Back to Journals » Drug Design, Development and Therapy » Volume 18
Recent Developments in Drug Design of Oral Synthetic Free Fatty Acid Receptor 1 Agonists
Authors Liu L ![]() , Zhang Q, Ma Y, Lin L, Liu W, Ding A, Wang C, Zhou S, Cai J, Tang H
, Zhang Q, Ma Y, Lin L, Liu W, Ding A, Wang C, Zhou S, Cai J, Tang H
Received 12 August 2024
Accepted for publication 12 November 2024
Published 11 December 2024 Volume 2024:18 Pages 5961—5983
DOI https://doi.org/10.2147/DDDT.S487469
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Tin Wui Wong
Lei Liu,1,2 Qinghua Zhang,1,2 Yichuan Ma,3 Ling Lin,2 Wenli Liu,2 Aizhong Ding,2 Chunjian Wang,2 Shuiping Zhou,1 Jinyong Cai,1 Hai Tang1,2
1Tasly Academy, Tasly Pharma Co., Ltd., Tianjin, People’s Republic of China; 2Tasly Academy Jiangsu Branch, Jiangsu Tasly Diyi Pharmaceutical Co., Ltd., Huaian, Jiangsu, People’s Republic of China; 3China Medical University (CMU)-The Queen’s University of Belfast (QUB) Joint College, Shenyang, Liaoning, People’s Republic of China
Correspondence: Hai Tang, Tasly Academy, Tasly Pharma Co., Ltd., No. 1 Tingjiang Road, Beichen District, Tianjin, 300410, People’s Republic of China, Email [email protected]
Abstract: Over the past two decades, synthetic FFAR1 agonists such as TAK-875 and TSL1806 have undergone meticulous design and extensive clinical trials. However, due to issues primarily related to hepatotoxicity, no FFAR1 agonist has yet received regulatory approval. Research into the sources of hepatotoxicity suggests that one potential cause lies in the molecular structure itself. These structures typically feature lipid-like carboxylic acid head groups, which tend to generate toxic metabolites. Strategies to mitigate these risks focus on optimizing chemical groups to reduce lipophilicity and prevent the formation of reactive metabolites. Recent studies have concentrated on developing low-molecular-weight compounds that more closely resemble natural products, with CPL207280 showing promising potential and liver safety, currently in Phase II clinical trials. Moreover, ongoing research continues to explore the potential applications of FFAR1 agonists in diabetes management, as well as in conditions such as non-alcoholic fatty liver disease (NAFLD) and cerebrovascular diseases. Utilizing advanced technologies such as artificial intelligence and computer-aided design, the development of compact molecules that mimic natural structures represents a hopeful direction for future research and development.
Keywords: antidiabetics, oral FFAR1 development, natural-inspired structure modification, hepatotoxicity, fatty acid-based lipotoxicity, artificial intelligence aided
Introduction
Global Diabetes Crisis
International Diabetes Federation (IDF) Diabetes Atlas reports that 537 million adults were living with diabetes in 2011 and predicts a rise to 783 million diabetics by 2045.1 Type two diabetes mellitus (T2DM), regarded as one of the most serious global health threats, is a pathological condition characterized by a multifaceted etiology involving insulin resistance and diminished insulin sensitivity, compounded by a decline in insulin secretion from the pancreatic beta cells.2–7 Uncontrolled hyperglycemia elevates the risk of and microvascular and macrovascular complications.8–10 Therefore, the therapeutic regulation of blood glucose levels is crucial for the clinical management and treatment of diabetes.
The Dual Role of Free Fatty Acids in Insulin Secretion and Pancreatic Beta Cell Function
Despite the availability of various treatments for T2DM, the majority of hypoglycemic drugs are limited by their adverse effects.11–14 Commonly used antidiabetic medications like sulfonylureas could cause hypoglycemia and potentially hasten the functional decline and apoptosis of pancreatic beta cells.15,16 Accordingly, developing new hypoglycemic drugs that avoid these drawbacks has become a primary goal. Free fatty acids (FFAs) are significant energy sources and signaling molecules in various cellular activities, especially in insulin secretion.17–20 On one hand, continual exposure to high concentrations of FFAs has been found to deteriorate the functionality and secretory capability of pancreatic beta cells, a state known as lipotoxicity.21–24 On the other hand, short-term administration of FFAs facilitates glucose-stimulated insulin secretion (GSIS).17,25,26
The Role of Free Fatty Acid Receptor 1 and Its Activation in Metabolic Regulation
Free fatty acid receptor 1 (FFAR1, also called G-protein-coupled receptor 40, GPR40) is highly expressed in the pancreatic beta cells of healthy individuals but is less so in diabetics.27–29 Studies have indicated that FFAR1, which has a seven-transmembrane domain, plays a pivotal role in amplifying GSIS induced by FFAs.30–37 For instance, 20-hydroxyeicosatetraenoic acid, the FFAR1 agonist produced by human pancreatic beta cells, can activate FFAR1 and form a positive feedback loop, enhancing GSIS.38 FFAR1 signaling may influence palmitate-stimulated insulin secretion by enhancing mitochondrial respiration.39,40 The amplification pathway activated by FFAR1 functions only when blood glucose reaches a certain concentration, thereby preventing excessive insulin secretion and avoiding the resultant hyperinsulinemia and hypoglycemia.41–43 Additionally, activation of the FFAR1 also stimulates glucagon-like peptide-1 (GLP-1) secretion, which better protects pancreatic beta cells and poses a lower risk of weight gain.30,33,44,45 As a result, FFAR1 is widely considered a promising target for anti-diabetes therapies.
The Role of FFAR1 Agonists in Liver Health
The liver serves a crucial role in maintaining lipid, glucose homeostasis and overall energy metabolism within the body. Excessive lipid deposition and genetic anomalies in hepatic tissue are primary etiological factors triggering conditions such as steatohepatitis, insulin resistance, and various other hepatic injuries.46 FFAR1 is expressed in various human tissues, including the heart, liver, skeletal muscle, pancreas, and brain, with a preference for pancreatic beta cells.47 Although FFAR1 levels are low in the liver, certain research has looked into FFAR1’s potential involvement in non-alcoholic fatty liver disease (NAFLD) due to the link between insulin resistance and NAFLD. Studies have shown that the FFAR1 agonist GW9508 can reduce sterol regulatory element-binding protein 1 through a p38-dependent pathway in human hepatocellular carcinomas (HepG2) cells, which helps improve liver fat accumulation. This suggests that FFAR1 could be a new therapeutic target for treating hepatic steatosis.48 Activating FFAR1 with the compound GW9508 significantly reduced liver fat in both mice and HepG2 cells.49 This fat accumulation, usually caused by the liver X receptor, was lessened through the activation of the AMP-activated protein kinase pathway. Using the partial FFAR1 agonist RLA8 in mouse models with methionine/choline-deficient or high-fat diets reduced liver FFAs and triglycerides, alleviated insulin resistance, reduced oxidative stress and inflammation, and improved liver fibrosis and fat accumulation.50 Targeting FFAR1 with specific agonists offers a promising adjunct therapeutic strategy for alleviating hepatic steatosis, insulin resistance, and associated liver pathologies in NAFLD.
Challenges and Innovations in the Molecular Design Development of FFAR1 Agonists
Over the past two decades, multifarious potent synthetic FFAR1 agonists such as TAK-875, TSL1806 and CPL207280 (Table 1) have been rigorously designed, investigated, and progressed to clinical trials.51–63 Unfortunately, no FFAR1 agonist has received approval from regulatory authorities for commercial distribution until now. Due to the hepatotoxicity issues, TAK-875 was halted at the final stage before market approval. Researchers have conducted extensive investigations into the hepatotoxic mechanisms induced by FAR1 agonists. One widely recognized cause of hepatotoxicity is related to the molecular design of these compounds. Lipid-like structures and carboxylic acid head groups are key features of most FFAR1 agonists, but it is well known that these can lead to idiosyncratic drug toxicity through the formation of reactive acyl glucuronide metabolites.64–66 Some FFAR1 agonist molecules contained benzyl ether structures, which were reported that these structures are metabolized to benzaldehyde and benzoic acid in the liver mitochondria, leading to hepatotoxicity.65,67,68 From a molecular design perspective in this review, we present the molecular design of two prominent clinical drugs and provide a review of recent three-year original research articles focused on drug design and compound optimizations of synthetic FFAR1 agonists. It is anticipated that our efforts will provide new insights and modest contributions to the development of oral administration synthetic FFAR1 agonists.
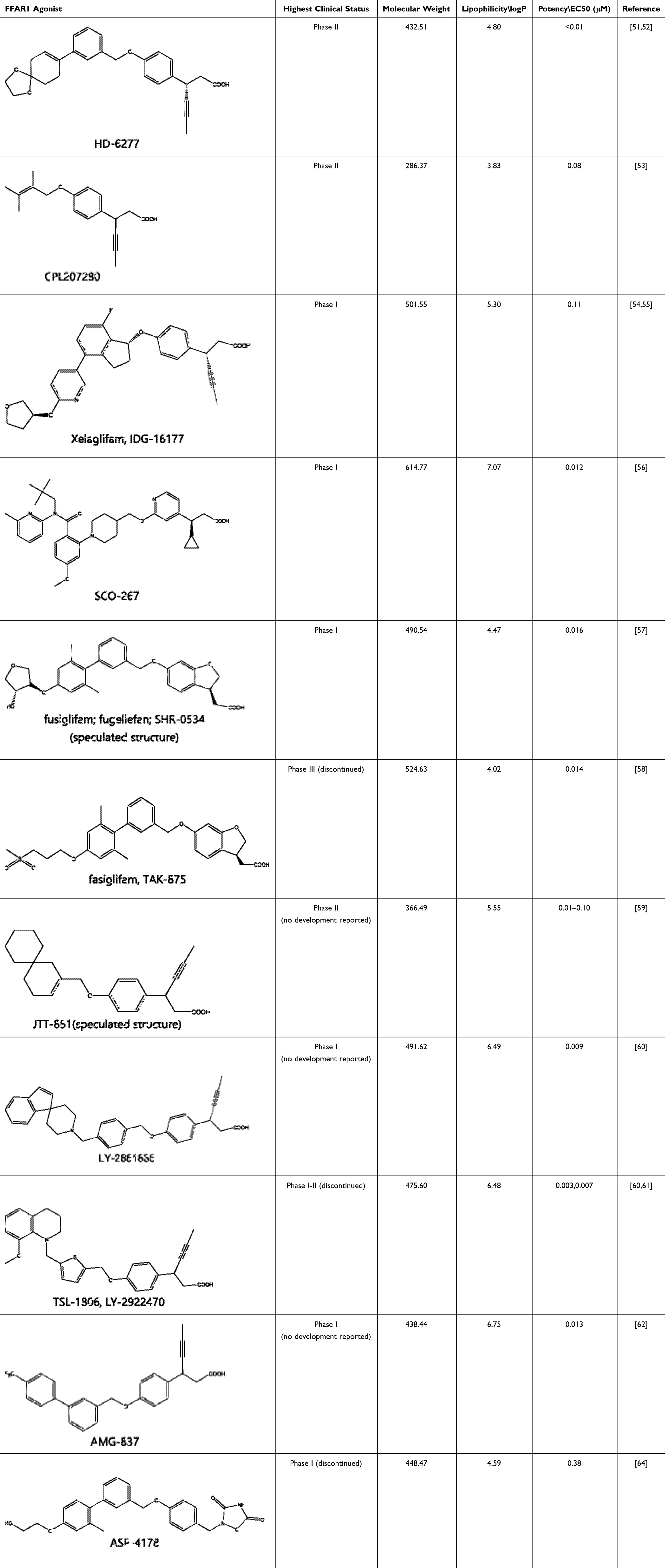 |
Table 1 Representatives of Reported Clinical Synthetic FFAR1 Agonists, with Molecular Weight and logP Calculated by ChemDraw and Clinical Status Derived from the Cortellis Database, a Resource Provided by Clarivate Analytics.69 |
Discovery Stories of Typical Clinical Drugs
The majority of FFAR1 drugs that have entered the clinical research stage are based on fatty acid-like structures, and the ongoing active clinical drug molecules like HD-6277 and CPL207280 are still within this category. Two representative FFAR1 agonists, TAK-875 and TSL-1806, were selected for a concise overview of their drug discovery stories due to their significant attributes. TAK-875 was chosen because it was once closest to achieving successful market approval but stopped by hepatotoxicity safety issue. The TSL-1806 was selected for its higher potency compared with clinical drug molecules in Table 1 and reproducible safety profiles demonstrated through multiple clinical trials.
Fasiglifam (TAK-875)
Figure 1 shows how Takeda scientists discovered fasiglifam (TAK-875). They refined the structure multiple times. Inspired by docosahexaenoic acid (DHA), a type of polyunsaturated fatty acid with strong FFAR1 agonist activity, and started with a commercial available compound 1 with certain agonist activity, improved higher activity from lead compound 2 to compound 3, but suffered high lipophilic and cytotoxic problems.70,71 After improving metabolic stability by cyclization handling the β-oxidation issue of phenylpropanoic acid moiety and reducing lipophilicity by sulfonylpropoxy part grafted, they eventually developed compound 4 (TAK-875), which had excellent potency, good physical chemical properties, favorable pharmacokinetics (PKs), and in-vitro safety.58,67 Since then, FFAR1 agonists have been recognized as having a three-segment fatty acid-like structure, consisting of an acid head, a middle linker, and an aromatic tail. This recognition had influenced various subsequent molecular designs. Additionally, the detailed crystal structure of FFAR1 with TAK-875, demonstrated in Figure 2, had been shared to offer a clear understanding of the binding pocket and assist in designing more effective FFAR1 agonist compounds. The carboxylic acid part of TAK-875 is nestled in a highly hydrophobic area, interacting with key residues like Arg 258, Arg 183, Tyr 240 and Tyr 91, while the other end of the TAK-875 molecule sticks out of the receptor.72
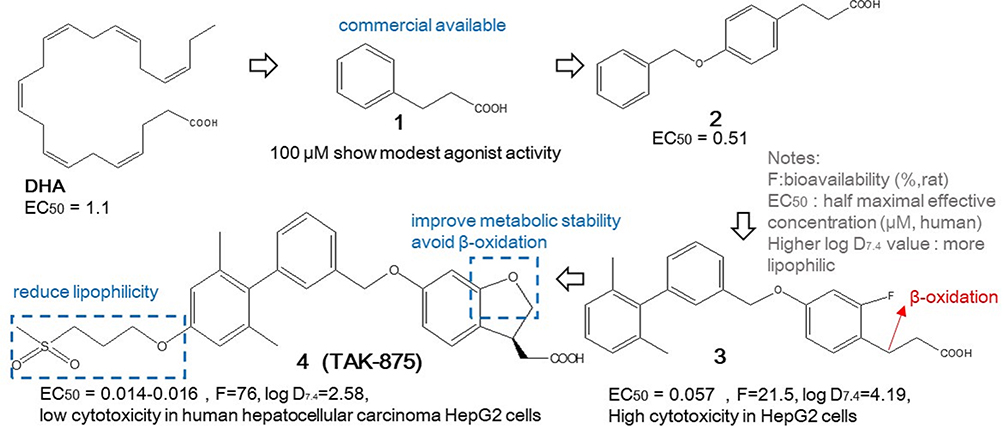 |
Figure 1 The discovery of TAK-875. |
 |
Figure 2 Structure of FFAR1 with TAK-875. (a) the common seven-helix bundle is shown as rainbow-colored ribbons, while TAK-875, positioned between helix 3 and helix 4, is displayed as a stick model; (b) the top view of FFAR1 and mesh pattern demonstrates binding pocket; (c) the computational model shows how TAK-875 interacts with FFAR1 in binding pocket. Notes: Figure 2 is adapted with permission from Srivastava A, Yano J, Hirozane Y, et al. High-resolution structure of the human GPR40 receptor bound to allosteric agonist TAK-875. Nature. 2014;513(7516):124–127. Copyright Springer Nature 2014.72 |
TAK-875, the most sophisticated compound from Takeda, once reached Phase III of clinical trials, which served as clinical proof-of-concept, demonstrating the efficacy of FFAR1 agonists in treating T2DM. Nevertheless, Takeda chose to cease the development of TAK-875 due to apprehensions regarding potential liver toxicity.73 It remains undisclosed whether the signal of liver toxicity was specifically tied to the structural make-up of TAK-875 or the mechanism by which it operates.74 It’s been found that compounds that are less polar and more lipophilic are more likely to bind to various targets and cross the blood–brain barrier (BBB), which can lead to harmful effects or even adverse toxicities in living organisms.75 Since FFAR1 is expressed at low levels in the human liver,76 it is assumed that the hepatotoxicity has a higher probability due to compound-related lipotoxicity. Certain researchers proposed that there should be a strong positive correlation between drug-induced liver injury and the lipophilicity of compound molecules, which can be controlled by parameters such as ligand efficacy (LE) and ligand lipophilicity efficacy (LLE) during drug discovery.65,77–79 LE increases as the number of non-hydrogen atoms in the molecule increases, while LLE is associated with lipophilicity. In brief, FFAR1 agonists should have a relatively small molecular weight and the low logP value to increase the LE and LLE parameters.
TSL-1806 (LY-2922470)
Drawing on the analysis of the endogenous FFAR1 agonist DHA and the characteristics of its binding pocket with FFAR1, a series of analogues were designed by researchers from Eli Lilly. As illustrated in Figure 3, compound 5, which features a head-linker-tail structure, emerged as a relatively ideal candidate due to its favorable activity. Liver metabolic studies revealed that the primary site of metabolism is β-oxidation in the head section and additional O-dealkylation observed in the linker section.60 For structural optimization and improve metabolic stability, efforts were concentrated on three main areas. In the acidic head section, introducing appropriate-sized substituents at the β position can effectively alleviate the aforementioned oxidation problems, where linear S-configuration propargyl of compound 6 exhibited optimal activity. But compound 6 suffered 38% metabolized, the further optimization dedicated to addressing this issue. In the intermediate linker section, substituting the para benzene ring with meta or ortho configurations might reduce the compound’s activity, likely due to disrupting its linear structure. Many efforts to replace this benzene ring with a heterocyclic or differently substituted benzene ring failed. Only the thiophene structure of compound 7 showed relative success and reduced O-dealkylation, but it introduced N-dealkylation problems, resulting in high metabolized ratio of 44%. In the lipophilic tail section, spirocyclic parts with relatively rigid and planar structure were replaced by nonspiropiperidine-related quinolinyl moiety to improve metabolic stability to 24% metabolized and PKs such as lower plasma clearance and higher polar surface area.60,61 The mentioned three optimizations consisted compound 8(LY-2922470) structure, it also called TSL-1806, as Tasly Pharmaceutical, under license from Eli Lilly, continued drug development in China.
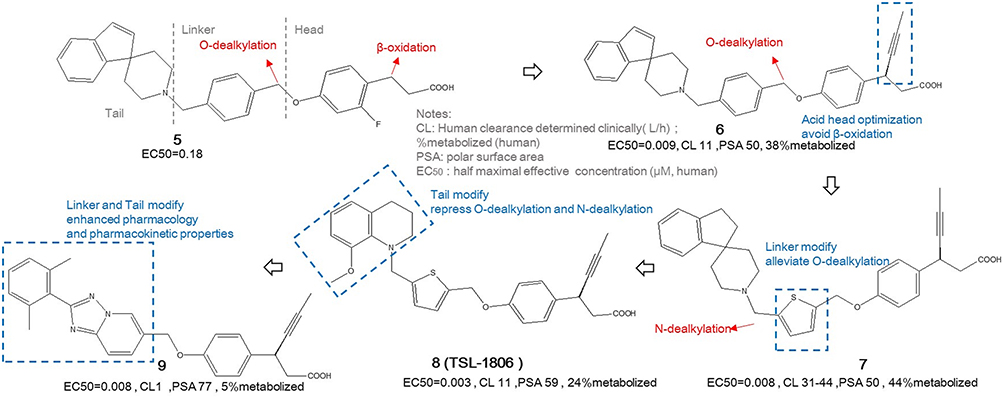 |
Figure 3 The discovered and optimized routine of TSL-1806. |
TSL-1806 has completed phases Ia, Ib, and Ic-II of clinical trials, demonstrating its viability as a potential oral glucose-lowering therapy.80–82 The TSL-1806 drug had good safety profile, but there was limited clarity on the dose-response relationship and therapeutic effect of the drug at different doses. The structure of TSL-1806 remains similar to fatty acid. It still faced the issue of rapid metabolism, and its pharmacodynamic duration is insufficient to support a once-daily oral treatment regimen in humans. Compound 9 with triazolopyridine-derivative-linker and phenyl groups substituted lipophilic tail was designed to improve pharmacological and PK properties.61 However, no further clinical studies of compound 9 can be found in the public official website of clinical trials. Owing to the great safety features and the additional benefits of mitigating brain injury and regulating metabolism of FFAR1 activation, TSL-1806 was also selected to explore new applications beyond its diabetic indications, such as ischemic stroke and endothelial inflammation treatments.83,84 The feasibility of FFAR1 agonists’ new indications needs to be further supported by research and clinical data.
Recent Efforts of Oral Synthetic FFAR1 Agonists
CPL207280
Scientists from Celon Pharma Sa constructed a small-molecule FFAR1 agonist compound 10 (CPL 207280) with the concern of long-term safety. As shown in Figure 4, introducing small non-cyclic structural fragments with natural inspiration of prenol and applying S-propargyl substation, similar as TSL-1806 and instead of cyclopropyl parts like TAK-875, CPL 207280 with smaller molecular weight maintains nearly 3 times higher potency and satisfactory PK properties such as improved higher LE and LLE as well as lower ligand efficiency dependent lipophilicity (LELP) compared with TAK-875.53 Optimized and salted with metformin free base solid, compound 11 of scale-up stability was applied in phase I clinical trial with little adverse effects,53,85 and is now continuing phase II clinical trial.86 The major benefits of using natural molecular substitutes are reducing production costs and decreasing potential toxicity, which can significantly enhance CPL 207280’s competitiveness in the subsequent market.
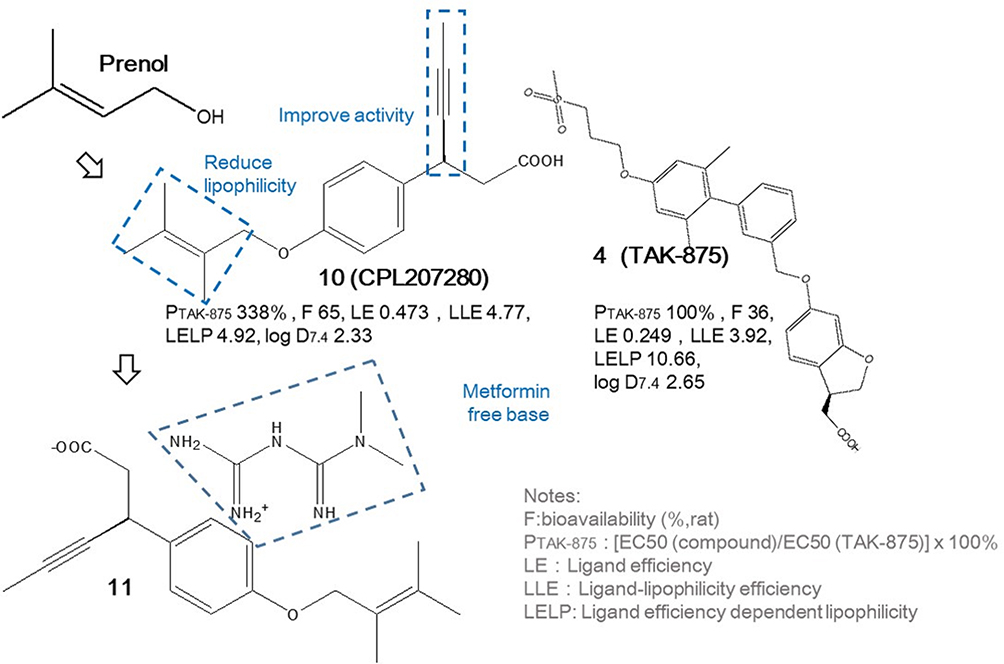 |
Figure 4 The design of CPL207280 and its clinical applied state with TAK-875 as reference. |
A comparative study of liver safety for CPL207280 and TAK-875 demonstrated that CPL207280 has superior safety in human hepatocytes, with less inhibition of bile acid transport proteins, lower mitochondrial damage, and absence of acyl glucuronide metabolites, due to its unique pharmacokinetics. In rats, 14 days of repeated dosing with CPL207280 showed high drug levels without accumulation, unlike TAK-875. In monkeys, high dose of CPL207280 for 14 days did not increase liver toxicity biomarkers.87 The study concluded that CPL207280 is safer than TAK-875 regarding liver toxicity, although further confirmation in late-stage clinical trials is needed, as TAK-875 exhibited liver toxicity in a small number of phase III trial patients.
LK1408
Based on the hypothesis that the toxicity of TAK-875 may stem from its high lipophilicity, a recent study undertook further structural optimizations of the TAK-875 molecule using structure-activity relationship (SAR) studies and computer-aided in silico molecular docking techniques. The research focused on retaining the phenylpropionic acid, identified as the pharmacophore core of TAK-875. As shown in Figure 5, compound 12, which excessively decreased lipophilic characteristics also lose its FFAR1 activation potency. Utilizing data from FFAR1 activation potency and human epithelial colorectal adenocarcinoma permeability screenings, compound 13 (LK1408), which balancing lipophilic and hydrophilic properties and demonstrating significant activity, was designed and considered as the lead compound. It demonstrated similar effects as both commercially available reference FFAR1 agonist GW9508 and nateglinide on insulin secretion. Further optimization involved substituting the pyrazole moiety with 2-alkoxypyridines and reducing bulky benzyl groups in compound 14, which improved activity possibly due to additional hydrophobic and π-stacking interactions with FFAR1.88
 |
Figure 5 The design of LK1408. |
Lipotoxicity-Free Indole Propanoic Acid Full FFAR1 Agonist
The reported signal transduction pathways of FFAR1 mainly include tissue-dependent Gq and Gs. In pancreatic beta cells, activation through Gq pathway increases intracellular Ca2+ levels, facilitating insulin secretion. In intestinal cells, simultaneous activation of Gq and Gs pathways increases Ca2+ and cyclic adenosine monophosphate (cAMP) levels, which stimulating the release of endocrine hormones like GLP-1, collectively promoting insulin secretion.89–91 Recently, a novel lipotoxicity-free full FFAR1 agonist with an indole propanoic acid frame was investigated based on structure-activity study and PK evaluations. As shown in Figure 6, compound 15 with halogen-methyl substitution enjoys higher both Gq and Gs activation activities than compound 16 (lead full agonist) with di-methyl substitution, but suffers from worse PK profiles like lower maximal plasma concentration and plasma exposure. In Figure 6b and c, the binding hydrophobic pocket and interactions between compound and FFAR1 is identified by docking investigation, and the designed compounds were proved as lipotoxicity-free and thus contributed to decrease the appearance of severe adverse effects. Additionally, compound 16 with unprotected β-position of propanoic acid also reported having great metabolic stability.92 The target molecule of this research is very simple, seemingly omitting the recognized intermediate connecting parts and the metabolic stability modifications at the beta position of the indole propionic acid part.
 |
Figure 6 (a) The discovery of full FFAR1 agonist; (b) The predicted binding pockets of compound 20/21 with FFAR1. (c) The lipotoxic evaluation (n=3) with statistical significance indicated as *p ≤ 0.05 and **p ≤ 0.01. Notes: Figure 6b and c are adapted with permission from Zhao X, Yoon D-O, Yoo J, Park H-J. Structure-Activity Relationship Study and Biological Evaluation of 2-(Disubstituted phenyl)-indole-5-propanoic Acid Derivatives as GPR40 Full Agonists. Journal of Medicinal Chemistry. 2021;64(7):4130–4149. Copyright © 2021, American Chemical Society.92 |
Phenylacetic Acid FFAR1 Agonist
The development story of the classic FFAR1 agonist TAK-875 reveals that compounds with a phenylpropionic acid structure are susceptible to β-oxidation, leading to metabolic instability and reduced plasma concentrations. Building upon phenylpropionic acid-based GW9508, researchers replaced the phenylpropionic acid head with a phenylacetic acid moiety and determined the structure of the intermediate linker, followed by a series of tail structure optimizations as shown in Figure 7a. Compound 17 with ortho-methyl substitution on the phenyl ring displayed weaker activity and affinity compared to compound 18 with meta-methyl substitution on the phenyl ring. Further optimization of the meta substitution led to the discovery of compound 19 as a lead compound, which exhibited better activity and affinity and which glucose-lowering effects surpassed traditional FFAR1 agonist GW9508 as reported. As predicted in Figure 7b, compound 19 has an ortho-tolyl substitution on phenyl ring tail, which is more favorable for molecular docking with residues (edge on interaction with Phe142) and binding pockets (bi-phenyl ring nestled) of FFAR1.93
 |
Figure 7 (a) The discovery of phenylacetic acid FFAR1 agonist; (b) The predicted binding pockets of compound 19 with FFAR1. Notes: Figure 7b is adapted with permission from Ge W, Yang B, Chen L, Zhou Z, Jin Y. Discovery of Novel G-Protein-Coupled Receptor 40 Agonist with Phenylacetic Acid Scaffold for the Treatment of Type 2 Diabetes. CHEMISTRYSELECT. 2021;6(29):7372–7375. Copyright © 2021, American Chemical Society.93 |
Phenoxyacetic Acid Dual FFAR1-PPARδ Agonist
Peroxisome proliferator-activated receptors (PPARs) belong to the nuclear receptor family and play important roles in regulating metabolism. PPARs consist of three subtypes: PPARα, PPARδ, and PPARγ. Among them, PPARδ is the most widely expressed and plays a key role in insulin resistance and improving insulin sensitivity, making it an emerging target in the treatment of T2DM. Activating PPARδ has beneficial effects on improving mitochondrial function in pancreatic beta cells, protecting them from apoptosis, and increasing GLP-1 receptor expression levels.94–96 Inspired by an interesting finding that a phenoxyacetic acid head can be observed in peer-recognized FFAR1 agonist (compound 4, TAK-875) and PPARδ agonist (compound 20, GW0742), researchers started with a lead compound (compound 21) that had similar FFAR1 activity as TAK-875 but little PPARδ activity. They designed a series of compounds based on the phenoxyacetic acid head (Figure 8a). Bulky substituents in the tail part, like a cyclohexane ring (compound 22), could bring PPARδ activity but sacrifice certain FFAR1 activity, while smaller substituents in the tail part (compound 23) contributed to higher dual activities. For the linker parts, a naphthalene ring, compared with a benzene ring, could increase PPARδ activity and maintain adequate FFAR1 activity. These three parts together comprised the dual agonist (compound 24), which exhibited balanced dual activities and included biphenyl parts, nestling in the hydrophobic pockets of either FFAR1 or PPARδ receptors (Figure 8b). The hypoglycemic effect of this dual agonist, despite its inferior potency, was better than that of the single FFAR1 agonist lead compound with superior potency, which certified the synergistic effects of activating both FFAR1 and PPARδ receptors in the treatment of T2DM.97
 |
Figure 8 (a) The design of dual FFAR1 and PPARδ agonists; (b) The predicted binding pockets of compound 24 with FFAR1 and PPARδ receptors. Notes: Figure 8b is adapted from Zhou Z, Cai Z, Zhang C, et al. Design, synthesis, and biological evaluation of novel dual FFA1 and PPARδ agonists possessing phenoxyacetic acid scaffold. Bioorganic & Medicinal Chemistry. 2022;56:116615. Copyright © 2022, with permission from Elsevier.97 |
Artificial Intelligence Aided Design of Reinventing Old Drugs as Lead FFAR1 Agonists
The researchers analyzed a dataset of 93 compounds using four artificial intelligence machine learning techniques, such as multilinear regression and Smola and Scholkopf’s regression algorithm. They employed the wrapper method in Weka 3.8 software for variable selection, which uses a classifier within a cross-validation loop to identify a set of relevant descriptors. The wrapper method was considered more accurate than filter methods, which rely solely on high or low relevance scores. The selected descriptors were then evaluated as the potential model based on the statistical parameters obtained. In the applicability domain analysis of the test set, the model achieved full coverage (100%). Thus, it was subsequently utilized to screen two associated DiaNat and DrugBank datasets. Based on the criteria of more than 7.4 half maximal effective concentration (EC50) as high activity and less than 3.83 LogP value as low lipophilicity, twenty-six compounds were selected as hits. Molecular docking and molecular dynamics tools based on the crystal structure of FFAR1 were employed for detailed analysis of receptor-ligand interactions. Figure 9a demonstrates the molecular features linked to their potency, evaluating their interaction and stability with the receptor. Interactions with Ala-83, Arg-183, Arg-258, Tyr-91, Leu-138, Phe-142, Leu-171 and Val-84 were found to be crucial for the activation of FFAR1. Finally, through comparative analysis of calculated molecular activities and binding affinity data, three potential marketed drugs fenofibric acid, bromfenac and bilastine with comparable good potency and binding energy of TAK-875 were screened as lead FFAR1 agonists (Figure 9b).98
 |
Figure 9 (a) Interactions between FFAR1 and selected compounds; (b) structures of three lead compounds and TAK-875. Notes: Figure 9a is adapted from Cabrera N, Cuesta SA, Mora JR, et al. In silico searching for alternative lead compounds to treat type 2 diabetes through a QSAR and molecular dynamics study. Pharmaceutics. 2022;14:232. © 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).98 |
Triazine Amine Dual FFAR1-FFAR4 Agonist
Free fatty acid receptor 4 (FFAR4, also called G-protein-coupled receptor 120, GPR120) is a receptor from the rhodopsin family primarily found in the intestine, macrophages, and adipose tissue. It has been shown to adjustable systemic insulin sensitivity and reduce inflammation. FFAR4 can influence GSIS by modulating GLP-1 release and improving insulin sensitivity by activating FFAR4 in fat cells and macrophages. The activation of FFAR4 has been linked to improved insulin sensitivity through its anti-inflammatory actions, while also aiding in the regulation of glucose levels. By targeting both FFAR1 and FFAR4, it is possible to address two of T2DM’s key issues, the dysfunction of beta cells and insulin resistance.99–104 As demonstrated in Figure 10a and b, a recently developed triazine amine-based dual FFAR1-FFAR4 agonist, featuring a non-lipophilic fatty acid skeleton, was designed with the intent to circumvent potential lipotoxicity. Building upon compound 28, the molecule was divided into three segments, left, core, and right. Researchers conducted four rounds of structural optimization using SAR and activation mechanism studies. In the first round, optimizing the left segment led to compound 29, which featured a 3-chloro-4-methyl substituted benzene ring. This compound exhibited high activity for both FFAR1 and FFAR4 but had poor solubility. The second round focused on the right segment, resulting in a phenylpyrrolidinone structure that significantly improved solubility by approximately 14-fold. Compound 30, with a 5-methyl substituted phenylpyrrolidinone, enhanced FFAR1 activation but had lower potency compared to TAK-875. Since bulk substitutions on the phenyl ring impaired FFAR4 activity, the third round revisited the left segment. The optimal result was compound 31, which included an ethylene linker between the left amino group and the unsubstituted benzene ring, enhancing FFAR1 activation potency without reducing activity. The final round involved small substitutions on the left benzene ring. Compound 32 emerged as the best candidate, featuring di-chloro substitutions at the 2 and 5 positions, demonstrating superior FFAR1 activation potency and efficacy over TAK-875, and the highest FFAR4 activation potency in this study.105 Further researches are still needed on the biopharmacology of this triazine amine structure, such as metabolic stability and toxicity.
 |
Figure 10 (a) The discovery of dual FFAR1 and FFAR4 agonists; (b) Left: compared FFAR1 activities and potencies of the mentioned compounds; Right: the predicted ligand−receptor interactions of compound 32 with FFAR1. Notes: Figure 10b is adapted from Luckmann M, Shenol A, Nissen TA, et al. Optimization of First-in-Class Dual-Acting FFAR1/FFAR4 Allosteric Modulators with Novel Mode of Action. ACS Medicinal Chemistry Letters. 2022;13(12):1839–1847. Copyright © 2022 The Authors. Published by American Chemical Society.105 |
“Three in One” Phenylsulfinyl Acetic Acid FFAR1 Agonist
Based on the hypothesis that the hepatotoxicity of the classical FFAR1 agonist TAK-875 stems from its intrinsic structure, enhancing the efficacy and selectivity of FFAR1 agonists to expand their therapeutic window is a viable strategy for drug design. Researchers recently proposed an innovative “three-in-one” pharmacophore strategy, which integrates beneficial features such as conformational constraint, polarity, and chirality into a single sulfoxide group. This group is inserted at the β-position of the classical phenylpropionic acid, leading to a new frame of agonists with a chiral 2-(phenylsulfinyl) acetic acid structure.
In compound 33 of Figure 11, compared to TAK-875, the sulfoxide group substituted at the β-position of the phenylpropionic acid, reducing the risk of metabolic instability and introducing conformational constraint that enhanced potency but decreased activity. Further improvement was achieved in compound 34, where the chiral substituent of the sulfoxide group significantly boosted agonist activity. The introduction of a polar functional group optimized compound 35, reducing lipophilicity as indicated by the decrease in Log P, thereby minimizing the risk of lipid toxicity (Figure 11b) and demonstrating higher cell viability under incubation at a relatively high concentration (100 μM). As evidenced in Figure 11a and b, both compounds 34 and 35 exhibited higher hypoglycemic efficiency than TAK-875. Therefore, the insertion of a conformational constraint, polarity, and a chiral sulfoxide group at the β-position of the phenylpropionic acid head simultaneously improved the efficacy and safety of the FFAR1 agonist, achieving multiple beneficial outcomes in a single modification.106
 |
Figure 11 (a) The design of 2-(phenylsulfinyl) acetic acid FFAR1 agonist; (b) Hypoglycemic effects and cell toxicities of compound 34 and 35 against human hepatocyte measured by WST-1 kit (n=6) with statistical significance indicated as *p < 0.05, **p < 0.01 and ***p < 0.001. Notes: Figure 11b is adapted from Chen C, Guo SM, Sun YJ, et al. Discovery of orally effective and safe GPR40 agonists by incorporating a chiral, rigid and polar sulfoxide into 8-position to the carboxylic acid. EUROPEAN JOURNAL OF MEDICINAL CHEMISTRY. 2023;251:115267. Copyright © 2023, with permission from Elsevier.106 |
ZLY50, Once-Weekly Oral FFA1 Agonist with BBB Penetration
As demonstrated in Figure 12a, researchers employed a drug repurposing strategy by selecting the small-molecule, already marketed drug fenbufen (compound 36) as the initial compound. Through SAR studies, they identified ZLY50 as a novel FFAR1 agonist. During the optimization process, the researchers primarily focused on the modifications and adjustments of the acidic head and the aromatic tail. For the acidic head optimization, they screened a series of reported fragments and found that a para-propinyl acid head group in compound 37, similar to TSL-1806, exhibited great activity and LE. Subsequently, they optimized the aromatic tail and discovered that para-trifluoromethyl tail and S isomered para-propinyl acid head substituted candidate (compound 38, ZLY50) demonstrated the highest activity and relative high LE. ZLY50 possessed favorable pharmacokinetic properties, including 87.91% high bioavailability and a long 74.4 h oral half-life, making it suitable for weekly oral administration. Additionally, ZLY50 significantly lowers blood glucose levels, reduces fat accumulation in pancreatic tissue, and exerts protective effects against inflammation in pancreatic cells and the liver. This discovery of ZLY50 offers a novel probability for the treatment of diabetes and fatty liver disease.107 Researchers also highlighted ZLY50 as first orally available FFA1 agonist with BBB penetration features and analgesic activity. This is similar to endogenous DHA, which demonstrates analgesic properties in both inflammatory and neuropathic pain states. Central nervous system activation of FFAR1 was reported critical for modulating the descending pain inhibitory pathways, thereby probably reducing nociceptive sensitivity of diabetics.108–110 Due to the restrictive nature of the BBB, most FFAR1 agonists utilized for analgesia are administered by intraventricular injection. The identification and development of orally FFAR1 agonist with BBB penetrating represent a significant potential therapeutic advancement in treatment of central post-stroke pain. However, the early-investigated FFAR1 agonist AMG837 did not progress beyond phase I clinical trials, which possible issue was the high brain exposure, leading to adverse central nervous system effects.111,112 Therefore, whether the high brain exposure following oral administration is suitable for clinical use still requires more comprehensive assessments of the FFAR1 agonist drug’s benefits and risks.
 |
Figure 12 (a) The discovery of ZLY50; (b) Left is the predicted ligand−receptor interactions of ZLY50 with FFAR1; while right is key advantages of ZLY50 reported. Notes: Figure 12b is adapted from Wang B, Cai Z, Yao H, et al. Discovery of a structurally novel, potent, and once-weekly free fatty acid receptor 1 agonist for the treatment of diabetes. EUROPEAN JOURNAL OF MEDICINAL CHEMISTRY. 2023;245:114883. Copyright © 2023, with permission from Elsevier.107 |
Conclusion
FFAR1 agonists, modulating GSIS and significantly reducing the risk of hypoglycemia, enhances the secretion of GLP-1, offering protection for pancreatic beta cells and mitigating weight gain. These attributes position FFAR1 agonists as promising candidates for diabetes management. Nonetheless, their molecular structures, such as lipid-like carboxylic acid heads, have been identified as one of the key factors contributing to hepatotoxicity. Therefore, designing chemical groups that avoid the formation of these reactive metabolites and reduce the lipophilicity of molecules could potentially decrease hepatotoxicity.
Lessons learned from clinical predecessors like TAK-875 and TSL-1806 have highlighted the challenges in developing FFAR1 agonists, attributed to their rapid liver metabolism and potential lipotoxicities linked to their chemical structures. Recent studies on synthetic FFAR1 agonists have focused on several key strategies (Figure 13). To enhance activity and reduce lipophilicity, certain researchers continue to delve into traditional SAR studies by connecting various fragments and inevitably increasing structural complexity, while other studies focus on screening natural small molecules and reducing the molecular weight of compounds. With the development of AI, the power of machine learning and computer-aided analysis cannot be ignored. Researchers perform modeling analyses on already approved drugs to explore their new applications, for example, the identification of several hit compounds of FFAR1 agonists. Additionally, FFAR1 agonists’ novel chemical frameworks that completely deviate from fatty acid analogs have been established. Among these four approaches, the fastest progressing one is the small, low-molecular-weight substituted class, particularly compound 10 (CPL207280), which is currently advancing into Phase II clinical trials. It was reported enjoyed superior safety in human hepatocytes with the absence of acyl glucuronide metabolites, but still required late clinical verification. Meanwhile, as T2DM competitors like dipeptidyl peptidase-4 inhibitors and GLP-1 receptor agonists are showing promise and already available on the market, FFAR1 agonists with simpler design also address growing concerns regarding the cost-effectiveness.
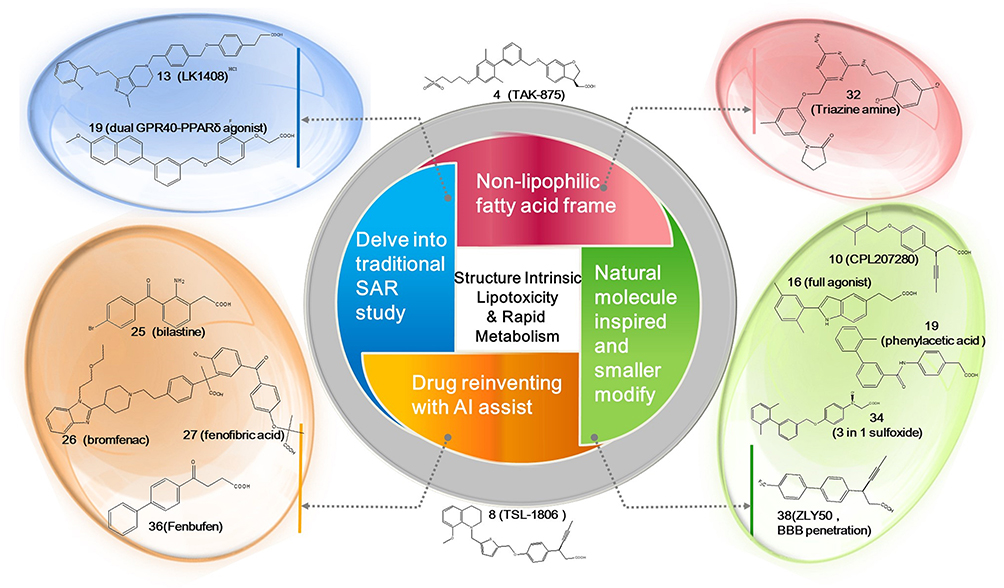 |
Figure 13 Recent developments of FFAR1 agonists with four directions. |
It is worth mentioning that the potential indications for FFAR1 agonists extend beyond T2DM, as current research is also exploring their efficacy in treating NAFLD, endothelial inflammation and cerebrovascular diseases. The feasibility of FFAR1 agonists for new indications needs to be further supported by research and clinical data. In summary, while the research on FFAR1 agonists may not be as fervent as it was in the last decade, there are still certain researchers and pharmaceutical companies advancing this field. With the significant development of computer-aided screening and artificial intelligence technologies, focusing on more compact structures that closely resemble natural molecules may herald a promising direction for the design of FFAR1 agonist drugs.
Abbreviations
AI, artificial intelligence; BBB, blood–brain barrier; cAMP, cyclic adenosine monophosphate; DHA, docosahexaenoic acid; EC50, half maximal effective concentration; FFAs, free fatty acids; FFAR1, free fatty acid receptor 1; FFAR4, free fatty acid receptor 4; GPR40, G-protein-coupled receptor 40; GPR120, G-protein-coupled receptor 120; GSIS, glucose-stimulated insulin secretion; GLP-1, glucagon-like peptide-1; HepG2, human hepatocellular carcinomas; IDF, International Diabetes Federation; LE, ligand efficacy; LLE, ligand lipophilicity efficacy; LELP, ligand efficiency-dependent lipophilicity; NAFLD, non-alcoholic fatty liver disease; PKs, pharmacokinetics; PPARs, peroxisome proliferator-activated receptors; T2DM, type two diabetes mellitus; SAR, structure-activity relationship.
Disclosure
The authors declare that there are no conflicts of interests. All authors have reviewed and approved the final manuscript.
References
1. IDF diabetes atlas-10th edition. Available from: www.diabetesatlas.org.
2. LeRoith D. β-cell dysfunction and insulin resistance in type 2 diabetes: role of metabolic and genetic abnormalities. Am J Med. 2002;113(6):3–11. doi:10.1016/S0002-9343(02)01276-7
3. Yabe D, Seino Y, Fukushima M, Seino S. β cell dysfunction versus insulin resistance in the pathogenesis of type 2 diabetes in East Asians. Current Diab Rep. 2015;15:1–9. doi:10.1007/s11892-015-0602-9
4. Kaur R, Kaur M, Singh J. Endothelial dysfunction and platelet hyperactivity in type 2 diabetes mellitus: molecular insights and therapeutic strategies. Cardiovasc Diabetol. 2018;17:1–17. doi:10.1186/s12933-018-0763-3
5. Kang GG, Francis N, Hill R, Waters D, Blanchard C, Santhakumar AB. Dietary polyphenols and gene expression in molecular pathways associated with type 2 diabetes mellitus: a review. Int J Mol Sci. 2019;21(1):140. doi:10.3390/ijms21010140
6. Galicia-Garcia U, Benito-Vicente A, Jebari S, et al. Pathophysiology of type 2 diabetes mellitus. Int J Mol Sci. 2020;21(17):6275. doi:10.3390/ijms21176275
7. Karmakar T, Chaki R, Ghosh N. Pharmacology and mechanisms of natural medicine in treatment of type 2 diabetes Mellitus. Evidence Based Validation Trad Med. 2021:1091–1127.
8. Fowler MJ. Microvascular and macrovascular complications of diabetes. Clin Diabetes. 2011;29(3):116–122. doi:10.2337/diaclin.29.3.116
9. van Wijngaarden RP, Overbeek JA, Heintjes EM, et al. Relation between different measures of glycemic exposure and microvascular and macrovascular complications in patients with type 2 diabetes mellitus: an observational cohort study. Diab Ther. 2017;8:1097–1109. doi:10.1007/s13300-017-0301-4
10. Dal Canto E, Ceriello A, Rydén L, et al. Diabetes as a cardiovascular risk factor: an overview of global trends of macro and micro vascular complications. Eur J Preventive Cardiol. 2019;26(2_suppl):25–32. doi:10.1177/2047487319878371
11. Murad MH, Coto-Yglesias F, Wang AT, et al. Drug-induced hypoglycemia: a systematic review. J Clin Endocrinol Metab. 2009;94(3):741–745. doi:10.1210/jc.2008-1416
12. Phung OJ, Scholle JM, Talwar M, Coleman CI. Effect of noninsulin antidiabetic drugs added to metformin therapy on glycemic control, weight gain, and hypoglycemia in type 2 diabetes. JAMA. 2010;303(14):1410–1418. doi:10.1001/jama.2010.405
13. Stein SA, Lamos EM, Davis SN. A review of the efficacy and safety of oral antidiabetic drugs. Expert Opin Drug Saf. 2013;12(2):153–175. doi:10.1517/14740338.2013.752813
14. May M, Schindler C. Clinically and pharmacologically relevant interactions of antidiabetic drugs. Therap Adv Endocrinol Metab. 2016;7(2):69–83. doi:10.1177/2042018816638050
15. Burge MR, Sood V, Sobhy TA, Rassam AG, Schade DS. Sulphonylurea-induced hypoglycaemia in type 2 diabetes mellitus: a review. Diabetes Obesity Metab. 1999;1:199–206. doi:10.1046/j.1463-1326.1999.00031.x
16. Maedler K, Carr RD, Bosco D, Zuellig RA, Berney T, Donath MY. Sulfonylurea induced β-cell apoptosis in cultured human islets. J Clin Endocrinol Metab. 2005;90:501–506. doi:10.1210/jc.2004-0699
17. Yaney G, Corkey B. Fatty acid metabolism and insulin secretion in pancreatic beta cells. Diabetologia. 2003;46:1297–1312. doi:10.1007/s00125-003-1207-4
18. Capurso C, Capurso A. From excess adiposity to insulin resistance: the role of free fatty acids. Vasc Pharmacol. 2012;57(2–4):91–97. doi:10.1016/j.vph.2012.05.003
19. Hara T, Kashihara D, Ichimura A, Kimura I, Tsujimoto G, Hirasawa A. Role of free fatty acid receptors in the regulation of energy metabolism. Biochim Biophys Acta-Mol Cell Biol Lipids. 2014;1841(9):1292–1300. doi:10.1016/j.bbalip.2014.06.002
20. Rachek LI. Free fatty acids and skeletal muscle insulin resistance. Progress Mol Biol Transl Sci. 2014;121:267–292.
21. McGarry J, Dobbins R. Fatty acids, lipotoxicity and insulin secretion. Diabetologia. 1999;42(2):128–138. doi:10.1007/s001250051130
22. Lupi R, Dotta F, Marselli L, et al. Prolonged exposure to free fatty acids has cytostatic and pro-apoptotic effects on human pancreatic islets: evidence that β-cell death is caspase mediated, partially dependent on ceramide pathway, and Bcl-2 regulated. Diabetes. 2002;51(5):1437–1442. doi:10.2337/diabetes.51.5.1437
23. Ježek P, Jabůrek M, Holendová B, Plecitá-Hlavatá L. Fatty acid-stimulated insulin secretion vs. lipotoxicity. Molecules. 2018;23(6):1483. doi:10.3390/molecules23061483
24. Lytrivi M, Castell A-L, Poitout V, Cnop M. Recent insights into mechanisms of β-cell lipo-and glucolipotoxicity in type 2 diabetes. J Mol Biol. 2020;432(5):1514–1534. doi:10.1016/j.jmb.2019.09.016
25. Amery CM, Nattrass M. Fatty acids and insulin secretion. Diabetes Obesity Metab. 2000;2(4):213–221. doi:10.1046/j.1463-1326.2000.00059.x
26. Manco M, Calvani M, Mingrone G. Effects of dietary fatty acids on insulin sensitivity and secretion. Diabetes Obesity Metab. 2004;6(6):402–413. doi:10.1111/j.1462-8902.2004.00356.x
27. Tomita T, Masuzaki H, Iwakura H, et al. Expression of the gene for a membrane-bound fatty acid receptor in the pancreas and islet cell tumours in humans: evidence for GPR40 expression in pancreatic beta cells and implications for insulin secretion. Diabetologia. 2006;49:962–968. doi:10.1007/s00125-006-0193-8
28. Bailey CJ. Could FFAR1 assist insulin secretion in type 2 diabetes? Lancet. 2012;379(9824):1370–1371. doi:10.1016/S0140-6736(12)60165-2
29. Ichimura A, Hasegawa S, Kasubuchi M, Kimura I. Free fatty acid receptors as therapeutic targets for the treatment of diabetes. Front Pharmacol. 2014;5:236. doi:10.3389/fphar.2014.00236
30. Itoh Y, Kawamata Y, Harada M, et al. Free fatty acids regulate insulin secretion from pancreatic β cells through GPR40. Nature. 2003;422:173–176. doi:10.1038/nature01478
31. Costanzi S, Neumann S, Gershengorn MC. Seven transmembrane-spanning receptors for free fatty acids as therapeutic targets for diabetes mellitus: pharmacological, phylogenetic, and drug discovery aspects. J Biol Chem. 2008;283(24):16269–16273. doi:10.1074/jbc.R800014200
32. Medina JC, Houze JB. GPR40 (FFAR1) Modulators. Annu Rep Med Chem. 2008;43:75–85.
33. Mancini AD, Poitout V. The fatty acid receptor FFA1/GPR40 a decade later: how much do we know? Trends Endocrinol Metab. 2013;24(8):398–407. doi:10.1016/j.tem.2013.03.003
34. Poitout V, Lin DC-H. Modulating GPR40: therapeutic promise and potential in diabetes. Drug Discovery Today. 2013;18(23–24):1301–1308. doi:10.1016/j.drudis.2013.09.003
35. Mancini A, Poitout V. GPR40 agonists for the treatment of type 2 diabetes: life after ‘TAKing’a hit. Diabetes Obesity Metab. 2015;17:622–629. doi:10.1111/dom.12442
36. Kimura I, Ichimura A, Ohue-Kitano R, Igarashi M. Free fatty acid receptors in health and disease. Physiol Rev. 2019;100:171–210. doi:10.1152/physrev.00041.2018
37. Barella LF, Jain S, Kimura T, Pydi SP. Metabolic roles of G protein-coupled receptor signaling in obesity and type 2 diabetes. FEBS J. 2021;288(8):2622–2644. doi:10.1111/febs.15800
38. Tunaru S, Bonnavion R, Brandenburger I, et al. 20-HETE promotes glucose-stimulated insulin secretion in an autocrine manner through FFAR1. Nat Commun. 2018;9(1):177. doi:10.1038/s41467-017-02539-4
39. Kristinsson H, Bergsten P, Sargsyan E. Free fatty acid receptor 1 (FFAR1/GPR40) signaling affects insulin secretion by enhancing mitochondrial respiration during palmitate exposure. Biochim Biophys Acta Mol Cell Res. 2015;1853(12):3248–3257. doi:10.1016/j.bbamcr.2015.09.022
40. Kristinsson H, Sargsyan E, Manell H, Smith D, Göpel S, Bergsten P. Basal hypersecretion of glucagon and insulin from palmitate-exposed human islets depends on FFAR1 but not decreased somatostatin secretion. Sci Rep. 2017;7(1):4657. doi:10.1038/s41598-017-04730-5
41. Henquin J-C. Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes. 2000;49(11):1751–1760. doi:10.2337/diabetes.49.11.1751
42. Petersen MC, Shulman GI. Mechanisms of insulin action and insulin resistance. Physiological Reviews. 2018;98:2133–2223. doi:10.1152/physrev.00063.2017
43. Arora A, Behl T, Sehgal A, et al. Free fatty acid receptor 1: a ray of hope in the therapy of type 2 diabetes mellitus. Inflammopharmacology. 2021:1–15.
44. Edfalk S, Steneberg P, Edlund H. Gpr40 is expressed in enteroendocrine cells and mediates free fatty acid stimulation of incretin secretion. Diabetes. 2008;57(9):2280–2287. doi:10.2337/db08-0307
45. Jin C, Chen H, Xie L, Zhou Y, Liu L-l, Wu J. GPCRs involved in metabolic diseases: pharmacotherapeutic development updates. Acta Pharmacol Sin. 2024:1–16.
46. Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. J Clin Invest. 2004;114(2):147–152. doi:10.1172/JCI200422422
47. Cartoni C, Yasumatsu K, Ohkuri T, et al. Taste preference for fatty acids is mediated by GPR40 and GPR120. J Neurosci. 2010;30(25):8376–8382. doi:10.1523/JNEUROSCI.0496-10.2010
48. Ou HorngYih OH, Wu HungTsung WH, Lu FengHwa LF, et al. Activation of free fatty acid receptor 1 improves hepatic steatosis through a p38-dependent pathway. J Mol Endocrinol. 2014;53(2):165–174. doi:10.1530/JME-14-0003
49. Li M, Meng X, Xu J, et al. GPR40 agonist ameliorates liver X receptor-induced lipid accumulation in liver by activating AMPK pathway. Sci Rep. 2016;6(1):25237. doi:10.1038/srep25237
50. Li MH, Chen W, Wang LL, et al. RLA8—a new and highly effective quadruple PPAR-α/γ/δ and Gpr40 agonist to reverse nonalcoholic steatohepatitis and fibrosis. J Pharmacol Exp Ther. 2019;369(1):67–77. doi:10.1124/jpet.118.255216
51. Kim D, Kim CH, Choi HS, et al. HD-6277, a novel GPR40 agonist, improves glucose homeostasis and enhanced glucose-dependent insulin secretion in beta cells and type 2 diabetic rats. Diabetes. 2018;67(Supplement_1). doi:10.2337/db18-1202-P.
52. Yang J, Kim JW, Lee HK, et al. Novel 3-(4-(benzyloxy)phenyl)hex-4-inoic acid derivative, method of preparing same and pharmaceutical composition for preventing and treating metabolic disease including same as effective ingredient. WO2014171762.
53. Mach M, Bazydło-Guzenda K, Buda P, et al. Discovery and development of CPL207280 as new GPR40/FFA1 agonist. Eur J Med Chem. 2021;226:113810. doi:10.1016/j.ejmech.2021.113810
54. Yoon J, Song H, An K-M, et al. In vitro pharmacodynamic studies of IDG-16177, a potent GPR40 agonist, for the treatment of type 2 diabetes. Paper presented at: Diabetes 2021.
55. Yoon JM, Lee DG, Je IG, et al. Pharmaceutical composition comprising gpr40 agonist and sglt-2 inhibitor. WO2022231357.
56. Furukawa H, Miyamoto Y, Hirata Y, et al. Design and identification of a GPR40 full agonist (SCO-267) possessing a 2-carbamoylphenyl piperidine moiety. J Med Chem. 2020;63(18):10352–10379. doi:10.1021/acs.jmedchem.0c00843
57. Yang F, Dong Q, Jihui HA, Chunfei WA, Zhang L, Wang Y. Polycyclic derivatives, preparation method and medical uses thereof. WO2013104257.
58. Negoro N, Sasaki S, Mikami S, et al. Discovery of TAK-875: a potent, selective, and orally bioavailable GPR40 agonist. ACS Med Chem Lett. 2010;1(6):290–294. doi:10.1021/ml1000855
59. Shimada T, Ueno H, Tsutsumi K, et al. Spiro-ring compound and use thereof for medical purposes. WO2009054479.
60. Hamdouchi C, Kahl SD, Patel Lewis A, et al. The discovery, preclinical, and early clinical development of potent and selective GPR40 agonists for the treatment of type 2 diabetes mellitus (LY2881835, LY2922083, and LY2922470). J Med Chem. 2016;59(24):10891–10916. doi:10.1021/acs.jmedchem.6b00892
61. Hamdouchi C, Maiti P, Warshawsky AM, et al. Discovery of LY3104607: a potent and selective G protein-coupled receptor 40 (GPR40) agonist with optimized pharmacokinetic properties to support once daily oral treatment in patients with type 2 diabetes mellitus. J Med Chem. 2018;61(3):934–945. doi:10.1021/acs.jmedchem.7b01411
62. Houze JB, Zhu L, Sun Y, et al. AMG 837: a potent, orally bioavailable GPR40 agonist. Bioorg Med Chem Lett. 2012;22(2):1267–1270. doi:10.1016/j.bmcl.2011.10.118
63. Kenji N, Yasuhiro Y, Koji Y, Yasuhiro M. Novel salt of oxadiazolidinedione, and crystal thereof. JP2013184934.
64. Lassila T, Hokkanen J, Aatsinki S-M, Mattila S, Turpeinen M, Tolonen A. Toxicity of carboxylic acid-containing drugs: the role of acyl migration and CoA conjugation investigated. Chem Res Toxicol. 2015;28(12):2292–2303. doi:10.1021/acs.chemrestox.5b00315
65. Otieno MA, Snoeys J, Lam W, et al. Fasiglifam (TAK-875): mechanistic investigation and retrospective identification of hazards for drug induced liver injury. Toxicol Sci. 2018;163(2):374–384. doi:10.1093/toxsci/kfx040
66. Ookawara M, Matsuda K, Watanabe M, Moritoh Y. The GPR40 full agonist SCO-267 improves liver parameters in a mouse model of nonalcoholic fatty liver disease without affecting glucose or body weight. J Pharmacol Exp Ther. 2020;375(1):21–27. doi:10.1124/jpet.120.000046
67. Negoro N, Sasaki S, Mikami S, et al. Optimization of (2, 3-dihydro-1-benzofuran-3-yl) acetic acids: discovery of a non-free fatty acid-like, highly bioavailable G protein-coupled receptor 40/free fatty acid receptor 1 agonist as a glucose-dependent insulinotropic agent. J Med Chem. 2012;55(8):3960–3974. doi:10.1021/jm300170m
68. Takano R, Yoshida M, Inoue M, et al. Discovery of DS-1558: a potent and orally bioavailable GPR40 agonist. ACS Med Chem Lett. 2015;6(3):266–270. doi:10.1021/ml500391n
69. Clarivate. Cortellis Competitive Intelligence database. Available from: https://access.cortellis.cn.
70. Mikami S, Kitamura S, Negoro N, et al. Discovery of phenylpropanoic acid derivatives containing polar functionalities as potent and orally bioavailable G protein-coupled receptor 40 agonists for the treatment of type 2 diabetes. J Med Chem. 2012;55(8):3756–3776. doi:10.1021/jm2016123
71. Sasaki S, Kitamura S, Negoro N, et al. Design, synthesis, and biological activity of potent and orally available G protein-coupled receptor 40 agonists. J Med Chem. 2011;54(5):1365–1378. doi:10.1021/jm101405t
72. Srivastava A, Yano J, Hirozane Y, et al. High-resolution structure of the human GPR40 receptor bound to allosteric agonist TAK-875. Nature. 2014;513(7516):124–127. doi:10.1038/nature13494
73. Hedrington MS, Davis SN. Discontinued in 2013: diabetic drugs. Expert Opin Invest Drugs. 2014;23(12):1703–1711. doi:10.1517/13543784.2014.964859
74. Lead GPR40 agonist bites the dust. Nat Rev Drug Discov. 2014;13(2). doi:10.1038/nrd4246
75. Arnott JA, Planey SL. The influence of lipophilicity in drug discovery and design. Expert Opin Drug Discov. 2012;7(10):863–875. doi:10.1517/17460441.2012.714363
76. Tomita T, Hosoda K, Fujikura J, Inagaki N, Nakao K. The G-protein-coupled long-chain fatty acid receptor GPR40 and glucose metabolism. Front Endocrinol. 2014;5:152. doi:10.3389/fendo.2014.00152
77. McEuen K, Borlak J, Tong W, Chen M. Associations of drug lipophilicity and extent of metabolism with drug-induced liver injury. Int J Mol Sci. 2017;18(7):1335. doi:10.3390/ijms18071335
78. Urano Y, Oda S, Tsuneyama K, Yokoi T. Comparative hepatic transcriptome analyses revealed possible pathogenic mechanisms of fasiglifam (TAK-875)-induced acute liver injury in mice. Chem Biol Interact. 2018;296:185–197. doi:10.1016/j.cbi.2018.09.011
79. Doerfler H, Botesteanu D-A, Blech S, Laux R. Untargeted metabolomic analysis combined with multivariate statistics reveal distinct metabolic changes in GPR40 agonist-treated animals related to bile acid metabolism. Front Mol Biosci. 2021;7:598369. doi:10.3389/fmolb.2020.598369
80. NCT01746017. A study of LY2922470 in healthy participants and participants with diabetes. Available from: www.ClincialTrials.gov.
81. NCT01867216. A study of multiple doses of LY2922470 in participants with diabetes. Available from: www.ClincialTrials.gov.
82. CTR20211213. Phase Ic-II clinical trial of TSL-1806 capsule in type 2 diabetes patients. Available from: www.chinadrugtrials.org.cn.
83. Lu YY, Zhou WL, Cui QH, Cui CM. G protein-coupled receptor 40 agonist LY2922470 alleviates ischemic-stroke-induced acute brain injury and functional alterations in mice. Int J Mol Sci. 2023;24(15):12244. doi:10.3390/ijms241512244
84. Kim JW, Roh E, Choi KM, Yoo HJ, Hwang HJ, Baik SH. GPR40 agonism modulates inflammatory reactions in vascular endothelial cells. Diabet Metabol J. 2022;46(3):506–511. doi:10.4093/dmj.2021.0092
85. NCT04622111. Safety and pharmacokinetics study of CPL207280 compound in healthy volunteers. Available from: www.ClincialTrials.gov.
86. NCT05248776. Efficacy, safety and pharmacokinetics study of CPL207280 after 2-weeks administration in subjects with type 2 diabetes. Available from: www.ClincialTrials.gov.
87. Bazydlo-Guzenda K, Buda P, Mach M, et al. Evaluation of the hepatotoxicity of the novel GPR40 (FFAR1) agonist CPL207280 in the rat and monkey. PLoS One. 2021;16(9):e0257477. doi:10.1371/journal.pone.0257477
88. Lukin A, Bakholdina A, Zhurilo N, et al. Exploration of the nitrogen heterocyclic periphery around the core of the advanced FFA1 agonist fasiglifam (TAK-875). Arch Pharm. 2021;354(4):2000275. doi:10.1002/ardp.202000275
89. Ferdaoussi M, Bergeron V, Zarrouki B, et al. G protein-coupled receptor (GPR) 40-dependent potentiation of insulin secretion in mouse islets is mediated by protein kinase D1. Diabetologia. 2012;55:2682–2692. doi:10.1007/s00125-012-2650-x
90. Hauge M, Vestmar MA, Husted AS, et al. GPR40 (FFAR1)–combined Gs and Gq signaling in vitro is associated with robust incretin secretagogue action ex vivo and in vivo. Mol Metabol. 2015;4(1):3–14. doi:10.1016/j.molmet.2014.10.002
91. Milligan G, Alvarez‐Curto E, Watterson K, Ulven T, Hudson B. Characterizing pharmacological ligands to study the long‐chain fatty acid receptors GPR 40/FFA 1 and GPR 120/FFA 4. Br J Pharmacol. 2015;172(13):3254–3265. doi:10.1111/bph.12879
92. Zhao X, Yoon D-O, Yoo J, Park H-J. Structure-activity relationship study and biological evaluation of 2-(Disubstituted phenyl)-indole-5-propanoic acid derivatives as GPR40 full agonists. J Med Chem. 2021;64(7):4130–4149. doi:10.1021/acs.jmedchem.1c00031
93. Ge W, Yang B, Chen L, Zhou Z, Jin Y. Discovery of novel G-protein-coupled receptor 40 agonist with phenylacetic acid scaffold for the treatment of type 2 diabetes. Chemistryselect. 2021;6(29):7372–7375. doi:10.1002/slct.202101589
94. Luquet S, Gaudel C, Holst D, et al. Roles of PPAR delta in lipid absorption and metabolism: a new target for the treatment of type 2 diabetes. Biochim Biophys Acta Mol Basis Dis. 2005;1740(2):313–317. doi:10.1016/j.bbadis.2004.11.011
95. Tang T, Abbott MJ, Ahmadian M, Lopes AB, Wang Y, Sul HS. Desnutrin/ATGL activates PPARδ to promote mitochondrial function for insulin secretion in islet β cells. Cell Metab. 2013;18(6):883–895. doi:10.1016/j.cmet.2013.10.012
96. Yang Y, Tong Y, Gong M, et al. Activation of PPARβ/δ protects pancreatic β cells from palmitate-induced apoptosis by upregulating the expression of GLP-1 receptor. Cell Signal. 2014;26(2):268–278. doi:10.1016/j.cellsig.2013.11.019
97. Zhou Z, Cai Z, Zhang C, et al. Design, synthesis, and biological evaluation of novel dual FFA1 and PPARδ agonists possessing phenoxyacetic acid scaffold. Bioorg Med Chem. 2022;56:116615. doi:10.1016/j.bmc.2022.116615
98. Cabrera N, Cuesta SA, Mora JR, et al. In silico searching for alternative lead compounds to treat type 2 diabetes through a QSAR and molecular dynamics study. Pharmaceutics. 2022;14(2):232. doi:10.3390/pharmaceutics14020232
99. Talukdar S, Bae EJ, Imamura T, et al. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell. 2010;142(5):687–698. doi:10.1016/j.cell.2010.07.041
100. Ichimura A, Hirasawa A, Poulain-Godefroy O, et al. Dysfunction of lipid sensor GPR120 leads to obesity in both mouse and human. Nature. 2012;483(7389):350–354. doi:10.1038/nature10798
101. Oh DY, Walenta E, Akiyama TE, et al. A Gpr120-selective agonist improves insulin resistance and chronic inflammation in obese mice. Nature Med. 2014;20(8):942–947. doi:10.1038/nm.3614
102. Bonnefond A, Lamri A, Leloire A, et al. Contribution of the low-frequency, loss-of-function p. R270H mutation in FFAR4 (GPR120) to increased fasting plasma glucose levels. J Med Genet. 2015;52(9):595–598. doi:10.1136/jmedgenet-2015-103065
103. Li A, Li Y, Du L. Biological characteristics and agonists of GPR120 (FFAR4) receptor: the present status of research. Future Med Chem. 2015;7(11):1457–1468. doi:10.4155/fmc.15.75
104. Satapati S, Qian Y, Wu MS, et al. GPR120 suppresses adipose tissue lipolysis and synergizes with GPR40 in antidiabetic efficacy. J Lipid Res. 2017;58(8):1561–1578. doi:10.1194/jlr.M075044
105. Luckmann M, Shenol A, Nissen TA, et al. Optimization of first-in-class dual-acting FFAR1/FFAR4 allosteric modulators with novel mode of action. ACS Med Chem Lett. 2022;13(12):1839–1847. doi:10.1021/acsmedchemlett.2c00160
106. Chen C, Guo SM, Sun YJ, et al. Discovery of orally effective and safe GPR40 agonists by incorporating a chiral, rigid and polar sulfoxide into 8-position to the carboxylic acid. Eur J Med Chem. 2023;251:115267. doi:10.1016/j.ejmech.2023.115267
107. Wang B, Cai Z, Yao H, et al. Discovery of a structurally novel, potent, and once-weekly free fatty acid receptor 1 agonist for the treatment of diabetes. Eur J Med Chem. 2023;245:114883. doi:10.1016/j.ejmech.2022.114883
108. Nakamoto K, Nishinaka T, Matsumoto K, et al. Involvement of the long-chain fatty acid receptor GPR40 as a novel pain regulatory system. Brain Res. 2012;1432:74–83. doi:10.1016/j.brainres.2011.11.012
109. Harada S, Haruna Y, Aizawa F, et al. Involvement of GPR40, a long-chain free fatty acid receptor, in the production of central post-stroke pain after global cerebral ischemia. Eur J Pharmacol. 2014;744:115–123. doi:10.1016/j.ejphar.2014.09.036
110. Nakamoto K, Nishinaka T, Sato N, et al. The activation of supraspinal GPR 40/FFA 1 receptor signalling regulates the descending pain control system. Br J Pharmacol. 2015;172(5):1250–1262. doi:10.1111/bph.13003
111. Stoddart LA, Smith NJ, Milligan G. International Union of Pharmacology. LXXI. Free fatty acid receptors FFA1,-2, and-3: pharmacology and pathophysiological functions. Pharmacol Rev. 2008;60(4):405–417. doi:10.1124/pr.108.00802
112. Osborn O, Olefsky JM. The cellular and signaling networks linking the immune system and metabolism in disease. Nature Med. 2012;18(3):363–374. doi:10.1038/nm.2627
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.