and the major mortality arises from infection related to immunosuppression therapies or pneumonia accompanied by laryngotracheal obstruction.8 Thus, long-term steroid therapy is needed to lessen the possibility of fatal adverse effects in patients with RP.
Conclusion
RP is rare, but a life-threatening disease. Although ocular and clinical manifestations are not specific, ophthalmologists should consider RP in patients with both ocular and auricular inflammation.
Acknowledgments
The authors thank Prof Duco Hamasaki for editing the manuscript and Dr Matsushima for supplying histopathologic pictures. This case series was supported by the grant from The Eye Research Foundation for the Aged and the Grant-in Aid from the Ministry of Education, Science, Sports, and Culture of the Japanese Government and Charitable Trust Fund for Ophthalmic Research in Commemoration of Santen Pharmaceutical’s Founder.
Author contributions
All authors were involved with the patients’ care, the clinical data collection, and the analysis. All authors contributed toward drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
McAdam LP, O’Hanlan MA, Bluestone R, Pearson CM. Relapsing polychondritis: prospective study of 23 patients and a review of the literature. Medicine. 1976;55(3):193–215. | |
Damiani JM, Levine HL. Relapsing polychondritis – report of ten cases. Laryngoscope. 1979;89(6 pt1):929–946. | |
Isaak BL, Liesegang TJ, Michet CJ Jr. Ocular and systemic findings in relapsing polychondritis. Ophthalmology. 1986;93(5):681–689. | |
Teo L, Choo CT. Orbital inflammatory disease in relapsing polychondritis. Orbit. 2014;33(4):298–301. | |
Ormerod AD, Clark LJ. Relapsing polychondritis – treatment with cyclosporin A. Br J Dermatol. 1992;127(3):300. | |
Ruhlen JL, Huston KA, Wood WG. Relapsing polychondritis with glomerulonephritis. Improvement with prednisone and cyclophosphamide. JAMA. 1981;245(8):847–848. | |
Yamaoka K, Saito K, Hanami K, et al. A case of life-threatening refractory polychondritis successfully treated with combined intensive immunosuppressive therapy with methotrexate. Mod Rheumatol. 2007; 17(2):144–147. | |
Michet CJ Jr, McKenna CH, Luthra HS, O’Fallon WM. Relapsing polychondritis. Survival and predictive role of early disease manifestations. Ann Intern Med. 1986;104(1):74–78. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.