Back to Archived Journals » Reports in Organic Chemistry » Volume 5
Regulation of protein–protein interactions using stapled peptides
Authors Robertson N, Jamieson A
Received 17 March 2015
Accepted for publication 7 May 2015
Published 12 August 2015 Volume 2015:5 Pages 65—74
DOI https://doi.org/10.2147/ROC.S68161
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Sean Kerwin
Naomi S Robertson, Andrew G Jamieson
Centre for Chemical Biology, Department of Chemistry, University of Leicester, Leicester, UK
Abstract: The targeting of protein–protein interactions (PPIs) that include secondary structure motifs such as the α-helix and ß-sheet is a challenge in chemical biology. A new class of compounds called stapled peptides have been developed that mimic α-helix secondary structures involved in PPIs. These conformationally constrained peptides also show favorable physicochemical properties and so are promising lead compounds for drug discovery. This review focuses on the design aspects of hydrocarbon constrained α-helical proteomimetics and provides examples in which they target biologically relevant PPIs.
Keywords: protein–protein interactions, stapled helix, proteomimetics, peptide
Introduction
Protein–protein interactions (PPIs) are becoming increasingly popular drug targets because the chemical space required for inhibition is underexplored and so the intellectual property landscape is relatively barren.
However, the development of novel, cell-permeable molecules that regulate PPIs remains one of the major challenges in chemical biology due to the relatively large (typically ~1600 Å2) and dynamic nature of the PPI interface. While these molecular recognition events involve interactions over large surface areas, the majority of the binding affinity and specificity has been found to originate from constellations of a few amino acid residues that are described as interaction “hotspots”.1 At a molecular level, these constellations of residues are frequently found to be specific protein secondary structures such as the α-helix or -strand. Although these motifs are usually thermodynamically favored in folded proteins, isolated peptides typically lack the ability to spontaneously adopt these bioactive conformations. Peptides also have a number of other therapeutically undesirable properties including poor bioavailability, limited stability toward peptidase proteolysis, and lack of membrane permeability that can potentially limit their use as drugs.
A challenge therefore exists to design molecules that occupy the chemical space large enough to modulate PPIs and yet small enough to have suitable drug-like physicochemical properties. Recent research efforts have focused on the design and synthesis of proteomimetics, molecules that mimic both the structure and/or function of proteins. Many different strategies have been used to interrogate PPIs and these have previously been reviewed.2,3 Proteomimetics, and stapled peptides in particular, are proving useful for targeting PPIs.4,5 The main focus of this review is the design, synthesis, and application of stapled peptides as molecular probes to disrupt PPIs.
Strategies for targeting PPIs
Many different strategies for targeting PPIs have been studied, including the use of small molecule peptidomimetic inhibitors, which include ABT-7376 and ABT-2637 from Abbott Laboratories, that were found to be inhibitors of the Bcl-xL/Bak PPI (Figure 1). Additionally, the Nutlin8 and benzodiazepinedione9 families of compounds, developed by Hoffman-La Roche and Johnson & Johnson pharmaceuticals, respectively, were found to be inhibitors of the p53/mDM2 PPI. As well as small molecules, nonpeptidic scaffolds can be used to target PPIs such as the terphenyl scaffolds first developed by the Hamilton group.10 Hara et al have developed polyproline-type helix peptoids that target the p53/mDM2 PPI.11
 | Figure 1 Structures of small molecule peptidomimetic inhibitors (A) ABT-737, (B) ABT-263 that target the Bcl-xL/Bak PPII, (C) Nutlin-2, and (D) benzodiazepinedione-1 that target the p53/mDM2 PPI. |
A large proportion of the PPIs that have been reported are mediated by α-helices.12 Particular emphasis has therefore been placed on the design of conformational constraints that induce α-helix structure. Examples of α-helix constraints include salt bridges between charged amino acid side chain residues,13,14 lactam bridges,15,16 disulfide bridges,17 hydrogen bond surrogates,18,19 hydrophobic interactions,20 metal ligation,21,22 triazole staples synthesized from alkenyl and azido side chain residues,23 photocontrollable macrocycles,24 introduction of α,α-disubstituted amino acids,25,26 and hydrocarbon staples27 (Figure 2). In addition, short α-helical peptides have been nucleated with the help of capping groups28–30 and using designed molecules to template the helix.31,32
 | Figure 2 Strategies to constrain α-helical peptides. |
Proteomimetic compounds of this type demonstrate a number of attractive physicochemical properties in comparison with small molecules, peptides, and biologics. For example, improved binding affinity is observed by overcoming the entropic penalty associated with peptide folding. Good target specificity relative to small molecules and comparable to biologics is achieved due to maintaining the original amino acid residues, with the correct orientation and spacing, to facilitate the molecular recognition event. Conformational constraints also prevent the required extended sheet conformation for recognition by peptidases and so proteomimetics generally have a significantly longer half-life in blood serum.
Of these techniques, hydrocarbon stapling is coming of age as a method to develop peptidomimetic drugs that regulate PPIs and so will be the main focus of this article.
Hydrocarbon peptide stapling
The first “stapled” peptide was reported by Miller et al33,34 and Blackwell et al35 in 1998. The effects of using ring closing metathesis (RCM) on peptide antibiotics containing two alkenyl side chains and rich in α-aminoisobutyric acid (Aib) residues are known to constrain 310-helices through the Thorpe–Ingold effect.25,36,37
More recently, Korsmeyer, Verdine, and Walensky have developed the rules for effective peptide stapling,27,38 thus making the technology transferable to other PPIs. The social and economic value of this approach became apparent in 2005 with the foundation of Aileron Therapeutics, Inc. to develop and commercialize a drug-discovery pipeline based on stapled peptides.39 In 2013, the company announced the successful completion of the first-in-human clinical trial of a stapled peptide drug, ALRN-5281 for the treatment of rare endocrine diseases such as adult growth hormone deficiency and HIV lipodystrophy. The main outcome of the trial showed ALRN-5281 had no incidence of adverse events in healthy subjects.40 Additionally, Aileron Therapeutics is also developing a new stapled peptide drug, ATSP-7041, that targets intracellular PPIs. The i, i+7 hydrocarbon staple feature of ATSP-7041 increases its helicity from 11% in its linear form to 70% helical in the stapled form at pH 7.0.41 The study showed that ATSP-7041 is a potent dual inhibitor of MDM2 and MDMX, which are proteins overexpressed in cancers and deactivate p53 function. Thus, ATSP-7041 can reactivate the p53 pathway, including the inducement of cell-cycle arrest and apoptosis.41
Since the early works of Korsmeyer, Verdine, and Walensky, many other PPIs have been studied using stapled peptides. These include cancer targets such as p53,42 MCL-1 BH3,43 and PUMA BH344 and other therapeutic targets ranging from infectious diseases45,46 to metabolism.47,48 Obtaining crystal structures of stapled peptides bound to their targets is not trivial. In fact, there are less than 15 stapled peptide crystal structures available in the literature including an estrogen receptor beta binding stapled peptide48 and a MCL-1 BH3 stapled peptide43 (Figure 3). Interestingly, in these solid-state structures, the hydrocarbon staple functionality appears to form hydrophobic interactions with the surface of the target proteins and has been proposed to provide additional binding affinity.43,47 Recent work by the Tate group has also revealed the crystal structures of stapled and hydrogen bond surrogate α-helical peptides in a fully buried binding site.49 The fully buried stapled peptides present polar and hydrophobic functionalities on all sides of the peptide helix. These results, along with crystallographic data from Chang et al where stapled peptide ATSP-7041 was bound to MDMX,41 suggest the hydrophobic hydrocarbon staple can help in target binding and not just act as a nonparticipating constraint.
 | Figure 3 Crystal structures of (A) a BH3 stapled peptide (blue) bound to MCL-1 (gray) (PDB: 3MK8) and (B) a nuclear receptor helix stapled peptide (red) bound to estrogen receptor beta (gray) (PDB: 2YJD). |
Stapled peptides have been used extensively to develop potential anticancer drugs through the regulation of PPIs involving the BCL-2 family of proteins implicated in cell apoptosis. Stapled peptide mimics of BH3 domain segments showed α-helical conformation when compared to the native sequences.38 It was also found that stapling of the BH3 peptides gave protease resistance both in vitro and in vivo in comparison to the unmodified peptides.38 This phenomenon is accounted for by the fact that proteases require peptides to be in an extended conformation in order to access the peptide backbone and hydrolyze the amide bond. However, by incorporation of a staple, the peptide is forced into the α-helical conformation and so the amide backbone is inaccessible.
Ring closing metathesis
Hydrocarbon stapling involves joining two alkenyl side chains to form a macrocycle using ruthenium-catalyzed RCM as the key step in the synthesis. On-resin peptide RCM was first carried out by Miller et al in 1996.34 Due to their good functional group tolerance, ruthenium-based catalysts required for RCM are particularly well-suited for use on peptide substrates.
Miller et al’s initial work involved the synthesis of cyclic peptides through incorporation of allyl groups onto the side chain of serine residues, followed by RCM to create peptides of varying ring sizes.34 Following this, the efficient cross-linking of an Aib-containing heptapeptide was achieved by replacing two alanine residues at i and i+4 positions with unbranched O-allyl serine or homoserine residues. The two terminal alkene moieties on the peptide were then treated with Grubbs’ first generation catalyst to afford the macrocyclic peptide in high yield.35 Catalytic hydrogenation then provided peptides with a 310-helix structure as determined by circular dichroism (CD) spectroscopy. The ease of RCM on these peptides has been attributed to the preorganization of a 310-helix on solid support facilitated by the Aib residues.
Structural requirements for stapling
The combined use of α,α-disubstituted alkenyl amino acid residues and RCM to produce all-hydrocarbon stapled peptides was first conceived by Schafmeister et al in 2000.27 Initial work investigated the structural requirements needed for effective peptide stapling, including the length of the hydrocarbon bridge and the α-carbon stereochemistry of the α,α-disubstituted unnatural alkenyl amino acids (Figure 4).
 | Figure 4 Representation of three types of stapled peptide constraints: (A) i, i+3; (B) i, i+4; (C) i, i+7; indicating the stereochemistry required for the stapled amino acids and the multiple stapling techniques used to constrain longer helices; (D) double staple; and (E) stitched staple. |
A strategically useful feature of peptide stapling is that the hydrocarbon constraint can be incorporated at different positions along the peptide sequence. The most common types of peptide staple form a side chain–side chain bridge between the i, i+3, i, i+4 or i, i+7 residues. With shorter peptides a constraint across one turn of the helix (ie, i, i+3 or an i, i+4 staple) is employed. The i, i+3 type staple requires one R- and one S-pentenyl alanine (Figure 4A), whereas two S-pentenyl alanine residues are required for an i, i+4 type staple (Figure 4B). An i, i+7 staple provides a hydrocarbon bridge spanning two turns of the helix and requires a S-pentenyl alanine and an R-octenyl alanine (Figure 4C). Again, the stereochemistry of these amino acids is very important to develop an effective constraint.
Staple scanning
When limited structural information is available regarding a specific PPI, a staple scan can be used to determine the most effective position for the hydrocarbon constraint. This involves positioning an appropriate staple sequentially along the length of the peptide sequence. Staple scanning echoes the alanine scanning technique by also helping to determine if a residue is required for binding affinity.50
This strategy has been used to identify a selective inhibitor of MCL-1, a human cancer resistance factor.43 Stewart et al found that the peptide, which had the highest α-helical character, as shown by CD, also had the highest binding affinity for MCL-1.43
An extension of the peptide stapling approach for longer helical peptide sequences involves the incorporation of multiple hydrocarbon bridges. These can take the form of multiple individual staples (Figure 4D) or “stitched” stapled peptides incorporating two alkenyl chains on one α-carbon of the central amino acid (Figure 4E).51–54 One staple constraint has been shown to be ineffective for longer peptides; however, two staple constraints can significantly increase the helicity.54 Stitched α-helical peptides can be thought to be the next generation in terms of hydrocarbon stapling, creating spiro-macrocycles, which are known to be stabilizing. Stitched peptides have been shown to give increased helicity, protease resistance, and cell permeability compared to the monostapled peptides.54
The size of the macrocycle formed upon RCM is important to achieve an increased degree of helicity in the constrained α-helical peptide. Different ring sizes are achieved depending on the length of the amino acid side chains used for the RCM. The macrocycle formed upon RCM of an i−i+4 stapled peptide consists of 21 atoms, eight of which form the hydrocarbon constraint. This macrocycle is large in size and would generally mean that it would be problematic to ring close. However, the RCM reaction on these peptide substrates is relatively straightforward. This is due, in part, to the α-methyl-substituent of the alkenyl amino acid inducing the Thorpe–Ingold effect55,56 on the peptide backbone, as well as the presence of a hydrophobic environment as a result of using nonpolar solvents, which promotes helix formation.57
Peptides incorporating the α,α-disubstituted amino acid Aib were first described by Karle and Balaram and were shown to adopt 310-helix structures.25 The α-carbon dimethyl substitution of Aib restricts the torsion angles of the peptide bonds to φ=−49° and ψ=−26° in comparison to that of α-helix torsion angles, φ=−57° and ψ =−47°.58 As a result of these bond angles, the hydrogen bonding network of the two types of helices differ (Figure 5). The α,α-disubstituted alkenyl amino acids required for peptide stapling also appear to provide this effect, resulting in the peptide substrates being folded ready to undergo RCM.
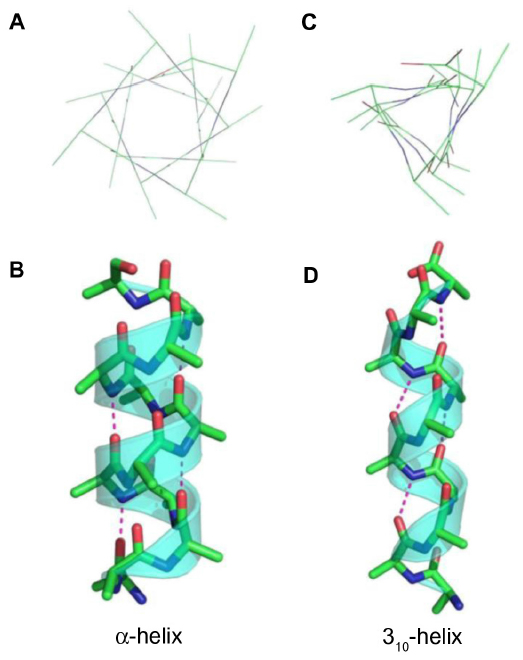 | Figure 5 Representations of the (A) top view, (B) side view of an α-helix, (C) top view, and (D) side view of a 310-helix, showing the hydrogen bonding pattern (pink dotted lines) of the two types of helices. |
Monosubstituted hydrocarbon staples have been successfully synthesized using RCM on some peptide sequences.43,59 However, it is preferable to incorporate an α,α-disubstituted amino acid, or some other helicity inducing substituent into the peptide to help promote the helical conformation and facilitate the RCM reaction. Although introduction of a hydrocarbon staple can improve the therapeutic properties of certain peptides,38,43,52,60 it has been found that the increased degree of helicity obtained through the incorporation of a hydrocarbon staple does not necessarily correlate to an increase in its binding affinity.61 This is partly because of the need for some flexibility to allow induced fit binding of some peptides to their binding partners.
Asymmetric α,α-disubstituted amino acid synthesis
The α,α-disubstituted alkenyl amino acids required for peptide stapling are commercially available, although they are expensive relative to standard fluorenylmethyloxycarbonyl (Fmoc) protected amino acids.
Asymmetric synthesis of the required alkenyl amino acids has been reported using Williams’ glycine enolate alkylation method and alkylation of Belokon’s nickel (II) Schiff base complex.62–65 The Belokon method has come to the fore as the most effective strategy due to the harsh conditions with the potential for over reduction of the alkene moiety using the Williams method. Aillard et al recently reported a robust and efficient asymmetric synthesis of unnatural alkenyl amino acids. A fluorine-modified NiII Schiff base complex gave improved diastereoselectivity of the key alkylation reaction (>95: 5 dr).66 This method now gives access to the required nonnative amino acids (Figure 6).
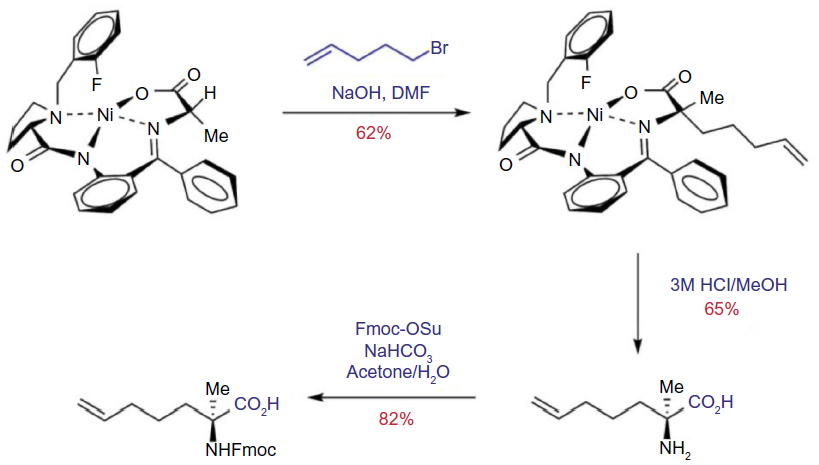 | Figure 6 Asymmetric synthesis of Fmoc-S5-OH via fluorine-modified nickel (II) Schiff base complex. |
Design of the macrocycle constraint
Following the initial studies on peptide stapling by Schafmeister et al,27 recent researches have focused on attempting to improve both the placement of the alkene in the staple and the lengths of the all-hydrocarbon staple. Notably, Pham et al have studied the placement of the alkene within the stapled macrocycle. A comparison of alkene position within the i, i+4 type stapled peptide was conducted using oct-2-enyl, oct-6-enyl, and the more usual oct-4-enyl macrocyclic constraint.57
Linear peptides incorporating a (S)-α-methyl, α-allylglycine (S3) at the i position and (S)-α-methyl, α-heptenylglycine (S7) at the i+4 position provided access to both the oct-2-enyl staple (S3–S7) (Figure 7C) and oct-6-enyl staple (S7–S3) (Figure 7E). Both these peptides failed to undergo RCM to provide the corresponding macrocyclic peptides at room temperature. In fact, RCM on the S3–S7 analog only proceeded at an elevated temperature of 60°C, compared to the original oct-4-enyl staples (S5–S5) (Figure 7D), which proceed at room temperature. RCM of the S7–S3 substrate was very poor, even at 60°C and resulted in decomposition of the substrate peptide at the higher temperatures. The S3–S7 analog gave improved helicity in comparison to the native sequence; however, it still did not match the helicity shown by the S5–S5 stapled peptide.57 These results demonstrate that the original oct-4-enyl i, i+4 type staple contains the alkene functionality in the optimal position on the hydrocarbon chain to provide an effective constraint. The i, i+3 all-hydrocarbon staple has also been studied to determine the optimal length of hydrocarbon bridge across one turn of a helix. In general, for an effective i, i+3 staple an S5 amino acid and its enantiomer, an R5 amino acid is required. However, Shim et al have studied the possibility of contracting the all-hydrocarbon bridge by removing two methylene units.67
 | Figure 7 Structures of (A) the three S-alkenyl amino acids: S3, S5, and S7; (B) the two R-alkenyl amino acids: R3 and R5; and the representations of the different positions possible for the staple in an i, i+4 staple; (C) oct-2-enyl; (D) oct-4-enyl; (E) oct-6-enyl; and in an i, i+3 staple; (F) hex-4-enyl; and (G) hex-2-enyl. |
Unsymmetrical contraction of the bridge was studied in order to analyze the differing positions of the alkene within the staple. Most of the linear peptide substrates failed to undergo RCM to give the corresponding macrocyclic peptide. However, two of the peptides did undergo metathesis, both of which had an R-configuration amino acid (R3 or R5) at the i position and an S-configuration amino acid (S5 or S3) at the i+3 position (Figure 7F and G). However, the difference in the position of the staple within the macrocycle led to a pronounced difference in the α-helicity of the peptides, with the hex-4-enyl staple (Figure 7F) being much more helical (55%) than the hex-2-enyl staple (Figure 7G), which was 15% helical.67
Shim et al have investigated the ruthenium catalysts required form RCM including the Grubbs first generation, Grubbs second generation, and Hoveyda–Grubbs catalysts. RCM was performed at both 25°C and 65°C using the three different catalysts. The results showed that Grubbs first generation catalyst was the most efficient at both temperatures, but at elevated temperatures Grubbs second generation and Hoveyda–Grubbs catalysts have a significant increase in efficiency.67
Proposed mechanism of membrane permeability
A notable physicochemical property of stapled peptides is their ability to potentially promote cell penetration as a result of the hydrophobic hydrocarbon staple. Evidence has recently been reported for stapled peptides traversing the membrane and entering Jurkat leukemia cells38 and human U2OS osteosarcoma cells68 through a clathrin- and caveolin-independent, ATP-dependent pinocytotic mechanism.5,38,42,68 Accumulation of the stapled peptides in ATP-depleted cells was reduced compared to normal cells. In addition, a significant reduction in the uptake of the stapled peptides into cells was observed after treatment of the cells with sodium chlorate, which blocks the sulfated proteoglycans presented on the cell surface. To confirm this finding, the researchers also treated proteoglycan deficient cells with the stapled peptides and found a reduction in cell uptake.68 It has also been reported that cellular uptake can be impeded by the presence of serum for some peptides,5 which could be as a result of competition with natural serum containing substrates for pinocytosis.69
Current and future directions
As with many new techniques, stapled peptides are surrounded by controversies, notably the recent works of two groups of researchers who contradict each other, and thus have initiated a debate on the effectiveness of peptide stapling in drug discovery. In 2012, LaBelle et al reported a stapled BIM peptide based on the BIM BH3 helix to target the BCL-2 family of proteins.70 The stapled peptide, BIM SAHBA, incorporated an i, i+4 all-hydrocarbon crosslink and was found to have increased binding in comparison to the native BIM BH3 helix. In addition, they found BIM SAHBA-induced cell apoptosis in leukemia by modulating the BCL-2 pathway.
Later that same year, Okamoto et al also reported a stapled peptide very similar to that of the previous study.71 However, in their hands, the stapling of the peptide did not enhance its affinity or biological activity. The BIM BH3 all-hydrocarbon stapled peptide did not in fact induce apoptosis much more than the native sequence and was also not inherently cell permeable.71 In addition, a lactam-stapled peptide based on the same helical sequence was found to have a greater percentage of helicity than the all-hydrocarbon staple.
Following this report, a rebuttal was published by Bird et al with the explanation that different lengths of BIM SAHBA were synthesized for specific studies.72 A weaker binding BIM SAHBA (145–164) stapled peptide with moderate helicity compared to the BIM SAHBA (146–166) peptide was used in the in vivo and nuclear magnetic resonance experiments to increase the peptide’s solubility and weaken its activation of BAX. BIM SAHBA (146–166) was three residues longer, had higher helicity and higher binding affinity than Bim SAHBA (145–164).
Okamoto et al subsequently reported data showing unstapled BIM SAHB peptides (containing two pentenylalanine residues at i and i+4 positions, but have not been subjected to RCM) were as active as the stapled peptides toward two particular cancer cell lines.73 Thus, it was concluded that the stapling was not responsible for the enhanced effect on activity, but rather, by altering the sequence of the peptides, the innate characteristics of that peptide are also altered.
The discrepancies in these studies demonstrate the subtleties in designing effectively constrained stapled peptides. Systematic rules for effective design are therefore urgently required and will be one of the main challenges for the community to address in coming years.
Recent peptide stapling techniques have returned to the original idea of using O-allyl homoserine residues, such as the stapled peptides used as inhibitors of the vitamin D receptor–coactivator interaction.74 In addition, the use of all- hydrocarbon stapling is still prevalent, with many more targets being studied. PPIs involving membrane-bound proteins have also been studied, such as the transmembrane (TM) helices of small multidrug resistant proteins. These TM helices dimerize and oligomerize, forming PPIs, to extrude relatively large toxic substrates through channels created in the membrane, giving the resistant nature of these small multidrug resistant proteins.75 Stapled peptide analogs of TM4 reduced the degradation of the peptides in blood plasma compared to a linear sequence. In addition, the stapled peptide also allowed the disruption of the dimerization domain between two TM4 helices, thus preventing efflux of the toxic substrates.75
Conclusion
Although still in its infancy, there is a bright future for stapled peptides. Current successes from stapled peptide clinical trials by Aileron Therapeutics are driving enthusiasm for the strategy in biomimetic drug development in the area of PPIs. In addition, stapled peptides are being used extensively as molecular probes in chemical biology to facilitate understanding of important biological mechanisms. In time, these examples are likely to lead to new drug discover efforts based on stapled peptides as lead compounds.
Future challenges include determining how stapled peptides penetrate the membrane, and establishing where they are located after entering the cell. A systematic set of rules are also required that outline the most effective position to locate a staple within a peptide sequence. The role, if any, of hydrocarbon staples on binding affinity has yet to be determined, and a study focusing on nonspecific binding of stapled peptides is therefore required.
Acknowledgments
The authors thank the Engineering and Physical Sciences Research Council (EPSRC) (EP/L018152/1) and University of Leicester for financial support.
Disclosure
The authors report no conflicts of interest in this work.
References
Keskin O, Ma B, Nussinov R. Hot regions in protein–protein interactions: the organization and contribution of structurally conserved hot spot residues. J Mol Biol. 2005;345(5):1281–1294. | |
Wilson AJ. Inhibition of protein-protein interactions using designed molecules. Chem Soc Rev. 2009;38(12):3289–3300. | |
Azzarito V, Long K, Murphy NS, Wilson AJ. Inhibition of α-helix-mediated protein-protein interactions using designed molecules. Nat Chem. 2013;5(3):161–173. | |
Verdine GL, Hilinski GJ. Stapled peptides for intracellular drug targets. Methods Enzymol. 2012;503:3–33. | |
Walensky LD, Bird GH. Hydrocarbon-stapled peptides: principles, practice, and progress. J Med Chem. 2014;57(15):6275–6288. | |
Lee EF, Czabotar PE, Smith BJ, et al. Crystal structure of ABT-737 complexed with Bcl-xL: implications for selectivity of antagonists of the Bcl-2 family. Cell Death Differ. 2007;14(9):1711–1713. | |
Tse C, Shoemaker AR, Adickes J, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68(9):3421–3428. | |
Vassilev LT, Vu BT, Graves B, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004; 303(5659):844–848. | |
Grasberger BL, Lu T, Schubert C, et al. Discovery and cocrystal structure of benzodiazepinedione HDM2 antagonists that activate p53 in cells. J Med Chem. 2005;48(4):909–912. | |
Orner BP, Ernst JT, Hamilton AD. Toward proteomimetics: terphenyl derivatives as structural and functional mimics of extended regions of an α-helix. J Am Chem Soc. 2001;123(22):5382–5383. | |
Hara T, Durell SR, Myers MC, Appella DH. Probing the structural requirements of peptoids that inhibit HDM2-p53 interactions. J Am Chem Soc. 2006;128(6):1995–2004. | |
Bullock BN, Jochim AL, Arora PS. Assessing helical protein interfaces for inhibitor design. J Am Chem Soc. 2011;133(36):14220–14223. | |
Marqusee S, Baldwin RL. Helix stabilization by Glu-…Lys+ salt bridges in short peptides of de novo design. Proc Natl Acad Sci U S A. 1987; 84(24):8898–8902. | |
Scholtz JM, Qian H, Robbins VH, Baldwin RL. The energetics of ion-pair and hydrogen-bonding interactions in a helical peptide. Biochemistry. 1993;32(37):9668–9676. | |
Chorev M, Roubini E, McKee RL, et al. Cyclic parathyroid hormone-related protein antagonists: lysine 13 to aspartic acid 17 [i to (i + 4)] side chain to side chain lactamization. Biochemistry. 1991;30(24):5968–5974. | |
Phelan JC, Skelton NJ, Braisted AC, McDowell RS. A general method for constraining short peptides to an α-helical conformation. J Am Chem Soc. 1997;119(3):455–460. | |
Jackson DY, King DS, Chmielewski J, Singh S, Schultz PG. General approach to the synthesis of short α-helical peptides. J Am Chem Soc. 1991;113(24):9391–9392. | |
Chapman RN, Dimartino G, Arora PS. A highly stable short alpha-helix constrained by a main-chain hydrogen-bond surrogate. J Am Chem Soc. 2004;126(39):12252–12253. | |
Patgiri A, Jochim AL, Arora PS. A hydrogen bond surrogate approach for stabilization of short peptide sequences in alpha-helical conformation. Acc Chem Res. 2008;41(10):1289–1300. | |
Albert JS, Hamilton AD. Stabilization of helical domains in short peptides using hydrophobic interactions. Biochemistry. 1995;34(3):984–990. | |
Ruan F, Chen Y, Hopkins PB. Metal ion-enhanced helicity in synthetic peptides containing unnatural, metal-ligating residues. J Am Chem Soc. 1990;112(25):9403–9404. | |
Ghadiri MR, Fernholz AK. Peptide architecture. Design of stable α-helical metallopeptides via a novel exchange-inert ruthenium(III) complex. J Am Chem Soc. 1990;112(26):9633–9635. | |
Kawamoto SA, Coleska A, Ran X, Yang C-Y, Wang S. Design of triazole-stapled BCL9 α-helical peptides to target the β-catenin/B-cell CLL/lymphoma 9 (BCL9) protein–protein interaction. J Med Chem. 2012;55(3):1137–1146. | |
Kneissl S, Loveridge EJ, Williams C, Crump MP, Allemann RK. Photocontrollable peptide-based switches target the anti-apoptotic protein Bcl-xL. Chembiochem. 2008;9(18):3046–3054. | |
Karle IL, Balaram P. Structural characteristics of alpha.-helical peptide molecules containing Aib residues. Biochemistry. 1990;29(29):6747–6756. | |
Balaram P. Non-standard amino acids in peptide design and protein engineering. Curr Opin Struct Biol. 1992;2(6):845–851. | |
Schafmeister CE, Po J, Verdine GL. An all-hydrocarbon cross-linking system for enhancing the helicity and metabolic stability of peptides. J Am Chem Soc. 2000;122(24):5891–5892. | |
Forood B, Feliciano EJ, Nambiar KP. Stabilization of alpha-helical structures in short peptides via end capping. Proc Natl Acad Sci U S A. 1993;90(3):838–842. | |
Zhou HX, Lyu PC, Wemmer DE, Kallenbach NR. Structure of a C-terminal.alpha.-helix cap in a synthetic peptide. J Am Chem Soc. 1994;116(3):1139–1140. | |
Yang J, Zhao K, Gong Y, Vologodskii A, Kallenbach NR. α-Helix nucleation constant in copolypeptides of alanine and ornithine or lysine. J Am Chem Soc. 1998;120(41):10646–10652. | |
Kemp DS, Curran TP, Boyd JG, Allen TJ. Studies of N-terminal templates for alpha.-helix formation. Synthesis and conformational analysis of peptide conjugates of (2S,5S,8S,11S)-1-acetyl-1,4-diaza-3-keto-5-carboxy-10-thiatricyclo[2.8.1.04,8]tridecane (Ac-Hel1-OH). J Org Chem. 1991;56(23):6683–6697. | |
Kemp DS, Curran TP, Davis WM, Boyd JG, Muendel C. Studies of N-terminal templates for alpha-helix formation. Synthesis and conformational analysis of (2S,5S,8S,11S)-1-acetyl-1,4-diaza-3-keto-5-carboxy-10-thiatricyclo[2.8.1.04,8]tridecane (Ac-Hel1-OH). J Org Chem. 1991;56(23):6672–6682. | |
Miller SJ, Grubbs RH. Synthesis of conformationally restricted amino acids and peptides employing olefin metathesis. J Am Chem Soc. 1995;117(21):5855–5856. | |
Miller SJ, Blackwell HE, Grubbs RH. Application of ring-closing metathesis to the synthesis of rigidified amino acids and peptides. J Am Chem Soc. 1996;118(40):9606–9614. | |
Blackwell HE, Grubbs RH. Highly efficient synthesis of covalently cross-linked peptide helices by ring-closing metathesis. Angew Chemie Int Ed. 1998;37(23):3281–3284. | |
Toniolo C, Benedetti E. Structures of polypeptides from α-amino acids disubstituted at the α-carbon. Macromolecules. 1991;24(14):4004–4009. | |
Demizu Y, Doi M, Kurihara M, et al. Conformational studies on peptides containing α,α-disubstituted α-amino acids: chiral cyclic α,α-disubstituted α-amino acid as an α-helical inducer. Org Biomol Chem. 2011;9(9):3303–3312. | |
Walensky LD, Kung AL, Escher I, et al. Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science. 2004;305(5689):1466–1470. | |
Sawyer TK. AILERON therapeutics. Chem Biol Drug Des. 2009;73(1):3–6. | |
ClinicalTrials.gov [Internet]. Phase 1 Safety Study of ALRN-5281 in Healthy Subjects. Bethesda, MD: National Library of Medicine (US); 2013. [Identifier NCT01775358, February 29, 2000]. | |
Chang YS, Graves B, Guerlavais V, et al. Stapled α-helical peptide drug development: a potent dual inhibitor of MDM2 and MDMX for p53-dependent cancer therapy. Proc Natl Acad Sci U S A. 2013;110(36):E3445–E3454. | |
Bernal F, Tyler AF, Korsmeyer SJ, Walensky LD, Verdine GL. Reactivation of the p53 tumor suppressor pathway by a stapled p53 peptide. J Am Chem Soc. 2007;129(9):2456–2457. | |
Stewart ML, Fire E, Keating AE, Walensky LD. The MCL-1 BH3 helix is an exclusive MCL-1 inhibitor and apoptosis sensitizer. Nat Chem Biol. 2010;6(8):595–601. | |
Edwards AL, Gavathiotis E, LaBelle JL, et al. Multimodal interaction with BCL-2 family proteins underlies the proapoptotic activity of PUMA BH3. Chem Biol. 2013;20(7):888–902. | |
Long YQ, Huang SX, Zawahir Z, et al. Design of cell-permeable stapled peptides as HIV-1 integrase inhibitors. J Med Chem. 2013;56(13):5601–5612. | |
Chapuis H, Slaninová J, Bednárová L, Monincová L, Buděšínský M, Čeřovský V. Effect of hydrocarbon stapling on the properties of α-helical antimicrobial peptides isolated from the venom of hymenoptera. Amino Acids. 2012;43(5):2047–2058. | |
Phillips C, Roberts LR, Schade M, et al. Design and structure of stapled peptides binding to estrogen receptors. J Am Chem Soc. 2011;133(25):9696–9699. | |
Szlyk B, Braun CR, Ljubicic S, et al. A phospho-BAD BH3 helix activates glucokinase by a mechanism distinct from that of allosteric activators. Nat Struct Mol Biol. 2014;21(1):36–42. | |
Douse CH, Maas SJ, Thomas JC, et al. Crystal structures of stapled and hydrogen bond surrogate peptides targeting a fully buried protein-helix interaction. ACS Chem Biol. 2014;9(10):2204–2209. | |
Jamieson AG, Boutard N, Sabatino D, Lubell WD. Peptide scanning for studying structure-activity relationships in drug discovery. Chem Biol Drug Des. 2013;81(1):148–165. | |
Bird GH, Madani N, Perry AF, et al. Hydrocarbon double-stapling remedies the proteolytic instability of a lengthy peptide therapeutic. Proc Natl Acad Sci U S A. 2010;107(32):14093–14098. | |
Rao T, Ruiz-Gómez G, Hill TA, Hoang HN, Fairlie DP, Mason JM. Truncated and helix-constrained peptides with high affinity and specificity for the cFos coiled-coil of AP-1. PLoS One. 2013;8(3):e59415. | |
Bird GH, Boyapalle S, Wong T, et al. Mucosal delivery of a double-stapled RSV peptide prevents nasopulmonary infection. J Clin Invest. 2014;124(5):2113–2124. | |
Hilinski GJ, Kim YW, Hong J, et al. Stitched α-helical peptides via bis ring-closing metathesis. J Am Chem Soc. 2014;136(35):12314–12322. | |
Beesley RM, Ingold CK, Thorpe JF. The formation and stability of spiro-compounds. Part I. spiro-compounds from cyclohexane. J Chem Soc Trans. 1915;107:1080–1106. | |
Fürstner A, Langemann K. A concise total synthesis of dactylol via ring closing metathesis. J Org Chem. 1996;61(25):8746–8749. | |
Pham TK, Yoo J, Kim Y-W. Comparison of Oct-2-enyl and Oct-4-enyl staples for their formation and α-helix stabilizing effects. Bull Korean Chem Soc. 2013;34(9):2640–2644. | |
Arnott S, Dover SD. Refinement of bond angles of an α-helix. J Mol Biol. 1967;30(1):209–212. | |
Yeo DJ, Warriner SL, Wilson AJ. Monosubstituted alkenyl amino acids for peptide “stapling”. Chem Commun (Camb). 2013;49(80):9131–9133. | |
Danial NN, Walensky LD, Zhang CY, et al. Dual role of proapoptotic BAD in insulin secretion and beta cell survival. Nat Med. 2008;14(2):144–153. | |
Giordanetto F, Revell JD, Knerr L, et al. Stapled Vasoactive Intestinal Peptide (VIP) derivatives improve VPAC2 agonism and glucose-dependent insulin secretion. ACS Med Chem Lett. 2013;4(12):1163–1168. | |
Williams RM, Sinclair PJ, Zhai D, Chen D. Practical asymmetric syntheses of α-amino acids through carbon-carbon bond constructions on electrophilic glycine templates. J Am Chem Soc. 1988;110(5):1547–1557. | |
Williams RM, Im MN. Asymmetric synthesis of monosubstituted and α,α-disubstituted α-amino acids via diastereoselective glycine enolate alkylations. J Am Chem Soc. 1991;113(24):9276–9286. | |
Belokon YN, Bakhmutov VI, Chernoglazova NI, et al. General method for the asymmetric synthesis of α-amino acids via alkylation of the chiral nickel(II) Schiff base complexes of glycine and alanine. J Chem Soc Perkin Trans 1. 1988;(2):305–312. | |
Belokon YN, Tararov VI, Maleev VI, Savel’eve TF, Ryzhov MG. Improved procedures for the synthesis of (S)-2-[N-(N′-benzylprolyl)amino]benzophenone (BPB) and Ni(II) complexes of Schiff’s bases derived from BPB and amino acids. Tetrahedron Asymmetry. 1998;9(23):4249–4252. | |
Aillard B, Robertson NS, Baldwin AR, Robins S, Jamieson AG. Robust asymmetric synthesis of unnatural alkenyl amino acids for conformationally constrained α-helix peptides. Org Biomol Chem. 2014;12(43):8775–8782. | |
Shim SY, Kim Y-W, Verdine GL. A new i, i + 3 peptide stapling system for α-helix stabilization. Chem Biol Drug Des. 2013;82(6):635–642. | |
Chu Q, Moellering RE, Hilinski GJ, et al. Towards understanding cell penetration by stapled peptides. MedChemCommun. 2014;6(1):111–119. | |
Commisso C, Davidson SM, Soydaner-Azeloglu RG, et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature. 2013;497(7451):633–637. | |
LaBelle JL, Katz SG, Bird GH, et al. A stapled BIM peptide overcomes apoptotic resistance in hematologic cancers. J Clin Invest. 2012;122(6):2018–2031. | |
Okamoto T, Zobel K, Fedorova A, et al. Stabilizing the pro-apoptotic BimBH3 helix (BimSAHB) does not necessarily enhance affinity or biological activity. ACS Chem Biol. 2013;8(2):297–302. | |
Bird GH, Gavathiotis E, Labelle JL, Katz SG, Walensky LD. Distinct BimBH3 (BimSAHB) stapled peptides for structural and cellular studies. ACS Chem Biol. 2014;9(3):831–837. | |
Okamoto T, Segal D, Zobel K, et al. Further insights into the effects of pre-organizing the BimBH3 helix. ACS Chem Biol. 2014;9(3):838–839. | |
Misawa T, Demizu Y, Kawamura M, Yamagata N, Kurihara M. Structural development of stapled short helical peptides as vitamin D receptor (VDR)–coactivator interaction inhibitors. Bioorg Med Chem. 2015;23(5):1055–1061. | |
Bellmann-Sickert K, Stone TA, Poulsen BE, Deber CM. Efflux by small multidrug resistance proteins is inhibited by membrane-interactive helix-stapled peptides. J Biol Chem. 2014;290(3):1752–1759. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.