Back to Archived Journals » Research and Reports in Biology » Volume 6
Recent insights into the pathophysiology of mTOR pathway dysregulation
Authors Gitto S, Altomare D
Received 25 October 2014
Accepted for publication 11 December 2014
Published 29 January 2015 Volume 2015:6 Pages 1—16
DOI https://doi.org/10.2147/RRB.S57088
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Zvi Kelman
Sarah B Gitto, Deborah A Altomare
Burnett School of Biomedical Sciences, College of Medicine, University of Central Florida, Orlando, FL, USA
Abstract: Mechanistic target of rapamycin (mTOR) dysregulation is present in a variety of human pathologies including neurological disease, cancer, diabetes, and cardiac disease. Hyperactivation leads to increased protein synthesis and cell growth, which are essential for growth, development, and cancer. Inhibition of mTOR results in induction of autophagy, a cell survival mechanism thought to be deficient in neurodegeneration. Counteracting the balance of mTOR signaling with target specific inhibitors is of interest in pathological conditions where mTOR signaling is upregulated. The US Food and Drug Administration (FDA) has approved the use of rapamycin for treatment of renal cell, pancreatic neuroendocrine, and hormone positive breast cancer. Many clinical trials are underway to determine the efficacy of mTOR inhibitors in other pathologies as monotherapies or combinational therapies with chemotherapeutics, tyrosine kinase inhibitors, molecular targeted therapies, and vascular endothelial growth factor (VEGF) inhibitors. Collectively, this review is an overview of the current practices and outcomes of pharmaceutically targeting this highly studied mediator of normal and aberrant cell function.
Keywords: signal transduction, small molecule inhibitors, translational impact, neurological diseases, cardiovascular disorders, diabetes, cancer
Introduction to mTOR signaling: biological significance, upstream modulators, and downstream effectors
Mechanistic target of rapamycin (mTOR) is amongst the most widely studied complexes due to its fundamental importance in cell biology and cell signaling mechanisms. mTOR is expressed in all cells as it plays a critical role in cell growth, proliferation and migration, and is essential in normal development.1 Dysregulation of mTOR signaling leads to various human pathological conditions including neurological diseases, cancer, diabetes, and cardiovascular complications. To date, numerous studies have been conducted to understand how to counterbalance this dysregulation in order to reverse or impede disease progression.
mTOR is a serine/threonine protein kinase associated in two distinct complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). As shown in Figure 1, the accessory proteins regulatory-associated protein of mTOR (RAPTOR) and rapamycin-insensitive companion of mTOR (RICTOR) define mTORC1 and mTORC2, respectively.2–4 mTORC1 also interacts with a negative regulator of AKT, named 40 kDa proline-rich AKT substrate 1 (AKT1S1 or PRAS40).5 Other proteins that are specific to mTORC2 include: the protein observed with RICTOR (PRR5 or PROTOR) which is involved with complex assembly, and stress-activated mitogen-activated protein kinase-interacting protein 1 (MAPKAP1 or mSIN) which plays a role in targeting mTORC2 to the membrane and is necessary in the phosphorylation of AKT (protein kinase B or PKB).6–8 Proteins common to both mTOR complexes include the negative regulator DEP domain-containing mTOR-interacting protein (DEPTOR) and the positive regulator mammalian lethal with SEC13 protein 8 (mLST8).9,10 A description of mTOR associated proteins and their functions are outlined in Table 1.
 | Figure 1 mTORC1 and mTORC2 domains. |
 | Table 1 mTOR associated proteins and their functions |
Great strides have been made in understanding mTOR signaling by using rapamycin and other pharmacological mTOR inhibitors. Rapamycin binds 12 kDa FK506-Binding Protein 1A (FKBP1A)-binding protein. Rapamycin-FKBP1A complex binds and inhibits RAPTOR-bound mTOR. Although rapamycin is an mTORC1 inhibitor, chronic exposure can inhibit mTORC2.11 These inhibitors have been important for delineating the upstream modulators and downstream effectors in the complex mTOR signaling cascade.
Upstream modulators of mTOR
mTOR is a modulator for protein synthesis, and regulates cell proliferation, cell growth, lipid synthesis, ribosomal biogenesis, and autophagy.12,13 mTORC1 acts as a primary sensor for nutrients, growth factors, oxygen, and energy by monitoring the abundance of resources available to determine if protein synthesis is feasible.
In response to amino acid uptake mTOR localizes to the lysosomal membrane surface where it can interact with Ras-related GTP binding protein B (Rag GTPase).14 Active Rag is recruited and anchored to the lysosomal membrane by the Ragulator complex (consisting of p18, p14, and MEK partner 1 or MP1) to interact with the GTP-bound RHEB (Ras homologue enriched in brain).15 The Ragulator–Rag complex acts as a docking site for mTORC1 to associate with the lysosome membrane. Both RHEB and the Rag–Ragulator complex are essential for the activation of mTOR in response to high levels of amino acids.15,16 When the Ragulator complex is inactive, Rag GTPase is released from the lysosomal surface into the cytoplasm and is no longer able to activate mTORC1.15 Leucine appears to be at the crux of cells amino acid uptake linked to mTOR activation.17
Growth factors are regulators of mTORC1 activity (Figure 2), and insulin signaling has been extensively studied in this context. Insulin binding to its cell surface receptor activates the phosphatidylinositol 3-kinase (PI3K) pathway either directly or indirectly through mediator proteins such as insulin receptor substrate-1 (IRS-1). PI3K phosphorylates phosphatidylinositol 4,5-bisphosphate (PIP2) into phosphatidylinositol 3,4,5-trisphosphate (PIP3). Phosphatase and tensin homologue (PTEN) encodes a phosphatidylinositol-3,4,5-triphosphate 3-phosphatase, which acts as a central negative regulator of PI3K, by acting on its substrate PIP3 to convert it to PIP2, and then further reducing PIP2 to phosphatidylinositol 5-monophosphate.13,18 PIP3 activates 3-phosphoinositide-dependent protein kinase-1 (PDPK1 or PDK1), which in turn activates the downstream effector AKT by phosphorylating threonine 308, while a second protein kinase (generally thought to be mTORC2) phosphorylates AKT Ser473. AKT then phosphorylates tuberous sclerosis complex (TSC) 2 and TSC2 binds and forms a complex with TSC1.19 The TSC1–TSC2 complex acts as a RHEB GTPase, and inhibits the RHEB activation of mTORC1.
 | Figure 2 The mTOR signaling pathway. |
Although the most relevant phosphorylation sites of TSC2 are unknown, AKT is predicted to phosphorylate TSC2 at five residues, Ser939, Ser981, Ser1130, Ser1132, and Thr1462.20 Phosphorylation of TSC2 attenuates mTOR activation and therefore downstream cellular processes including cell growth, proliferation, mRNA translation, and lipid synthesis. A more extensive description of TSC2 phosphorylation and its effects on mTOR activation can be reviewed elsewhere.21,22 Other proteins have been shown to interact with TSC2 having an either inhibitory or stimulatory affect. ERK phosphorylation of TSC2 at Ser540 and Ser664 inhibits the protein’s activity while other proteins, such as AMPK and GSK3, activate TSC2.23–25 A comprehensive description of TSC activation and inactivation by various proteins can be found elsewhere.21
Insulin and other growth factors also effectively activate the RAS/RAF pathway, hence activating mTOR in PI3K independent pathways. The RAS/RAF/MAPK pathway stimulates mTOR in a two-pronged mechanism. Activated extracellular signal-regulated kinase (ERK) 1/2 and its substrate p90 ribosomal protein S6 kinase (RSK) can inhibit TSC2 to activate mTORC1, or RSK phosphorylates RAPTOR, thereby activating mTORC1 directly.26,27
Altered metabolic state, DNA damage, hypoxia, and increased AMP:ATP ratio triggers AMP-activated protein kinase (AMPK) accumulation and activation.25 AMPK directly and indirectly inhibits mTORC1 by phosphorylation of RAPTOR and TSC2, respectively. Hypoxia can also inhibit mTOR signaling by an AMPK-independent mechanism. Hypoxia can induce expression of proteins regulated in development and DNA damage response 1 (REDD1).28 REDD1 promotes TSC complex assembly having an inhibitory effect on mTOR.28,29
As another regulator of mTORC1, PRAS40 acts as both a component and a substrate of mTORC1. PRAS40 and DEPTOR act as inhibitors of mTORC1 activity.13 AKT and mTORC1 both phosphorylate PRAS40, dissociating it from mTORC1 and relieving the inhibitory restraint it has on mTORC1.30 Dissociated PRAS40 is free to bind with 14-3-3, sequestering it from interactions with mTOR. PIM-1 (provirus integration site for Moloney murine leukemia virus) regulates mTOR activity by phosphorylating PRAS40 and dissociating it from mTORC1, while promoting mTORC1 phosphorylation of 4EBP1.31
Downstream effectors of mTOR
Modulation of downstream mTORC1 effectors promotes protein synthesis, cell proliferation and inhibits autophagy, whereas mTORC2 is known to regulate cytoskeleton organization. Both mTOR complexes are major effector kinases of eukaryotic translation initiation factor 4E (eIF4E)-binding protein 1 and p70 ribosomal S6 kinase 1 (p70S6K1), each regulators of mRNA translation.
p70S6K plays a crucial role in controlling cell cycle, growth, and survival. Phosphorylated p70S6K dissociates from eEF2K and phosphorylates elF4B. Phosphorylated 4E-BP1 dissociates from elF4E allowing the elF4 trimer to form (consisting of eIF4A, eIF4E, and eIF4G). p70S6K also phosphorylates elF4B, thereby activating binding to the trimer to allow the final elF4 complex to bind mRNA and affect translation initiation and elongation. p70S6K promotes cell cycle progression from G1 to S phase by translational regulation of cyclin-D1 and possibly p21 cyclin-dependent kinase inhibitor 1A (p21 or Cip1).32 p70S6K regulates cell survival through the intrinsic apoptosis mechanism. Bcl-2-associated death promoter (BAD) is hyperphosphorylated in the absence of p70S6K and acts to induce apoptosis. However, active p70S6K blocks this effect.33 p70S6K1 works via negative feedback to inhibit both mTOR complexes. It is a negative regulator of mTORC2 as it is both required and sufficient to promote phosphorylation of RICTOR phosphorylation, primarily on the Thr1135 residue.34 It also inhibits IRS-1 in the insulin-signaling pathway directly or indirectly through growth factor receptor-bound adaptor protein 10 (GRB10).
mTOR also plays a pivotal role in lipid homeostasis. Treatment with rapamycin blocks the transcription of genes associated with lipid synthesis. Although the exact mechanism is unknown, mTORC1 phosphorylates Lipin 1. Lipin 1 is then exported from the nucleus, reducing the overall nuclear concentration of sterol responsive element binding protein (SREBP), and promotes lipogenesis.35,36 Lipin 1 also induces transcription of peroxisome proliferator-activated receptors gamma (PPARγ) for adipogenesis.36,37
mTOR is important in the regulation of various transcription factors including STAT3 (signal transducers and activator of transcription 3), c-Myc, and FOXO (Forkhead box O) proteins. Phosphorylation of STAT3 by mTOR regulates energetic output of mitochondria.38 c-Myc and FOXO proteins are involved in a variety of physiological processes including cell differentiation, proliferation, and cell growth. c-Myc inhibits anti-apoptotic proteins and activates pro-apoptotic proteins.39 c-Myc relies on mTOR phosphorylation by 4EBP1, and acts as a negative regulator of TSC1, S6, and IRS-1. FOXO proteins are phosphorylated by AKT and SGK1 (serum and glucocorticoid-induced kinase 1). Activated FOXO1 inhibits TSC2, and thus favors activation of the mTOR pathway.
In addition, mTORC2 is a main modulator of cell growth and survival. mTORC2 is directly upstream of AKT and phosphorylates it at the residue Ser473. mTORC2 activation of AKT stimulates downstream mTORC1. mTORC2 also phosphorylates and activates effector molecule protein kinase C (PKC). PKC is a member of the MAPK/ERK signaling pathway and controls cell growth, cell polarity, and migration.
mTOR signaling in neurological diseases: review of recent data
mTOR is present in all tissues but has the highest expression levels in brain and skeletal muscles. Knockout of mTOR is an embryonically lethal mutation emanating from its importance in early embryo cell size regulation, cell proliferation, and overall neurological development.1 Dysregulation of mTOR has been associated with a variety of neurological pathologies including TSC, neuronal malignancies, neurofibromatosis type I (NF1), epilepsy, neurodevelopmental disorders including autism spectrum disorder (ASD), as well as numerous neurodegenerative diseases. More specifically, the role of mTORC1 signaling changes throughout development. Active expression of mTOR in embryonic presynaptic cortical neurons leads to neuronal apoptosis in mice. Conversely, activated mTORC1 in postmitotic neurons leads to cortical hypertrophy and epilepsy. Chronic mTOR activation leads to accumulation of neuronal cytosolic inclusion bodies indicative of neuronal degeneration.40
Tuberous sclerosis complex (TSC)
TSC is a rare autosomal dominant disorder where benign tumors grow throughout the body most commonly in the brain, skin, lung, heart, and kidneys. The central nervous system is also usually affected in TSC resulting in seizures, developmental, and behavior problems, subependymal nodules, and subependymal giant cell astrocytomas (SEGAs). Genetic or spontaneous mutations in either TSC1 or TSC2 genes, which encode for hamartin or tuberin, respectively, inhibit the TSC from forming. By blocking TSC formation the mTOR pathway is hyperactivated resulting in cell growth and proliferation, and leading to tumorigenesis. Rapamycin has been reported to reduce renal tumors, astrocyte proliferation, and epilepsy in TSC preclinical mouse models.41,42 Clinical trials have been conducted to determine the effectiveness of mTOR inhibitors in human patients suffering from TSC. The mTOR inhibitor everolimus (RAD001) has been shown to yield significant reduction in TSC related SEGA and renal angiomyolipoma tumor burden leading to treatment approval.43
Epilepsy
Epilepsy is a chronic and sometimes progressive neurological disease characterized by recurrent seizures. Antiseizure medications are only symptom moderating, not molecularly modulating treatments. Antiepileptic medications are preventative therapies for those who have not already developed the disease. By definition, neither of these treatments are cures and new drug therapies are important for future epilepsy treatment.
Epileptic seizures are a common symptom of TSC. TSC provides the clearest association between mTOR dysregulation and epilepsy. Moreover, TSC mouse models highlight the importance of mTOR signaling in epilepsy. Treatment with mTOR inhibitors before symptomatic seizure onset prevents the development of epilepsy and premature death in TSC1 conditional knockout mice.42 TSC mouse models with altered protein activation or expression upstream of mTOR, including PTEN knockout mice, provide evidence of mTOR activation in epilepsy. A single dose of rapamycin was sufficient to prolong epilepsy recurrence for several weeks in PTEN conditional knockout mice. Intermittent prolonged treatment also increased overall survival.44 Non-TSC related epilepsy models using mTOR inhibitors have also shown promising results for reducing seizures.45,46
Developmental disorders
mTOR is involved with many critical neurodevelopment processes. mTOR regulates neuronal axon and dendrite formation. Furthermore, synthesis of proteins that modulate synaptic plasticity and long-term memory are regulated by mTORC1.47,48 In particular, ASD are a group of neurodevelopmental disorders characterized by difficulties in social interaction, verbal and nonverbal communication, and repetitive behavioral disorders. These disorders have a morphological and functional deficiency in neurons. It is now known that dysregulation of mTOR is associated with ASD including Fragile X, Rett, and Down syndrome.49
Fragile X syndrome (FXS) is the leading cause of inherited mental disability, and the leading cause of autism. FXS is caused by a trinucleotide CGG expansion in the fragile X mental retardation gene (FMR1) arresting fragile X mental retardation protein (FMRP). In terms of the mTOR signaling pathway, FMRP is a repressor for PIKE (phosphoinositide 3-kinase enhancer), an upstream activator of PI3K,50 and Fmr knockout mice exhibit upregulation of PIKE-dependent mTOR pathway.51,52
Rett syndrome is a postnatal neurological disorder caused by mutations on the methyl-CpG binding-protein 2 (MECP2) gene. mTOR signaling is responsible for MECP2 synthesis and unlike other neurological disorders discussed, Rett syndrome patients have decreased mTOR signaling.53
Down syndrome is the most common genetic neurodevelopmental disorder causing mental retardation. Central mTOR pathway proteins are hyperactivated in human Down’s syndrome brain tissues, while IRS-1 activity is decreased.54 The AKT/mTOR signaling pathway is overexpressed in early hippocampal development in patients with Down syndrome, and there is increased expression of phosphorylated proteins such as S6, S6 kinase, eIF4E-binding protein 1, and mTOR.55 Rapamycin has been shown to restore normal levels of phosphorylated mTOR in a Down syndrome murine model.56
Non-TSC related brain malignancies
The severity of gliomas correlates to the mTOR pathway activation.57 Mutations resulting in constitutively active PI3K or loss PTEN are found in nearly all glioblastomas.57–60 Inhibition of mTOR is a compelling treatment option patients who suffer from non-TSC related neoplasias, such as gliomas. The mTOR inhibitor CCI-779 (temsirolimus) reached Stage II clinical trials. Temsirolimus was well tolerated in patients with recurrent glioblastoma multiform (GBM) when given a weekly dose of 250 mg, albeit there was no sign of efficacy in these patients.61 Temsirolimus administered weekly at the dose of 75 mg/m2 also did not show efficacy in children with high-grade gliomas.62 Despite the lack of clinical efficacy, the high tolerability of temsirolimus has made it desirable for studies in combination with chemotherapeutics, vascular endothelial growth factor (VEGF) inhibitors, and other molecular targeted therapies. However, clinical trials have not shown promising combinational therapies of temsirolimus with bevacizumab (VEGF inhibitor), sorafenib (Raf inhibitor), erlotinib (EGFR inhibitor), or radiation therapy.63–66 Most of these studies have failed on account that temsirolimus doses in combination with other therapies have a lower maximum tolerated dose then what is clinically advantageous.
Inhibition of mTOR may result in a positive feedback resulting in increased transcription and/or activation of PI3K. This upregulation may explain the inefficiency of traditional mTORC1 inhibitors in clinical trials. Studies using dual PI3K/mTOR and mTORC1/2 inhibitors alone or in combination with other drugs in clinical trial for GBM treatment are underway. In patient derived cell lines, treatment with ERBB inhibitor PF-00299804 (dacomitinib) causes GBM apoptosis, but does not alter PI3K/mTOR hyperactivation or the cell proliferation profile. Cells treated with a combination of dacomitinib and the dual PI3K/mTOR inhibitor PF-05212384 inhibited cellular proliferation of GBM cells and had a synergistic effect, increasing the induction of GBM apoptosis compared to dacomitinib alone.67 NVP-BEZ235 binds to the ATP-binding site and inhibits both PI3K and mTOR and has been shown to specifically block dysfunctional PI3K signaling in cancer cells.68
In a human xenograft GBM nude mouse model, human luciferase expressing tumors were grown intracranially. Newly discovered mTOR/PI3K inhibitor XL765 treatment resulted in a 12-fold reduction in tumor burden as determined by bioluminescence, and Temozolomide (TMZ) treatment of mice resulted in a 30-fold decrease in bioluminescence. The combination of XL765 and TMZ yielded a 140-fold reduction and increased median survival.69 A second newly discovered dual mTOR kinase ATP-competitive inhibitor, CC214-2, has been shown to inhibit rapamycin-resistant glioblastoma cell growth in vivo.70 Clinical studies are needed to address the tolerance and efficacy of the combination of PI3K/mTOR or mTORC1/mTORC2 and ERBB inhibition or chemotherapeutics in patients suffering from GBM.
Neurodegeneration
The hallmark of classical neurodegenerative diseases such as Alzheimer, Parkinson, and Huntington diseases is the accumulation of misfolded proteins. These proteins have a toxic effect on the surrounding neurons leading to cell death. mTOR signaling pathway blocks apoptosis to inhibit cell death, and is suspected to inhibit mechanisms related to clearing these unwanted misfolded protein aggregates.
Alzheimer disease (AD), the most common neurodegenerative disease, is characterized by progressive cognitive impairments associated with accumulation of amyloid-β plaques and neurofibrillary tangles. Upregulation of mTOR signaling is shown in both human tissue samples and murine models of AD.71 In a murine model, suppression of mTOR signaling reduced the formation of amyloid-β plaques and restored memory deficits.72 Inhibition of mTOR with rapamycin has been shown to prevent cognitive impairment in the PDAPP transgenic mouse model. Temsirolimus reduced the accumulation of neurofibrillary tangles when mutant tau mice were treated before or after the initial signs of motor function impairment.73 Interestingly, Rheb GTPase overexpression has been shown to decrease the amyloid-β aggregation, independent of mTOR. The AD brain has a reduced level of Rheb GTPase and may be a key regulator in plaque formation.74 Modulation of mTOR signaling has potential clinical implications for patients suffering from AD.
Parkinson disease (PD) is defined by death of dopaminergic neurons located in the substantia nigra. Stress-related protein REDD1, is elevated in substantia nigra neurons from PD model and induction of cell death is through the inhibition of mTOR.75 Treatment with rapamycin in both in vivo and in vitro models disrupts REDD1 expression associated with neuron survival, thereby supporting mTOR’s role in PD.76
Huntington disease is an autosomal dominant mutation caused by a trinucleotide repeat expansion in the Huntington protein. Accumulation of misfolded mutant Huntington protein leads to neurodegeneration, affecting mainly the basal ganglia and the cerebral cortex. Catalytic mTORC1 and mTORC2 inhibitors both induce autophagy and aggregation in a Huntington disease neuronal cell model with inducible expression of Htt fragments.77 Rapamycin protects neurodegeneration in a fly model with polyglutamine expansions, and CCI-779 reduces protein aggregates and improves behavioral deficiencies.78,79
Recent studies also demonstrate a link between hyperactivation of mTOR and misfolded SOD1 accumulation in the progressive motor neuron degenerative disease Amyotrophic lateral sclerosis (ALS) or Lou Gehrig’s disease. mTOR activity is critical for delayed disease progression in ALS mice models involving increased sensitivity to FASL-induced cell death.80
Role of mTOR signaling in cancer: review of recent data
PI3K/AKT/mTOR pathway regulation is disrupted in the majority of cancer types. Protein kinases associated with the activation of the pathway are generally overexpressed, hyperactivated, or constitutively active in cancer. Conversely, proteins that inhibit the pathway may be downregulated or contain mutations ablating their function. Often this trend in protein activity and abundance correlates to the stage and aggressiveness of the malignancy. Current investigations of mTOR inhibitors are underway to determine efficacy, potency, and adverse effects in clinical and preclinical trials. A brief summary of these clinical trials are outlined in Table 2.
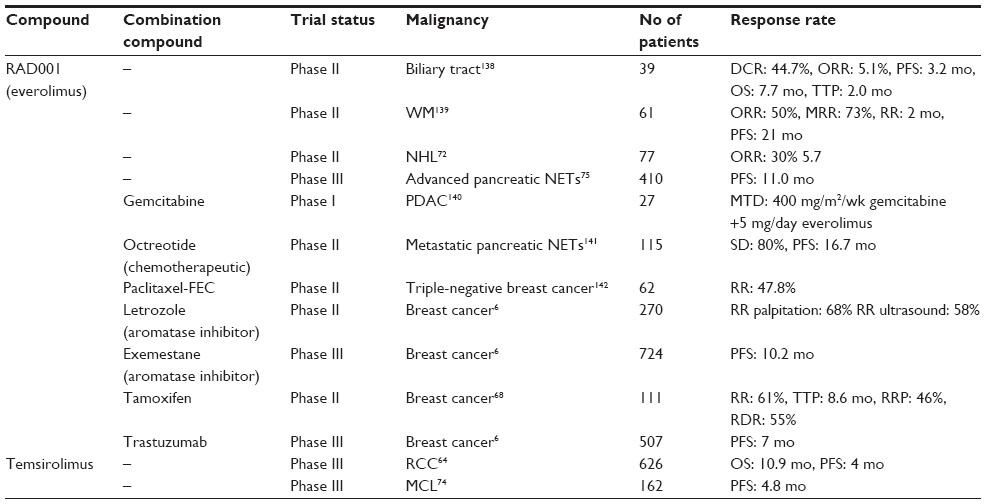 | Table 2 Representative cancer clinical trials using mTOR inhibitors |
Renal cell carcinoma
Temsirolimus is a Food and Drug Administration (FDA) approved drug for the treatment of renal cell carcinoma (RCC). In a Phase III trial with 626 poor-prognostic RCC patients enrolled, temsirolimus significantly increased overall survival to 10.9 months.81 It also increases progression-free survival from 1.9 to 4 months in patients diagnosed with metastatic RCC and with presentation of only mild or moderate adverse effects.81
Although, mTOR inhibitors increase the progression free survival (PFS), overall survival rates remain stunted and concerning. Combinational therapies of mTOR inhibitors and other common cancer therapies are proving to be a challenge due to toxicity. In a Phase I clinical trial, the combination of everolimus and sunitinib is associated with toxicities in patients with metastatic RCC.82 Also, in a Phase II clinical trial combining bevacizumab and everolimus has a toxic effect.83
Breast cancer
Multiple clinical trials have been conducted using mTOR inhibitors in woman diagnosed with receptor-positive breast cancer, and many show promising results. In 2009, a Phase II clinical trial concluded that everolimus had a synergistic effect with the aromatase inhibitor letrozole in postmenopausal woman. Patients who received everolimus had increased antitumor response rate determined by palpitation (59%–68%), ultra sound (47%–58%), and Ki67 histological staining.84 The 2012, GINECO Phase II study concluded that combination of tamoxifen and everolimus results in increased clinical benefit (42%–61%), increased time to progression (4.5–8.6 months) and a reduced risk of progression and death by 46% and 55%, respectively.85 In a randomized Phase III clinical trial, combination of everolimus and the aromatase inhibitor exemestane increased PFS in postmenopausal women diagnosed with receptor-positive breast cancer, compared to patients receiving the exemestane alone.86 Medium PFS in patients receiving the combinational therapy was 10.2 months compared to only 4.1 months for patient’s receiving exemestane alone.86 The promising results of this study preceded the FDA’s approval of everolimus and exemestane combinational therapy for metastatic hormone receptor-positive breast cancer. More recently, the BOLERO-3 Phase III clinical trial, studied the efficacy of everolimus, and trastuzumab plus vinorelbine. Combinational therapy resulted in a significant increase in the time of PFS from 5.78 to 7 months.87 Adverse side effects in the everolimus treated group were increased by 22%, and would need to be considered and reduced in future clinical applications.87
Triple negative breast cancer is defined by the absence of estrogen receptor, progesterone receptor, and HER2/neu, and is resistant to common effective therapies for other types of breast cancer. Women diagnosed with triple negative breast cancer have increased recurrence and mortality rates. Currently, clinical trials are underway to combine chemotherapeutics or platinum-based drugs with mTOR inhibitors to help increase drug efficacy and PFS.
Neuroendocrine tumors
Neuroendocrine tumors (NETs) are a rare subset of hormone secreting tumors that can be divided into two subtypes, pancreatic or carcinoid. Upregulation of the mTOR pathway is prevalent in almost all pancreatic NETs. In large-scale protein expression profiling studies performed by various groups, a direct correlation was found linking the expression abundance for endogenous mTORC1 regulator mediators, such as PTEN and TSC, and tumor proliferation rate, malignancy stage, and aggressiveness.88 These studies also show hyperactivation and/or overexpression of AKT, mTOR, 4EBP1, p70S6K, and eIF4E present in the majority of NETs.89–91 The prevalence of dysregulation of mTOR signaling protein abundance and activation state makes this pathway a promising target for NET therapies.
Everolimus increases PFS and has been approved for the treatment of patients with advanced pancreatic NET in the US and Europe.92 Currently additional clinical trials are underway to determine if mTOR inhibition is more effective in combination therapies. RADIANT-1 was a Phase II clinical trial conducted to determine the efficacy of everolimus in metastatic pancreatic NETs patients with a history of failed chemotherapy. Overall, everolimus alone had an antitumor response in patients and the effects were exacerbated with the addition of chemotherapeutic octreotide. The well-tolerated combinational therapy results in 80% of the patients with stable disease and 16.7 month PFS.93
Carcinoid tumors are a group of gastrointestinal tumors found in the stomach, small intestine, appendix, colon, and rectum or may be found in the lung. Similar to pancreatic NETs, the mTOR pathway is overactive in a variety of carcinoid tumors and inhibition of this activation has clinical advantages when treating the disease. Dual PI3K/mTOR inhibition with BEZ235 decreases carcinoid growth and induces apoptosis when compared to PI3K inhibition alone.94 With addition of the MEK inhibitor PD0325901 secretion of hormones from the tumor cells are also decreased, providing enhanced therapeutic benefit.94
Leukemia and lymphoma
Members of the PI3K/AKT/mTOR pathway are often hyperactivated in tumor specimens from patients with non-Hodgkin lymphoma (NHL). A Phase II clinical trial using everolimus in relapsed aggressive NHL has shown promising antitumor effects. Seventy-seven patients were enrolled to receive the usual dose of 10 mg everolimus daily. Overall, 30% of patients responded to the treatment with a 5.7-month median duration of response. Twenty of these patients underwent partial remission and three achieved complete remission.95 These results promise an increased benefit for patients with aggressive NHL receiving everolimus.
Patients diagnosed with large B-cell lymphoma and follicular lymphoma receiving temsirolimus have a significant antitumor response.96 A randomized Phase III trial in relapsed mantel cell lymphoma (MCL), a subtype of B-cell lymphoma and one of the rarest forms of non-Hodgkin leukemia, demonstrated a higher overall response rate with temsirolimus compared with standard chemotherapy.97
In preclinical studies of acute myeloid leukemia (AML), the most common type of leukemia, AZD8055 treatment has antitumor benefits. Mice treated with AZD8055 showed reduced AML blast cell proliferation, inhibition of cell cycle progression, induction of caspase-dependent apoptosis and autophagy, as well as increased survival.98 Conversely, a recent study claims AML cells undergo autophagy as a survival mechanism, and suggests that using an inhibitor of autophagy in combination with dual mTORC1/mTORC2 inhibitors will provide a useful treatment for AML patients.99
In addition, an attempt was made to correlate specific mechanisms for constitutive activation of signaling pathways in acute lymphoblastic leukemia (ALL) and response to mTOR inhibition. In this study, treatment with RAD001 had a moderate antiproliferative effect and no apoptosis in TEL-ABL expressing ALL cells, whereas inhibition of upstream PI3K resulted in both antiproliferation effects and cell death. Thus, RAD001 treatment alone is unlikely to be effective in ALL, and a dual inhibitor drug targeting approach is likely to be more effective in a broader range of ALL cells.100
Dysregulated mTOR signaling in other human diseases: review of recent data
Cardiovascular disorders
mTOR plays a fundamental role in cardiomyocyte growth, development, and function. Cardiomyocyte specific deletion of mTOR is a lethal mutation affecting 92% of mice by the end of gestation.101 Ablation of mTORC1 or RAPTOR in cardiac specific knockout mice yields decreased cardiomyocyte mitochondrial content, apoptosis, and ultimately death.102–104 Genetic knockout mice for proteins that regulate mTOR activity show a cardiac phenotype similar to mTOR knockout mice. Cardiac specific RHEB deficient mice present with symptoms of cardiomyocyte hypertrophy, exhibit sarcomere maturation defects, reduced translation, and death at 8–10 days postnatal development.105
In stress conditions and aging, pharmacological inhibition of mTOR may provide therapeutic benefit for cardiomyocyte hypertrophy. Inducible genetic knockdown and pharmacological inhibition of mTOR has been shown to prolong survival by attenuating age-related cardiovascular changes.106,107 Cardiac hypertrophy is the thickening of the ventricular walls in the apex of the heart often caused by stress of cardiac sarcomere proteins. Rapamycin abates isoproterenol-induced cardiac hypertrophy in adult rats by maintaining mitochondria structure and functionality.108
Recently, cardiomyocyte autophagy has also been shown to play a role in cardiac homeostasis. Cardiomyocyte autophagy must be tightly regulated to maintain a balance between the beneficial effect of proper heart function and damaged organelle elimination, without the detrimental outcome of heart failure.109,110 Cardiomyocyte specific PTEN knockout mice present with established hypertrophic cardiomyopathy and treatment with 2 mg/kg/day rapamycin repaired autophagy dysregulation.111
Atherosclerosis is a cardiovascular disease where lipid plaques build up in the lumen of arteries. Chronic plaque buildup causes hardening of the arteries, reducing oxygen-rich blood flow throughout the body. Vascular calcification is a major risk factor for atherosclerosis and is utilized as a predictor of coronary heart disease.112 Vascular smooth muscle cell differentiation into osteoblast-like cells is a pivotal step in vascular calcification. Recently it has been shown that overexpression of mTOR is observed in these osteoblast-like cells. Differentiation is inhibited by the downregulation of mTOR by siRNA or rapamycin.113 Lipid accumulation in atherosclerosis plaques causes an immune response and recruits monocytes. At the site of the lesion monocytes are differentiated into macrophages, and act to clear plaques by phagocytosis. Upon phagocytosis, macrophages undergo cell death and release their intracellular lipids enhancing plaque formation.114 Bone marrow transplant from mice deficient for macrophage specific RAPTOR resulted in reduced atherosclerosis.115
Diabetes
Chronic activation of mTORC1 contributes to obesity by promoting the storage and deposition of excess fat causing insulin resistance. Upon overfeeding, lipids are stored as triglycerides in white adipose tissue. High levels of triglycerides are common in people with high blood cholesterol levels, obesity, diabetes, and heart problem. Synthesis of triglycerides and differentiation of white adipose are mediated by mTORC1. RAPTOR knockout mice have improved insulin sensitivity, resistance to diet-induced obesity and hypercholesterolemia, and increased quantities of brown fat as opposed to white fat.116 Also, mTORC1 indirectly upregulates the translation of PPARγ, the transcription factor responsible for the differentiation of preadipocytes.117,118
mTORC1 phosphorylates S6 kinase, which leads to a negative feedback loop affecting the activity of IRS-1 through the phosphorylation of Ser307 and Ser636/Ser639 sites that are associated with insulin resistance, whereas loss of S6 kinase is protective against obesity and provokes insulin sensitivity.119 Type 2 diabetic mice treated with rapamycin significantly reduced body weight, heart weight, plasma glucose, triglyceride, insulin levels, and oxidative stress suggesting increased cardiac function in these mice.120
Recent studies have also demonstrated the role of mTORC2 on lipid and glucose metabolism. Acute pharmacological inhibition of mTOR causes insulin resistance, glucose intolerance, and increased lipid oxidation in vivo.121 More specifically, AZD8055 stimulates GLUT4 translocation to the cell membrane in muscle tissues to allow intracellular glucose transport for metabolic utilization.121 Liver specific RICTOR knockout in mice led to hyperinsulinemia, hyperglycemia, hypolipidemia, glucose intolerance, deregulated glycolysis and gluconeogenesis through activation of AKT in an mTORC1-independent mechanism.122
Clinically, the antidiabetic drug Metformin is the first line therapy for treatment of diabetes mellitus Type 2. It inhibits the cells response to amino acid intake by blocking Rag heterodimer binding with mTORC1.123 Metformin reduces hyperglycemia primarily by reducing glucose production and improving insulin sensitivity. Drug resistance is developed with chronic metformin intake and currently treatment strategies are being developed to combat this caveat. One example is the use of AMPK activator R118 in preclinical models. R118 treatment results in increased skeletal muscle glycolysis and lipolysis, but does not supersede Metformin in liver glucose and fat metabolism regulation.124
Therapeutic perspectives: compounds currently in development that target the mTOR pathway, potential mTOR targets
The mTOR inhibitor rapamycin (sirolimus) has a macrocyclic lactone structure and was first approved as an immunosuppressant for patients of solid organ transplants. Besides immunosuppressive properties, sirolimus also has fungicidal and antiproliferative characteristics. Second generation structural derivatives of sirolimus include temsirolimus (42-[2,2-bis(hydroxymethyl)] rapamycin), everolimus (42-O-(2-hydroxyethyl) rapamycin) and ridaforolimus (macrolide dimethylphophinic acid rapamycin-40-O-yl ester derivative of sirolimus, also known as deforolimus). These rapalogs inhibit mTOR by binding to the cytosolic protein FKBP-12.125
Although rapalogs have a similar mechanism of action, the drug pharmacokinetic/pharmacodynamic (PK/PD) profiles are a result of differences in metabolism, as well as drug formulation and dosing. Temsirolimus is an inactive soluble ester with low oral bioavailability, but can be administered intravenously, whereby it is metabolized to an active sirolimus compound that has anticancer properties with improved pharmacokinetics and no immunosuppressant characteristics. Everolimus is orally bioavailable and with no active metabolites. Both temsirolimus and everolimus are can be used for RCC, although recommendations are for temsirolimus use in treatment-naïve patients with metastatic RCC, whereas everolimus is recommended for patients with progressive metastatic RCC following VEGF receptor-tyrosine kinase inhibitor therapy.125 New studies also have identified several mTOR hyperactivating mutations that increase solid tumor sensitivity to sirolimus or everolimus.126,127
Because rapalogs have a primary effect as antiproliferative drugs to delay tumor growth, newer third generation mTOR inhibitors (TORkinibs, Table 3) were developed to block the mTOR kinase ATP binding site, and thereby target both mTORC1 and mTORC2. This strategy was undertaken as an alternative to rapalogs, which target mTORC1 but leave mTORC2 intact to activate a feedback loop to phosphorylate AKT and prevent apoptosis. Another strategy was to use inhibitors that act on the active site of PI3K and mTORC1/2 (Table 3).
 | Table 3 Representative classes of mTOR inhibitors |
Since cell survival and cytoskeletal organization can be regulated through increased mTORC2 kinase activity in some tumors and phosphorylation of its substrates, inhibition of both mTOR complexes may result in better antitumor effects. For example, FOXO1 signaling was highly activated in cells that were resistant to EGFR tyrosine kinase inhibitor (TKI), whereas dual mTOR inhibition resulted in proliferative defects and G1-cell cycle arrest in a broader range of sensitive and resistant cells.128 Thus, PI3K pathway inhibitors have emerged as a possible solution to the problem of EGFR TKI resistance, and mTORC1/2 inhibition may be more effective for tumors that have acquired resistance to therapeutics. Moreover, preclinical experiments suggest that in some patients PI3K inhibitors may need to be combined with other pharmaceutical agents for effectiveness against aggressive tumors.129
Colon cancer stem cells also have been found to exhibit elevated mTORC2 expression. Moreover, SGK1 was implicated as central to mTORC2 signaling because of the negative effect on tumor characteristics following its knockdown. The mTORC1/mTORC2 inhibitor Torin-1 impeded growth, motility, invasion, and survival of colon cancer stem cells in vitro, and inhibited tumor growth and reduced vessel formation in vivo. Torin-1 was specific for tumor cells since it did not affect the survival of normal colon stem cells in vivo. Rather, Torin-1 affected the expression of markers for cell proliferation, angiogenesis, lymphogenesis, and stemness of colon cancer cells, including Ki67, DLL1, DLL4, Notch, Lgr5, and CD44.130 The differential effect of mTOR inhibition on cancer stem cells may influence tumor recurrence.
Also in support of a differential response to mTOR inhibitors, RAW 264.7 macrophages were stimulated with community-acquired-MRSA isolate in the presence of Vancomycin, and rapamycin or mTORC1/2 inhibitors (ie, Torin-1 or KU63794) were added alone and in various combinations. Cell supernatants were collected and assayed for TNF, IL-1, IL-6, IFN, and NO. Rapamycin was found to exhibit a significant induction–suppression biphasic response, whereas mTORC1/2 inhibitors did not exhibit induction and cytokine production was suppressed 50%–60%.131 Similarly, another recent study has shown that mTORC1/2 inhibition can inhibit inflammatory activity in lipopolysaccharide-activated RAW 264.7 cells.132 The differential effect of different mTOR inhibitors on inflammatory response could indirectly have an effect on tumor response.
ATP-competitive mTOR inhibitors, such as Torin-2, potently target mTORC1 and mTORC2.133 Torin-2 is also a potent inhibitor of ATR (ataxia telangiectasia and Rad3 related), ATM (ataxia telangiectasia mutated), and DNA-PK (DNA-dependent protein kinase), and had more effective antitumor activity compared to rapalogs.134 Dose dependent cytotoxic activity of Torin-2 was observed in a panel of B-pre ALL cell lines, with an IC50 in the nanomolar range. Torin-2 resulted in apoptosis and autophagy, induced G0/G1 cell cycle arrest, and affected both mTORC1 and mTORC2 activities. It suppressed feedback activation of PI3K/AKT, whereas RAD001 required the addition of the AKT inhibitor MK-2206 to achieve the same effect. Strategies targeting PI3K/AKT/mTOR at different points of the signaling cascade might result in improved treatment of B-pre ALL patients.135
Dual mTORC1/2 inhibition with Torin-2 also was found to be effective for papillary thyroid carcinoma (PTC). mTORC1 and mTORC2 activity was observed in 81% and 39% of PTC samples, respectively. Coexpression of mTORC1/2 activity was identified in 32.5% (164/504) of PTC, and was linked with activated AKT and 4E-BP1. Torin-2 or gene silencing of mTOR expression resulted in inactivation of P70S6, 4E-BP1, AKT, and Bad, as well as downregulation of cyclin D1. Torin-2 diminished cell viability and induced caspase-dependent apoptosis in PTC cells, and blocked xenografted tumors. Collectively, dual targeting of mTORC1/2 activity is likely to be a therapeutic strategy for PTC.136
A new study suggests that glycogen synthase kinase-3 (GSK3) is important for tumor response to mTORC1/2 inhibitors. Disruption of GSK3 suppressed growth of cancer cells, constitutively activated GSK3β sensitized cancer cells to mTOR inhibition, and mTOR inhibitors reduced cyclin D1 levels in a GSK3β-dependent manner. Inhibition of mTORC2 resulted in proteasome-mediated cyclin D1 degradation, suggesting that mTORC2 inhibition mediates GSK3-dependent reduction of cyclin D1. In contrast, expression of ubiquitin E3 ligase FBX4 rescued this cyclin D1 reduction, implicating FBX4 in mediating this effect of mTOR inhibition. The findings represent a novel mechanism by which mTORC2 promotes cell growth, and provides justification for understanding the clinical action of mTOR inhibitors.137
Conclusion
PI3K/AKT/mTOR activation is frequently implicated in resistance to antitumor strategies. Inhibitors of the PI3K/AKT/mTOR pathway are being evaluated in preclinical studies and in clinical trials to determine which classes of pathway inhibitors can restore therapeutic sensitivity when administered in combination.
Rapamycin and the rapalogs are allosteric inhibitors of mTORC1 and typically have weak activity against mTORC2. mTOR inhibitors have clinical benefit for patients with metastatic RCC and other cancer types. Rapalogs, such as everolimus, are approved by the US FDA for the treatment of advanced renal cell cancer and pancreatic NETs. However, the single-agent activity of rapalogs in most other tumor types is frequently described as moderate.
Rapamycin and its derivatives are generally cytostatic rather than cytotoxic. Multiple feedback loops regulate cell survival. In one of the primary feedback mechanisms, mTORC1 phosphorylation of S6K1 can promote turnover of IRS and attenuation of PI3K signaling. Inhibition of mTORC1 is able to accentuate PI3K signaling by blocking the negative feedback. Also as part of its regulatory function, mTORC1 signaling can inhibit mTORC2 by phosphorylation of RICTOR. Rapamycin may leave mTORC2-mediated AKT phosphorylation and activation operational. Thus, targeting multiple components within this signaling pathway or across different cross-talking pathways may provide better tumor control and overcome resistance mechanisms.
Currently several newer ATP-competitive mTOR inhibitors are in the pharmaceutical pipeline and are being tested in clinical trials. TORKinibs inhibit both mTORC1 and mTORC2 and generally are more effective than rapamycin at inhibiting protein synthesis, AKT phosphorylation, and at inducing G1 arrest and/or apoptosis. TORKinibs are also valuable research tools for understanding the biology of mTORCs.
Increased toxicity can become a problem with more potent pan-kinase blockades. Overall, additional studies are needed to evaluate if inhibiting multiple components of the PI3K/AKT/mTOR signaling cascade is more effective than blockade at a single mediator, and if mTORC1/2 or PI3K/mTORC1/mTORC2 inhibitors are more effective at establishing a more favorable balance for antitumor efficacy and drug tolerability.
Acknowledgments
DAA is supported in part by University of Central Florida (UCF), Burnett School of Biomedical Sciences institutional startup funds. SBG is supported in part by a UCF College of Graduate Studies Research Excellence Fellowship.
Disclosure
The authors report no conflicts of interest in this work.
References
Murakami M, Ichisaka T, Maeda M, et al. mTOR is essential for growth and proliferation in early mouse embryos and embryonic stem cells. Mol Cell Biol. 2004;24(15):6710–6718. | |
Hara K, Maruki Y, Long X, et al. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell. 2002;110(2):177–189. | |
Kim DH, Sarbassov DD, Ali SM, et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110(2):163–175. | |
Sarbassov DD, Ali SM, Kim DH, et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14(14):1296–1302. | |
Sancak Y, Thoreen CC, Peterson TR, et al. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell. 2007; 25(6):903–915. | |
Pearce LR, Huang X, Boudeau J, et al. Identification of Protor as a novel Rictor-binding component of mTOR complex-2. Biochem J. 2007;405(3):513–522. | |
Yang Q, Inoki K, Ikenoue T, Guan KL. Identification of Sin1 as an essential TORC2 component required for complex formation and kinase activity. Genes Dev. 2006;20(20):2820–2832. | |
Frias MA, Thoreen CC, Jaffe JD, et al. mSin1 is necessary for Akt/PKB phosphorylation, and its isoforms define three distinct mTORC2s. Curr Biol. 2006;16(18):1865–1870. | |
Peterson TR, Laplante M, Thoreen CC, et al. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell. 2009;137(5):873–886. | |
Loewith R, Jacinto E, Wullschleger S, et al. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell. 2002;10(3):457–468. | |
Sarbassov DD, Ali SM, Sengupta S, et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22(2):159–168. | |
Kapahi P, Chen D, Rogers AN, et al. With TOR, less is more: a key role for the conserved nutrient-sensing TOR pathway in aging. Cell Metab. 2010;11(6):453–465. | |
Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12(1):21–35. | |
Sancak Y, Peterson TR, Shaul YD, et al. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008;320(5882):1496–1501. | |
Sancak Y, Bar-Peled L, Zoncu R, et al. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010;141(2):290–303. | |
Smith EM, Finn SG, Tee AR, Browne GJ, Proud CG. The tuberous sclerosis protein TSC2 is not required for the regulation of the mammalian target of rapamycin by amino acids and certain cellular stresses. J Biol Chem. 2005;280(19):18717–18727. | |
Lynch CJ. Role of leucine in the regulation of mTOR by amino acids: revelations from structure-activity studies. J Nutr. 2001;131(3):861S–865S. | |
Cully M, You H, Levine AJ, Mak TW. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat Rev Cancer. 2006;6(3):184–192. | |
Potter CJ, Pedraza LG, Xu T. Akt regulates growth by directly phosphorylating Tsc2. Nat Cell Biol. 2002;4(9):658–665. | |
Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell. 2002;10(1):151–162. | |
Huang J, Manning BD. The TSC1-TSC2 complex: a molecular switchboard controlling cell growth. Biochem J. 2008;412(2):179–190. | |
Huang J, Manning BD. A complex interplay between Akt, TSC2 and the two mTOR complexes. Biochem Soc Trans. 2009;37(Pt 1):217–222. | |
Ma L, Chen Z, Erdjument-Bromage H, Tempst P, Pandolfi PP. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell. 2005;121(2):179–193. | |
Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115(5):577–590. | |
Inoki K, Ouyang H, Zhu T, et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell. 2006;126(5):955–968. | |
Carriere A, Cargnello M, Julien LA, et al. Oncogenic MAPK signaling stimulates mTORC1 activity by promoting RSK-mediated raptor phosphorylation. Curr Biol. 2008;18(17):1269–1277. | |
Carriere A, Romeo Y, Acosta-Jaquez HA, et al. ERK1/2 phosphorylate Raptor to promote Ras-dependent activation of mTOR complex 1 (mTORC1). J Biol Chem. 2011;286(1):567–577. | |
Burugarolas J, Lei K, Hurley RL, et al. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004;18(23):2893–2904. | |
De Young MP, Horak P, Sofer A, Sgori A, Ellisen L. Hypoxia regulates TSC1/2 - mTOR signaling and tumor suppression through REDD1-mediated 14-3-3 shuttling. Genes Dev. 2008;22(2):239–251. | |
Wang H, Zhang Q, Wen Q, et al. Proline-rich Akt substrate of 40 kDa (PRAS40):a novel downstream target of PI3k/Akt signaling pathway. Cell Signal. 2012;24(1):17–24. | |
Vander Haar E, Lee SI, Bandhakavi S, Griffin TJ, Kim DH. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol. 2007;9(3):316–323. | |
Vinals F, Chambard JC, Pouyssegur J. p70 S6 kinase-mediated protein synthesis is a critical step for vascular endothelial cell proliferation. J Biol Chem. 1999;274(38):26776–26782. | |
del Peso L, Gonzalez-Garcia M, Page C, Herrera R, Nunez G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science. 1997;278(5338):687–689. | |
Julien LA, Carriere A, Moreau J, Roux PP. mTORC1-activated S6K1 phosphorylates Rictor on threonine 1135 and regulates mTORC2 signaling. Mol Cell Biol. 2010;30(4):908–921. | |
Porstmann T, Santos CR, Griffiths B, et al. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 2008;8(3):224–236. | |
Peterson TR, Sengupta SS, Harris TE, et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell. 2011;146(3):408–420. | |
Zhang P, Takeuchi K, Csaki LS, Reue K. Lipin-1 phosphatidic phosphatase activity modulates phosphatidate levels to promote peroxisome proliferator-activated receptor gamma (PPARgamma) gene expression during adipogenesis. J Biol Chem. 2012;287(5):3485–3494. | |
Yokogami K, Wakisaka S, Avruch J, Reeves SA. Serine phosphorylation and maximal activation of STAT3 during CNTF signaling is mediated by the rapamycin target mTOR. Curr Biol. 2000;10(1):47–50. | |
Hoffman B, Liebermann DA. Apoptotic signaling by c-MYC. Oncogene. 2008;27(50):6462–6472. | |
Kassai H, Sugaya Y, Noda S, et al. Selective activation of mTORC1 signaling recapitulates microcephaly, tuberous sclerosis, and neurodegenerative diseases. Cell Rep. 2014;7(5):1626–1639. | |
Lee L, Sudentas P, Donohue B, et al. Efficacy of a rapamycin analog (CCI-779) and IFN-gamma in tuberous sclerosis mouse models. Genes Chromosomes Cancer. 2005;42(3):213–227. | |
Zeng LH, Xu L, Gutmann DH, Wong M. Rapamycin prevents epilepsy in a mouse model of tuberous sclerosis complex. Ann Neurol. 2008;63(4):444–453. | |
Kingswood JC, Jozwiak S, Belousova ED, et al. The effect of everolimus on renal angiomyolipoma in patients with tuberous sclerosis complex being treated for subependymal giant cell astrocytoma: subgroup results from the randomized, placebo-controlled, Phase 3 trial EXIST-1. Nephrol Dial Transplant. 2014;29(6):1203–1210. | |
Sunnen CN, Brewster AL, Lugo JN, et al. Inhibition of the mammalian target of rapamycin blocks epilepsy progression in NS-Pten conditional knockout mice. Epilepsia. 2011;52(11):2065–2075. | |
Russo E, Citraro R, Donato G, et al. mTOR inhibition modulates epileptogenesis, seizures and depressive behavior in a genetic rat model of absence epilepsy. Neuropharmacology. 2013;69:25–36. | |
Huang X, Zhang H, Yang J, et al. Pharmacological inhibition of the mammalian target of rapamycin pathway suppresses acquired epilepsy. Neurobiol Dis. 2010;40(1):193–199. | |
Tang SJ, Reis G, Kang H, et al. A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proc Natl Acad Sci U S A. 2002;99(1):467–472. | |
Stoica L, Zhu PJ, Huang W, et al. Selective pharmacogenetic inhibition of mammalian target of Rapamycin complex I (mTORC1) blocks long-term synaptic plasticity and memory storage. Proc Natl Acad Sci U S A. 2011;108(9):3791–3976. | |
Zhou J, Parada LF. PTEN signaling in autism spectrum disorders. Curr Opin Neurobiol. 2012;22(5):873–879. | |
Sawicka K, Zukin RS. Dysregulation of mTOR signaling in neuropsychiatric disorders: therapeutic implications. Neuropsychopharmacology. 2012;37(1):305–306. | |
Hoeffer CA, Sanchez E, Hagerman RJ, et al. Altered mTOR signaling and enhanced CYFIP2 expression levels in subjects with fragile X syndrome. Genes Brain Behav. 2012;11(3):332–341. | |
Wong M. Mammalian target of rapamycin (mTOR) pathways in neurological diseases. Biomed J. 2013;36(2):40–50. | |
Ricciardi S, Boggio EM, Grosso S, et al. Reduced AKT/mTOR signaling and protein synthesis dysregulation in a Rett syndrome animal model. Hum Mol Genet. 2011;20(6):1182–1196. | |
Perluigi M, Pupo G, Tramutola A, et al. Neuropathological role of PI3K/Akt/mTOR axis in Down syndrome brain. Biochim Biophys Acta. 2014;1842(7):1144–1153. | |
Iyer AM, van Scheppingen J, Milenkovic I, et al. mTOR Hyperactivation in down syndrome hippocampus appears early during development. J Neuropathol Exp Neurol. 2014;73(7):671–683. | |
Troca-Marin JA, Alves-Sampaio A, Montesinos ML. An increase in basal BDNF provokes hyperactivation of the Akt-mammalian target of rapamycin pathway and deregulation of local dendritic translation in a mouse model of Down’s syndrome. J Neurosci. 2011;31(26):9445–9455. | |
Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321(5897):1807–1812. | |
Li XY, Zhang LQ, Zhang XG, et al. Association between AKT/mTOR signalling pathway and malignancy grade of human gliomas. J Neurooncol. 2011;103(3):453–458. | |
Riemenschneider MJ, Betensky RA, Pasedag SM, Louis DN. AKT activation in human glioblastomas enhances proliferation via TSC2 and S6 kinase signaling. Cancer Res. 2006;66(11):5618–5623. | |
Fine B, Hodakoski C, Koujak S, et al. Activation of the PI3K pathway in cancer through inhibition of PTEN by exchange factor P-REX2a. Science. 2009;325(5945):1261–1265. | |
Chang SM, Wen P, Cloughesy T, et al. Phase II study of CCI-779 in patients with recurrent glioblastoma multiforme. Invest New Drugs. 2005;23(4):357–361. | |
Geoerger B, Kieran MW, Grupp S, et al. Phase II trial of temsirolimus in children with high-grade glioma, neuroblastoma and rhabdomyosarcoma. Eur J Cancer. 2012;48(2):253–262. | |
Lee EQ, Kuhn J, Lamborn KR, et al. Phase I/II study of sorafenib in combination with temsirolimus for recurrent glioblastoma or gliosarcoma: North American Brain Tumor Consortium study 05-02. Neuro Oncol. 2012;14(12):1511–1518. | |
Lassen U, Sorensen M, Gaziel TB, Hasselbalch B, Poulsen HS. Phase II study of bevacizumab and temsirolimus combination therapy for recurrent glioblastoma multiforme. Anticancer Res. 2013;33(4):1657–1660. | |
Scaringi C, Enrici RM, Minniti G. Combining molecular targeted agents with radiation therapy for malignant gliomas. Onco Targets Ther. 2013;6:1079–1095. | |
Wen PY, Chang SM, Lamborn KR, et al. Phase I/II study of erlotinib and temsirolimus for patients with recurrent malignant gliomas: North American Brain Tumor Consortium trial 04-02. Neuro Oncol. 2014;16(4):567–578. | |
Zhu Y, Shah K. Multiple lesions in receptor tyrosine kinase pathway determine glioblastoma response to pan-ERBB inhibitor PF-00299804 and PI3K/mTOR dual inhibitor PF-05212384. Cancer Biol Ther. 2014;15(6):815–822. | |
Maira SM, Stauffer F, Brueggen J, et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther. 2008;7(7):1851–1863. | |
Prasad G, Sottero T, Yang X, et al. Inhibition of PI3K/mTOR pathways in glioblastoma and implications for combination therapy with temozolomide. Neuro Oncol. 2011;13(4):384–392. | |
Gini B, Zanca C, Guo D, et al. The mTOR kinase inhibitors, CC214-1 and CC214-2, preferentially block the growth of EGFRvIII-activated glioblastomas. Clin Cancer Res. 2013;19(20):5722–5732. | |
Oddo S. The role of mTOR signaling in Alzheimer disease. Front Biosci (Schol Ed). 2012;4:941–952. | |
Caccamo A, De Pinto V, Messina A, Branca C, Oddo S. Genetic reduction of mammalian target of rapamycin ameliorates Alzheimer’s disease-like cognitive and pathological deficits by restoring hippocampal gene expression signature. J Neurosci. 2014;34(23):7988–7998. | |
Frederick C, Ando K, Leroy K, et al. Rapamycin ester analog CCI-779/temsirolimus alleviates tau pathology and improves motor deficit in mutant tau transgenic mice. J Alzheimers Dis. Epub November 14, 2014. | |
Shahani N, Pryor W, Swarnkar S, et al. Rheb GTPase regulates beta-secretase levels and amyloid beta generation. J Biol Chem. 2014;289(9):5799–5808. | |
Malagelada C, Ryu EJ, Biswas SC, Jackson-Lewis V, Greene LA. RTP801 is elevated in Parkinson brain substantia nigral neurons and mediates death in cellular models of Parkinson’s disease by a mechanism involving mammalian target of rapamycin inactivation. J Neurosci. 2006;26(39):9996–10005. | |
Malagelada C, Jin ZH, Jackson-Lewis V, Przedborski S, Greene LA. Rapamycin protects against neuron death in in vitro and in vivo models of Parkinson’s disease. J Neurosci. 2010;30(3):1166–1175. | |
Roscic A, Baldo B, Crochemore C, Marcellin D, Paganetti P. Induction of autophagy with catalytic mTOR inhibitors reduces huntingtin aggregates in a neuronal cell model. J Neurochem. 2011;119(2):398–407. | |
Ravikumar B, Vacher C, Berger Z, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36(6):585595. | |
Saxena S, Roselli F, Singh K, et al. Neuroprotection through excitability and mTOR required in ALS motoneurons to delay disease and extend survival. Neuron. 2013;80(1):80–96. | |
Petri S, Kiaei M, Wille E, Calingasan NY, Flint Beal M. Loss of Fas ligand-function improves survival in G93A-transgenic ALS mice. J Neurol Sci. 2006;251(1–2):44–49. | |
Hudes G, Carducci M, Tomczak P, et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med. 2007;356(22):2271–2281. | |
Molina AM, Feldman DR, Voss MH, et al. Phase 1 trial of everolimus plus sunitinib in patients with metastatic renal cell carcinoma. Cancer. 2012;118(7):1868–1876. | |
Harshman LC, Barbeau S, McMillian A, Srinivas S. A phase II study of bevacizumab and everolimus as treatment for refractory metastatic renal cell carcinoma. Clin Genitourin Cancer. 2013;11(2):100–106. | |
Baselga J, Semiglazov V, van Dam P, et al. Phase II randomized study of neoadjuvant everolimus plus letrozole compared with placebo plus letrozole in patients with estrogen receptor-positive breast cancer. J Clin Oncol. 2009;27(16):2630–2637. | |
Bachelot T, Bourgier C, Cropet C, et al. Randomized phase II trial of everolimus in combination with tamoxifen in patients with hormone receptor-positive, human epidermal growth factor receptor 2-negative metastatic breast cancer with prior exposure to aromatase inhibitors: a GINECO study. J Clin Oncol. 2012;30(22):2718–2724. | |
Baselga J, Campone M, Piccart M, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. 2012;366(6):520–529. | |
Andre F, O’Regan R, Ozguroglu M, et al. Everolimus for women with trastuzumab-resistant, HER2-positive, advanced breast cancer (BOLERO-3):a randomised, double-blind, placebo-controlled phase 3 trial. Lancet Oncol. 2014;15(6):580–591. | |
Missiaglia E, Dalai I, Barbi S, et al. Pancreatic endocrine tumors: expression profiling evidences a role for AKT-mTOR pathway. J Clin Oncol. 2010;28(2):245–255. | |
Shah T, Hochhauser D, Frow R, et al. Epidermal growth factor receptor expression and activation in neuroendocrine tumours. J Neuroendocrinol. 2006;18(5):355–360. | |
Catena L, Bajetta E, Milione M, et al. Mammalian target of rapamycin expression in poorly differentiated endocrine carcinoma: clinical and therapeutic future challenges. Target Oncol. 2011;6(2):65–68. | |
Kasajima A, Pavel M, Darb-Esfahani S, et al. mTOR expression and activity patterns in gastroenteropancreatic neuroendocrine tumours. Endocr Relat Cancer. 2011;18(1):181–192. | |
Yao JC, Shah MH, Ito T, et al. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med. 2011;364(6):514–523. | |
Yao JC, Lombard-Bohas C, Baudin E, et al. Daily oral everolimus activity in patients with metastatic pancreatic neuroendocrine tumors after failure of cytotoxic chemotherapy: a phase II trial. J Clin Oncol. 2010;28(1):69–76. | |
Valentino JD, Li J, Zaytseva YY, et al. Cotargeting the PI3K and RAS pathways for the treatment of neuroendocrine tumors. Clin Cancer Res. 2014;20(5):1212–1222. | |
Witzig TE, Reeder CB, LaPlant BR, et al. A phase II trial of the oral mTOR inhibitor everolimus in relapsed aggressive lymphoma. Leukemia. 2011;25(2):341–347. | |
Smith SM, van Besien K, Karrison T, et al. Temsirolimus has activity in non-mantle cell non-Hodgkin’s lymphoma subtypes: the University of Chicago phase II consortium. J Clin Oncol. 2010;28(31):4740–4746. | |
Hess G, Herbrecht R, Romaguera J, et al. Phase III study to evaluate temsirolimus compared with investigator’s choice therapy for the treatment of relapsed or refractory mantle cell lymphoma. J Clin Oncol. 2009;27(23):3822–3829. | |
Willems L, Chapuis N, Puissant A, et al. The dual mTORC1 and mTORC2 inhibitor AZD8055 has anti-tumor activity in acute myeloid leukemia. Leukemia. 2012;26(6):1195–1202. | |
Altman JK, Szilard A, Goussetis DJ, et al. Autophagy is a survival mechanism of acute myelogenous leukemia precursors during dual mTORC2/mTORC1 targeting. Clin Cancer Res. 2014;20(9):2400–2409. | |
Badura S, Tesanovic T, Pfeifer H, et al. Differential effects of selective inhibitors targeting the PI3K/AKT/mTOR pathway in acute lymphoblastic leukemia. PLoS One. 2013;8(11):e80070. | |
Zhu Y, Pires KM, Whitehead KJ, et al. Mechanistic target of rapamycin (Mtor) is essential for murine embryonic heart development and growth. PLoS One. 2013;8(1):e54221. | |
Zhang D, Contu R, Latronico MV, et al. MTORC1 regulates cardiac function and myocyte survival through 4E-BP1 inhibition in mice. J Clin Invest. 2010;120(8):2805–2816. | |
Shende P, Plaisance I, Morandi C, et al. Cardiac raptor ablation impairs adaptive hypertrophy, alters metabolic gene expression, and causes heart failure in mice. Circulation. 2011;123(10):1073–1082. | |
Sciarretta S, Volpe M, Sadoshima J. Mammalian target of rapamycin signaling in cardiac physiology and disease. Circ Res. 2014;114(3):549–564. | |
Tamai T, Yamaguchi O, Hikoso S, et al. Rheb (Ras homologue enriched in brain)-dependent mammalian target of rapamycin complex 1 (mTORC1) activation becomes indispensable for cardiac hypertrophic growth after early postnatal period. J Biol Chem. 2013;288(14):10176–10187. | |
Flynn JM, O’Leary MN, Zambataro CA, et al. Late-life rapamycin treatment reverses age-related heart dysfunction. Aging Cell. 2013;12(5):851–862. | |
Wu JJ, Liu J, Chen EB, et al. Increased mammalian lifespan and a segmental and tissue-specific slowing of aging after genetic reduction of mTOR expression. Cell Rep. 2013;4(5):913–920. | |
Chen X, Zeng S, Zou J, et al. Rapamycin attenuated cardiac hypertrophy induced by isoproterenol and maintained energy homeostasis via inhibiting NF-kappaB activation. Mediators Inflamm. 2014;2014:868753. | |
Cao DJ, Wang ZV, Battiprolu PK, et al. Histone deacetylase (HDAC) inhibitors attenuate cardiac hypertrophy by suppressing autophagy. Proc Natl Acad Sci U S A. 2011;108(10):4123–4128. | |
Gottlieb RA, Gustafsson AB. Mitochondrial turnover in the heart. Biochim Biophys Acta. 2011;1813(7):1295–1301. | |
Xu X, Roe ND, Weiser-Evans MC, Ren J. Inhibition of mammalian target of rapamycin with rapamycin reverses hypertrophic cardiomyopathy in mice with cardiomyocyte-specific knockout of PTEN. Hypertension. 2014;63(4):729–739. | |
Greenland P, Bonow RO, Brundage BH, et al. ACCF/AHA 2007 clinical expert consensus document on coronary artery calcium scoring by computed tomography in global cardiovascular risk assessment and in evaluation of patients with chest pain: a report of the American College of Cardiology Foundation Clinical Expert Consensus Task Force (ACCF/AHA Writing Committee to Update the 2000 Expert Consensus Document on Electron Beam Computed Tomography). Circulation. 2007;115(3):402–426. | |
Zhan JK, Wang YJ, Wang Y, et al. The mammalian target of rapamycin signalling pathway is involved in osteoblastic differentiation of vascular smooth muscle cells. Can J Cardiol. 2014;30(5):568–575. | |
Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145(3):341–355. | |
Ai D, Jiang H, Westerterp M, et al. Disruption of mammalian target of rapamycin complex 1 in macrophages decreases chemokine gene expression and atherosclerosis. Circ Res. 2014;114(10):1576–1584. | |
Polak P, Cybulski N, Feige JN, et al. Adipose-specific knockout of raptor results in lean mice with enhanced mitochondrial respiration. Cell Metab. 2008;8(5):399–410. | |
Le Bacquer O, Petroulakis E, Paglialunga S, et al. Elevated sensitivity to diet-induced obesity and insulin resistance in mice lacking 4E-BP1 and 4E-BP2. J Clin Invest. 2007;117(2):387–396. | |
Tontonoz P, Hu E, Spiegelman BM. Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a lipid-activated transcription factor. Cell. 1994;79(7):1147–1156. | |
Um SH, Frigerio F, Watanabe M, et al. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature. 2004;431(7005):200–205. | |
Das A, Durrant D, Koka S, et al. Mammalian target of rapamycin (mTOR) inhibition with rapamycin improves cardiac function in type 2 diabetic mice: potential role of attenuated oxidative stress and altered contractile protein expression. J Biol Chem. 2014;289(7):4145–4160. | |
Kleinert M, Sylow L, Fazakerley DJ, et al. Acute mTOR inhibition induces insulin resistance and alters substrate utilization in vivo. Mol Metab. 2014;3(6):630–641. | |
Hagiwara A, Cornu M, Cybulski N, et al. Hepatic mTORC2 activates glycolysis and lipogenesis through Akt, glucokinase, and SREBP1c. Cell Metab. 2012;15(5):725–738. | |
Kalender A, Selvaraj A, Kim SY, et al. Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab. 2010;11(5):390–401. | |
Jenkins Y, Sun TQ, Li Y, et al. Global metabolite profiling of mice with high-fat diet-induced obesity chronically treated with AMPK activators R118 or metformin reveals tissue-selective alterations in metabolic pathways. BMC Res Notes. 2014;7(1):674. | |
Pal SK, Quinn DI. Differentiating mTOR inhibitors in renal cell carcinoma. Cancer Treat Rev. 2013;39(7):709–719. | |
Grabiner BC, Nardi V, Birsoy K, et al. A diverse array of cancer-associated MTOR mutations are hyperactivating and can predict rapamycin sensitivity. Cancer Discov. 2014;4(5):554–563. | |
Wagle N, Grabiner BC, Van Allen EM, et al. Activating mTOR mutations in a patient with an extraordinary response on a phase I trial of everolimus and pazopanib. Cancer Discov. 2014;4(5):546–553. | |
Fei SJ, Zhang XC, Dong S, et al. Targeting mTOR to overcome epidermal growth factor receptor tyrosine kinase inhibitor resistance in non-small cell lung cancer cells. PLoS One. 2013;8(7):e69104. | |
Gadgeel SM, Wozniak A. Preclinical rationale for PI3K/Akt/mTOR pathway inhibitors as therapy for epidermal growth factor receptor inhibitor-resistant non-small-cell lung cancer. Clin Lung Cancer. 2013;14(4):322–332. | |
Francipane MG, Lagasse E. mTOR pathway in colorectal cancer: an update. Oncotarget. 2014;5(1):49–66. | |
Shappley RK, Spentzas T. Differential Role of Rapamycin and Torin/KU63794 in Inflammatory Response of 264.7 RAW Macrophages Stimulated by CA-MRSA. Int J Inflam. 2014;2014:560790. | |
Pan H, Xu LH, Ouyang DY, et al. The second-generation mTOR kinase inhibitor INK128 exhibits anti-inflammatory activity in lipopolysaccharide-activated RAW 264.7 cells. Inflammation. 2014;37(3):756–765. | |
Liu Q, Wang J, Kang SA, et al. Discovery of 9-(6-aminopyridin-3-yl)-1-(3-(trifluoromethyl)phenyl)benzo[h][1,6]naphthyridin-2(1H)-one (Torin2) as a potent, selective, and orally available mammalian target of rapamycin (mTOR) inhibitor for treatment of cancer. J Med Chem. 2011;54(5):1473–1480. | |
Liu Q, Xu C, Kirubakaran S, et al. Characterization of Torin2, an ATP-competitive inhibitor of mTOR, ATM, and ATR. Cancer Res. 2013;73(8):2574–2586. | |
Simioni C, Cani A, Martelli AM, et al. Activity of the novel mTOR inhibitor Torin-2 in B-precursor acute lymphoblastic leukemia and its therapeutic potential to prevent Akt reactivation. Oncotarget. 2014;5(20):10034–11047. | |
Ahmed M, Hussain AR, Bavi P, et al. High prevalence of mTOR complex activity can be targeted using Torin2 in papillary thyroid carcinoma. Carcinogenesis. 2014;35(7):1564–1572. | |
Koo J, Yue P, Gal AA, Khuri FR, Sun SY. Maintaining glycogen synthase kinase-3 activity is critical for mTOR kinase inhibitors to inhibit cancer cell growth. Cancer Res. 2014;74(9):2555–2568. | |
Buzzoni R, Pusceddu S, Bajetta E, et al. Activity and safety of RAD001 (everolimus) in patients affected by biliary tract cancer progressing after prior chemotherapy: a phase II ITMO study. Ann Oncol. 2014;25(8):1597–1603. | |
Ghobrial IM, Witzig TE, Gertz M, et al. Long-term results of the phase II trial of the oral mTOR inhibitor everolimus (RAD001) in relapsed or refractory Waldenstrom Macroglobulinemia. Am J Hematol. 2014;89(3):237–242. | |
Joka M, Boeck S, Zech CJ, et al. Combination of antiangiogenic therapy using the mTOR-inhibitor everolimus and low-dose chemotherapy for locally advanced and/or metastatic pancreatic cancer: a dose-finding study. Anticancer Drugs. 2014;25(9):1095–1101. | |
Bajetta E, Catena L, Fazio N, et al. Everolimus in combination with octreotide long-acting repeatable in a first-line setting for patients with neuroendocrine tumors: an ITMO group study. Cancer. 2014;120(16):2457–2463. | |
Gonzalez-Angulo AM, Akcakanat A, Liu S, et al. Open-label randomized clinical trial of standard neoadjuvant chemotherapy with paclitaxel followed by FEC versus the combination of paclitaxel and everolimus followed by FEC in women with triple receptor-negative breast cancer. Ann Oncol. 2014;25(6):1122–1127. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.