Back to Journals » Therapeutics and Clinical Risk Management » Volume 22
Real-World Evidence on Statin Use for Traumatic Brain Injury Associated Degenerative Neurological Disorders
Authors Lin YH, Tsai IJ ![]() , Chang YA, Huang CH, Chang CC
, Chang YA, Huang CH, Chang CC ![]()
Received 17 January 2026
Accepted for publication 23 March 2026
Published 26 March 2026 Volume 2026:22 591742
DOI https://doi.org/10.2147/TCRM.S591742
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
Yu-Hui Lin,1,* I-Ju Tsai,2,* Yu-Ang Chang,3 Ching-Hui Huang,4– 6,* Chia-Chu Chang7,8,*
1Division of Integrated Dementia Care, Department of Neurology, Kuang Tien General Hospital, Taichung, Taiwan; 2Department of Medical Research, Kuang Tien General Hospital, Taichung, Taiwan; 3Department of Medical Education, Taipei Medical University Hospital, Taipei, Taiwan; 4Division of Cardiology, Department of Internal Medicine, Changhua Christian Hospital, Changhua, Taiwan; 5Department of Mathematics, National Changhua University of Education, Changhua, Taiwan; 6Department of Beauty Science and Graduate Institute of Beauty Science Technology, Chienkuo Technology University, Changhua, Taiwan; 7Department of Internal Medicine, Kuang Tien General Hospital, Taichung, Taiwan; 8Department of Nutrition, Hungkuang University, Taichung, Taiwan
*These authors contributed equally to this work
Correspondence: Chia-Chu Chang, Department of Internal Medicine, Kuang Tien General Hospital, No. 127, Sec. 7, Xiangshang Road, Shalu District, Taichung City, 433, Taiwan, Tel +886 4 2662 5111, Email [email protected] Ching-Hui Huang, Division of Cardiology, Department of Internal Medicine, Changhua Christian Hospital, No. 135, Nian-Siau Street, Changhua City, Changhua, 500, Taiwan, Email [email protected]
Background: Traumatic brain injury (TBI) is a major risk factor for subsequent neurodegenerative disorders. Emerging evidence suggests that statins may mitigate neuroinflammation and lipid dysregulation after TBI. This study aimed to evaluate the association between statin use and the risk of post-TBI degenerative neurological disorders in a global, real-world population.
Methods: We conducted a retrospective cohort study using the TriNetX global research network and a 1:1 propensity score matching procedure to balance demographic, clinical, and medication covariates. Patients with the first TBI diagnosis from 2000 onward were included. Individuals with continuous statin use every year from 1 year before to 5 years after the index date were classified as the statin treated group, whereas those with no statin prescriptions during the same period were categorized as the untreated group. Patients younger than 18 years and those with pre-index dementia, stroke, or CNS infection were excluded. A 1:1 propensity score matching procedure was applied to balance demographic, clinical, and medication covariates. Cox proportional hazards models estimated hazard ratios (HRs) for vascular dementia, non-vascular dementia, stroke, depression, and Parkinson’s disease during the 5-year follow-up.
Results: After matching, 21,427 patients were included in each group. Statin-treated patients demonstrated higher risks of all neurologic outcomes compared with untreated individuals, including vascular dementia (HR 2.47; 95% CI, 1.83– 3.34), non-vascular dementia (HR 1.45; 95% CI, 1.30– 1.61), stroke (HR 1.80; 95% CI, 1.70– 1.89), depression (HR 1.91; 95% CI, 1.79– 2.04), and Parkinson’s disease (HR 1.63; 95% CI, 1.34– 1.98). Subgroup analyses showed consistent risk elevations across sex and age categories.
Conclusion: The current Real-World data do not provide evidence supporting the use of statins primarily for neuroprotection after TBI. These findings underscore the importance of pursuing alternative therapeutic strategies that directly target TBI-associated neurodegenerative mechanisms.
Keywords: traumatic brain injury, TBI, statins, neurodegenerative disease, real-world data, TriNetX
Introduction
Traumatic brain injury (TBI) represents a significant and growing global public health challenge, often referred to as the “silent epidemic” because of its high incidence, long-term morbidity, and limited public recognition.1 It is estimated that approximately 69 million people experience TBI annually worldwide, leading to substantial mortality and disability.1,2 In the United States alone, TBI contributes to nearly 56,000 deaths per year, and more than 5 million individuals live with TBI-related disability.1,3 Beyond the immediate neurological consequences, TBI has emerged as an important risk factor for a spectrum of chronic neurodegenerative diseases, including Alzheimer’s disease (AD), Parkinson’s disease (PD), and chronic traumatic encephalopathy (CTE).4 Severe or repetitive head injuries during early or midlife increase the risk of late-life dementia by up to fivefold compared with the general population.5,6
The pathophysiology of TBI encompasses both primary and secondary injury mechanisms. The primary mechanical insults, such as blunt impact or acceleration-deceleration, induces immediate structural damage. The secondary injury cascade evolves over hours to years and involves a complex interplay of neuroinflammatory, excitotoxic, oxidative, and metabolic processes.7 Microglial activation, astrocytic gliosis, mitochondrial dysfunction, and blood–brain barrier (BBB) breakdown collectively contribute to persistent neuroinflammation and progressive neuronal loss.8 Proinflammatory cytokines such as tumor necrosis factor-alpha (TNF-α), interleukin (IL)-1β, and IL-6 perpetuate synaptic dysfunction and neuronal apoptosis.7 Chronic microglial activation has been documented decades after a single TBI, supporting a sustained inflammatory response that may drive neurodegeneration and cognitive decline.9,10 Moreover, TBI-induced dysregulation of lipid metabolism has recently been recognized as a contributing factor to neurodegeneration. Altered levels of sphingomyelin and very long-chain fatty acids (VLCFAs) within the injured brain are associated with glial activation and oxidative stress.11 The elongation of very long-chain fatty acid protein 1 (ELOVL1), a key enzyme in VLCFA synthesis, plays a role in astrocyte-mediated neurotoxicity, suggesting that targeting lipid metabolic pathways could mitigate secondary neuronal damage.12,13 In the study, statin use correlated with improved functional outcomes, enhanced neuronal plasticity, and reduced risk of post-traumatic dementia.13 Although some heterogeneity exists regarding optimal dosing and timing, the overall evidence strongly supports a protective association.
Statins, or 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors, are widely used lipid-lowering agents with well-established cardiovascular benefits. However, beyond cholesterol reduction, statins exert multiple pleiotropic effects including anti-inflammatory, antioxidative, endothelial-stabilizing, and neuroprotective properties.14,15 These effects are mediated through the inhibition of isoprenoid synthesis, modulation of nitric oxide pathways, and suppression of inflammatory signaling cascades such as NF-κB and mTOR.16,17 Preclinical studies have demonstrated that both early and delayed administration of statins after experimental TBI improve neurological outcomes by promoting neurogenesis, angiogenesis, and synaptic repair.18 Simvastatin, in particular, has been shown to enhance motor recovery and inhibit glial scar formation even when treatment is initiated up to one week after injury.12 Multiple mechanisms have been proposed to explain the neuroprotective actions of statins after TBI. Statins attenuate microglial activation and cytokine release, thereby reducing neuroinflammation.18 They also enhance endothelial nitric oxide synthase (eNOS) expression, improving cerebral blood flow and microvascular integrity.19 Additionally, statins modulate glutamate transporter activity, lowering excitotoxic neuronal injury by reducing extracellular glutamate levels.20 On a molecular level, statins upregulate the Nrf2-antioxidant response element (ARE) pathway, conferring resistance to oxidative stress.21 Through these pleiotropic effects, statins may stabilize neuronal homeostasis and prevent the long-term neurodegenerative processes that follow TBI.
Clinical studies corroborate the neuroprotective potential of statins in TBI patients. In a large retrospective cohort of older Medicare beneficiaries, post-TBI statin use was associated with significant reductions in mortality and in the incidence of stroke, depression, and Alzheimer’s disease and related dementias (ADRD).11 A systematic review encompassing 18 clinical and experimental studies further supported these observations, demonstrating that statins—particularly simvastatin—consistently improved cognitive outcomes.22
In contrast, large-scale observational and interventional studies have repeatedly failed to demonstrate meaningful reductions in cognitive decline or dementia incidence among statin users.23,24 In a well-designed cohort study of community-dwelling older adults aged 70–79 years, Rea et al reported no significant difference in cognitive outcomes or dementia risk over approximately six years of follow-up between statin users and non-users.25 Sensitivity analyses addressing confounding by indication, medication adherence, and vascular comorbidities produced comparable results.25,26 To date, no large-scale global EHR-based study has systematically evaluated the long-term risk of dementia, stroke, depression, and Parkinson’s disease after TBI in patients with continuous statin exposure.
This study aimed to evaluate the association between post-TBI statin use and the risk of developing degenerative neurological disorders in a global, real-world population.
Methods
Study Design and Patients
We conducted a retrospective cohort study using the TriNetX global research network, which contains de-identified electronic health records from participating healthcare organizations. This study was conducted in accordance with the principles of the Declaration of Helsinki and was approved by the Institutional Review Board (IRB) of Kuang Tien General Hospital (ktgh 11501). The requirement for informed consent was waived by the IRB because this was a retrospective, non-interventional study using de-identified secondary data obtained from the TriNetX global research network. The data were obtained from the TriNetX Global Research Network, a commercially licensed real-world data platform and are not publicly available. All data are in compliance with the Health Insurance Portability and Accountability Act (HIPAA) and applicable data protection regulations, and access is restricted to authorized users for research purposes. The study involved minimal risk to participants, and all data were analyzed and reported in aggregate to ensure confidentiality and privacy protection.
Patients with TBI diagnosed between 2000 and the most recent data cutoff were eligible for inclusion. TBI was defined using ICD-10 codes for intracranial injury and skull fractures (S02.0, S02.1, S02.8, S02.91, S04.02–S04.04, S06, S07.1, T74.4). The index date was defined as the date of first qualifying TBI diagnosis. To ensure capture of complete baseline information, individuals with a TBI diagnosis prior to 2000 were excluded.
Exposure Definition
The index date was defined as the date of the first TBI diagnosis. Patients were categorized into two exposure groups based on their statin utilization patterns. The statin therapy group consisted of individuals with documented prescriptions for statins, including atorvastatin, rosuvastatin, simvastatin, pravastatin, lovastatin, fluvastatin, or pitavastatin, and evidence of continuous statin use every year from 1 year before to 5 years after the index date. In contrast, the non–statin therapy group included patients with no recorded statin prescriptions in any year during the same period relative to the index date.
Baseline Exclusions
We excluded individuals who were <18 years old, those with missing sex information, and those with any diagnosis of dementia, stroke (I60–I69), or central nervous system (CNS) infection (ICD-10 G00–G09) prior to the index date. Patients with baseline comorbidities were identified using validated ICD-10 code groups, including diabetes (E08–E13), chronic kidney disease (N18), alcohol-related disorders (F10), cardiovascular disease (I00–I99), and stroke. Baseline use of cardiovascular medications was characterized using ATC codes C02A/C/D/K, C03A–D, C07A, C08C/D, and C09A–D.
Outcomes
Patients were followed for five years from the index date for the occurrence of the following incident outcomes: Vascular dementia (F01), Non-vascular dementia, including Alzheimer’s disease (F02, F03, G30), Stroke (I60–I69), Depression (F32, F33), Parkinson’s disease (G20, G21).
Statistical Analysis
Only variables available within the TriNetX database were included in the analyses; therefore, potential unmeasured confounders, such as smoking status, body mass index, socioeconomic status, and TBI severity, could not be adjusted for. Propensity score matching (PSM) was performed using 1:1 greedy nearest neighbor matching with a caliper of 0.1 standard deviations of the logit of the propensity score. Age, sex, baseline comorbidities (including diabetes mellitus, chronic kidney disease, and alcohol-related disorders), and cardiovascular medication use (including agents acting on the renin–angiotensin system, beta-blockers, and calcium channel blockers) were included in the propensity score model to balance baseline characteristics between the exposure groups. P-values and standardized mean differences are reported for baseline characteristics before and after matching. Incidence of outcomes within five years was compared using Cox proportional hazards models to estimate hazard ratios (HRs) with 95% confidence intervals (CIs). Five-year cumulative incidence was calculated as the proportion of patients experiencing each outcome during follow-up. Death was treated as a censoring event in the Cox proportional hazards models, and competing risk analyses were not performed. Prespecified subgroup analyses included sex and age categories using cut-points of 65 years. All analyses were conducted on the TriNetX platform.
Results
Study Population
A total of 21,427 patients met criteria for the TBI with statin therapy group, and 1,384,489 were included in the TBI without statin therapy group prior to matching (Figure 1). After propensity score matching, 21,427 individuals remained in each group with well-balanced baseline characteristics. Before matching, the treatment group was significantly older (Table 1; mean age 60.6 vs. 40.4 years, P < 0.0001) and had higher prevalence of diabetes, CKD, and cardiovascular medication use. After matching, all covariates were well balanced with no statistically significant differences.
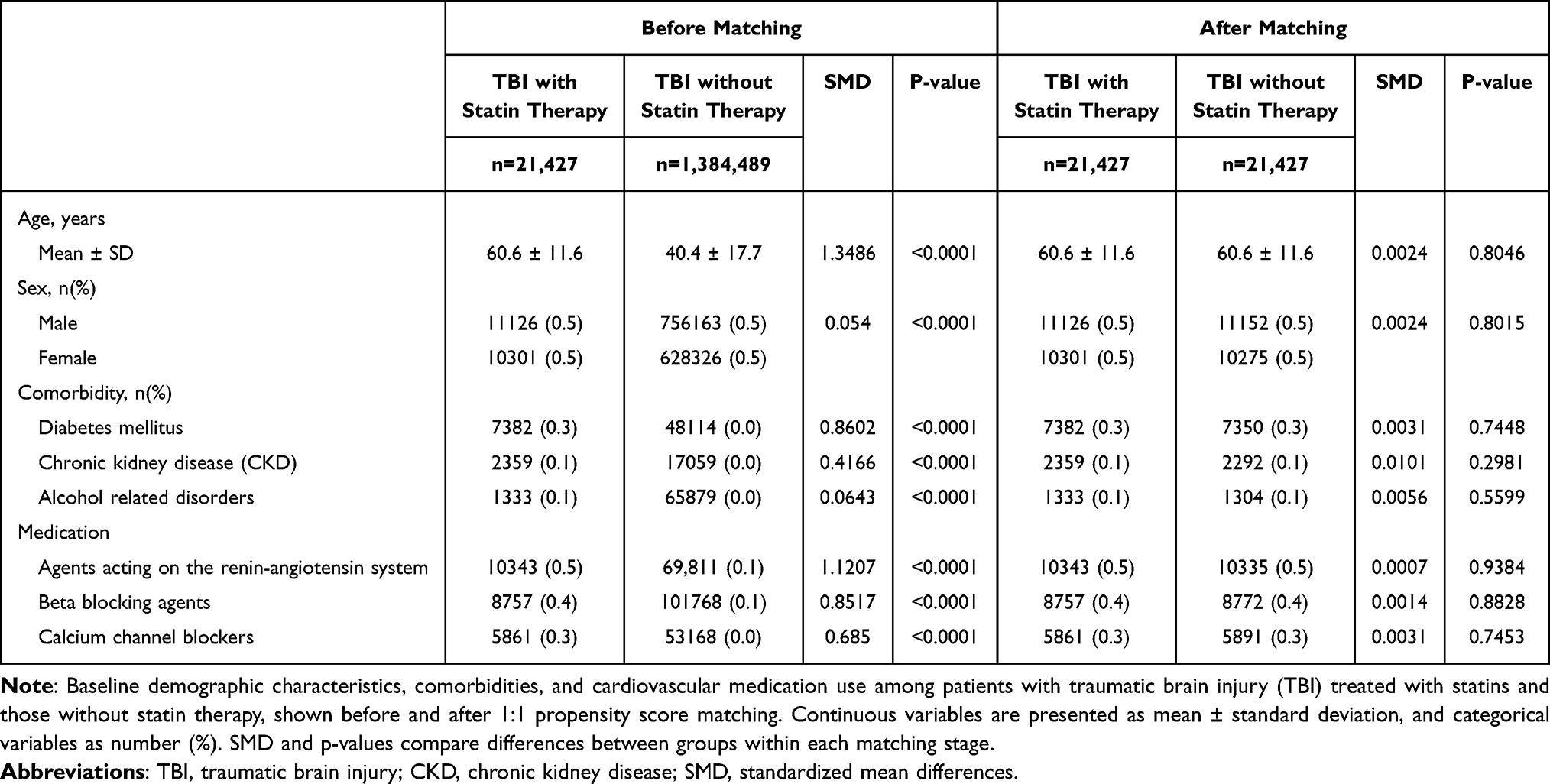 |
Table 1 Baseline Characteristics of Patients with Traumatic Brain Injury Before and After Propensity Score Matching |
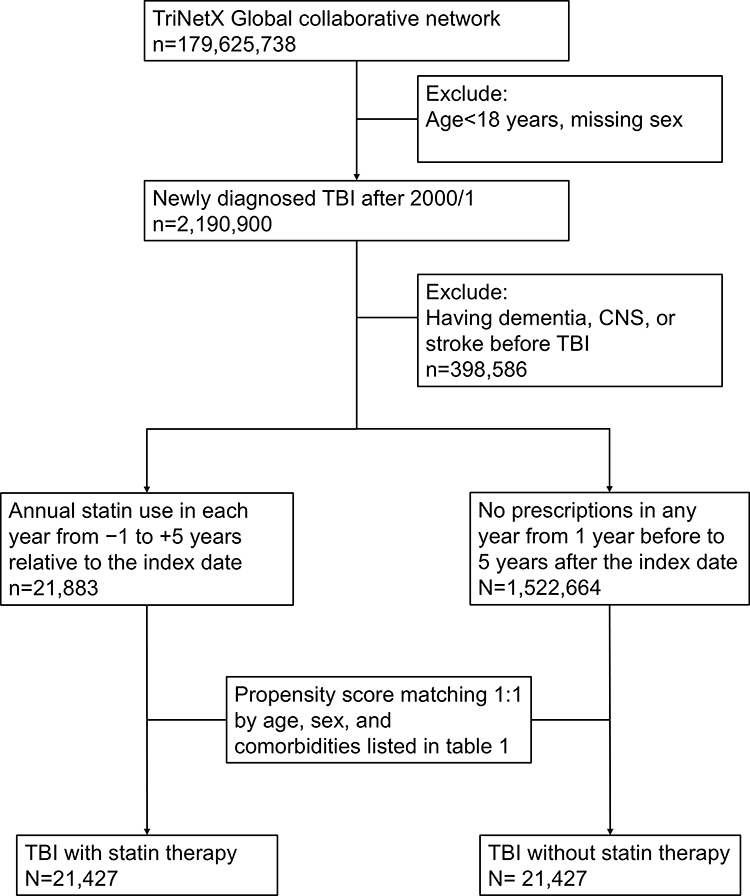 |
Figure 1 Flow diagram of study cohort selection. Flow diagram illustrating patient selection from the TriNetX research network. Exclusion criteria included age <18 years, missing sex information, and pre-index diagnoses of dementia, stroke, or central nervous system infection. Eligible patients were classified according to statin exposure and included in the 1:1 propensity score–matched analysis. “Before matching” characteristics are restricted to patients eligible (n=21,883) for matching in TriNetX; unmatched individuals are automatically excluded (n=21,427) prior to generation of the PSM report. |
Primary Outcomes
During the 5-year follow-up, patients in the statin-treated TBI cohort exhibited consistently higher incidence rates of all neurologic outcomes compared with their matched counterparts without statin exposure (Table 2). In the propensity score–matched analysis, statin use was associated with significantly increased risks of vascular dementia (HR 2.47; 95% CI, 1.83–3.34), non-vascular dementia (HR 1.45; 95% CI, 1.30–1.61), stroke (HR 1.80; 95% CI, 1.70–1.89), depression (HR 1.91; 95% CI, 1.79–2.04), and Parkinson’s disease (HR 1.63; 95% CI, 1.34–1.98).
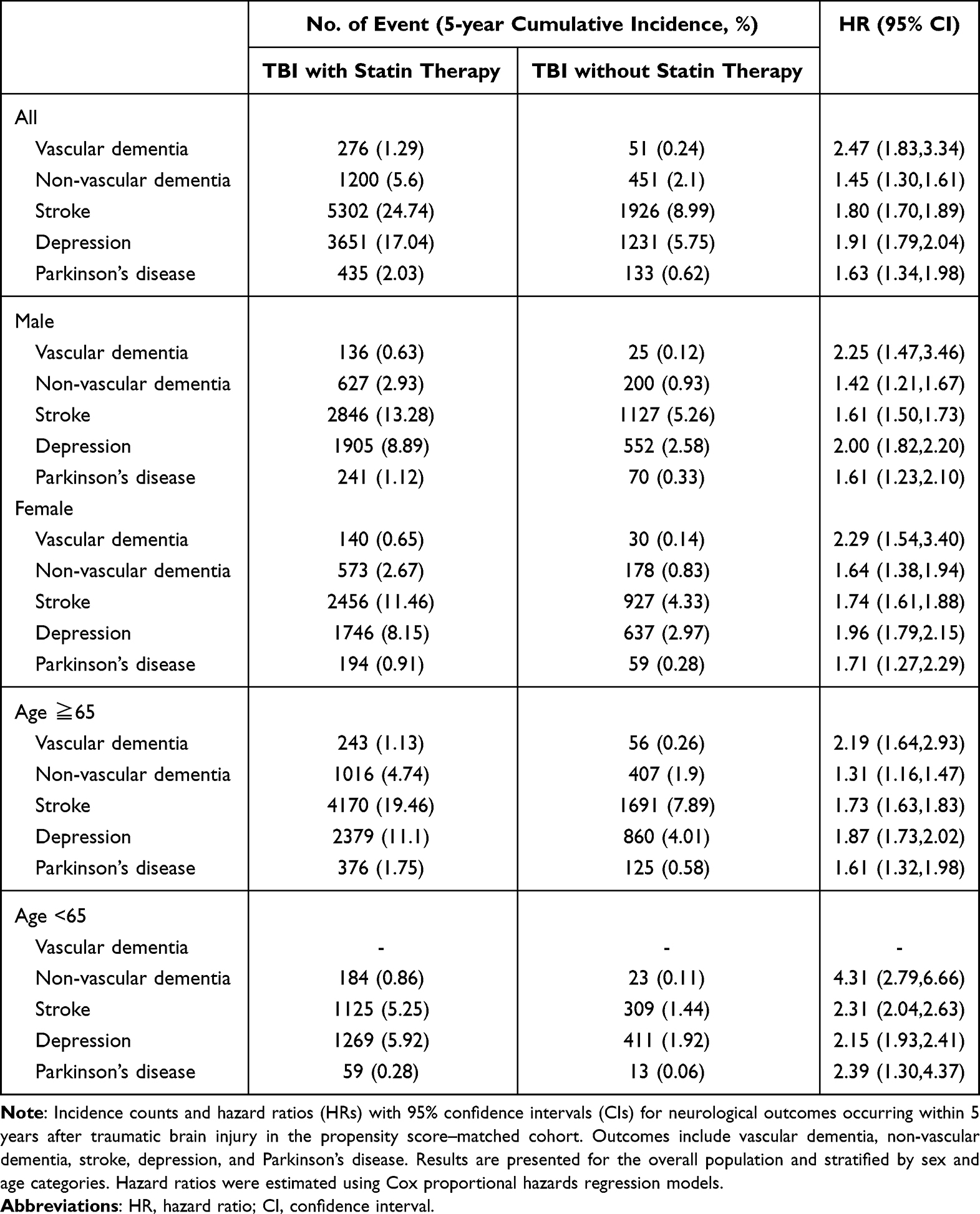 |
Table 2 Neurological Outcomes Associated with Statin Use After Traumatic Brain Injury: in the Propensity Score–Matched Cohort |
Sex-Stratified Analyses
In sex-stratified analyses, elevated risks associated with statin exposure persisted in both men and women. Among male patients, statin use was associated with higher risks of vascular dementia (HR 2.25; 95% CI, 1.47–3.46), non-vascular dementia (HR 1.42; 95% CI, 1.21–1.67), stroke (HR 1.61; 95% CI, 1.50–1.73), depression (HR 2.00; 95% CI, 1.82–2.20), and Parkinson’s disease (HR 1.61; 95% CI, 1.23–2.10). Similar patterns were observed among female patients, for whom statin exposure was likewise associated with increased risks of vascular dementia (HR 2.29; 95% CI, 1.54–3.40), non-vascular dementia (HR 1.64; 95% CI, 1.38–1.94), stroke (HR 1.74; 95% CI, 1.61–1.88), depression (HR 1.96; 95% CI, 1.79–2.15), and Parkinson’s disease (HR 1.71; 95% CI, 1.27–2.29).
Age-Stratified Analyses
Age-stratified analyses revealed broadly consistent associations among older adults. In individuals aged ≥65 years, statin use was associated with elevated risks of vascular dementia (HR 2.19; 95% CI, 1.64–2.93), non-vascular dementia (HR 1.31; 95% CI, 1.16–1.47), stroke (HR 1.73; 95% CI, 1.63–1.83), depression (HR 1.87; 95% CI, 1.73–2.02), and Parkinson’s disease (HR 1.61; 95% CI, 1.32–1.98). In contrast, among patients younger than 65 years, the number of vascular dementia events was insufficient for analysis; however, significantly elevated risks were observed for non-vascular dementia (HR 4.31; 95% CI, 2.79–6.66), stroke (HR 2.31; 95% CI, 2.04–2.63), depression (HR 2.15; 95% CI, 1.93–2.41), and Parkinson’s disease (HR 2.39; 95% CI, 1.30–4.37). These findings suggest that the increased risks associated with statin exposure after TBI are evident across age groups.
Overall, Figure 2 demonstrates that statin use after TBI was not associated with a neuroprotective benefit, including dementia (both vascular and non-vascular types), stroke, depression, and Parkinson’s disease. All hazard ratios (HRs) are greater than 1, indicating an increased relative risk among statin users compared with matched non-statin users. The subgroup analysis suggests that these associations were evaluated across stratified variables including sex, age, laboratory parameters, and medical history, indicating that the elevated risks were not confined to a single demographic subgroup.
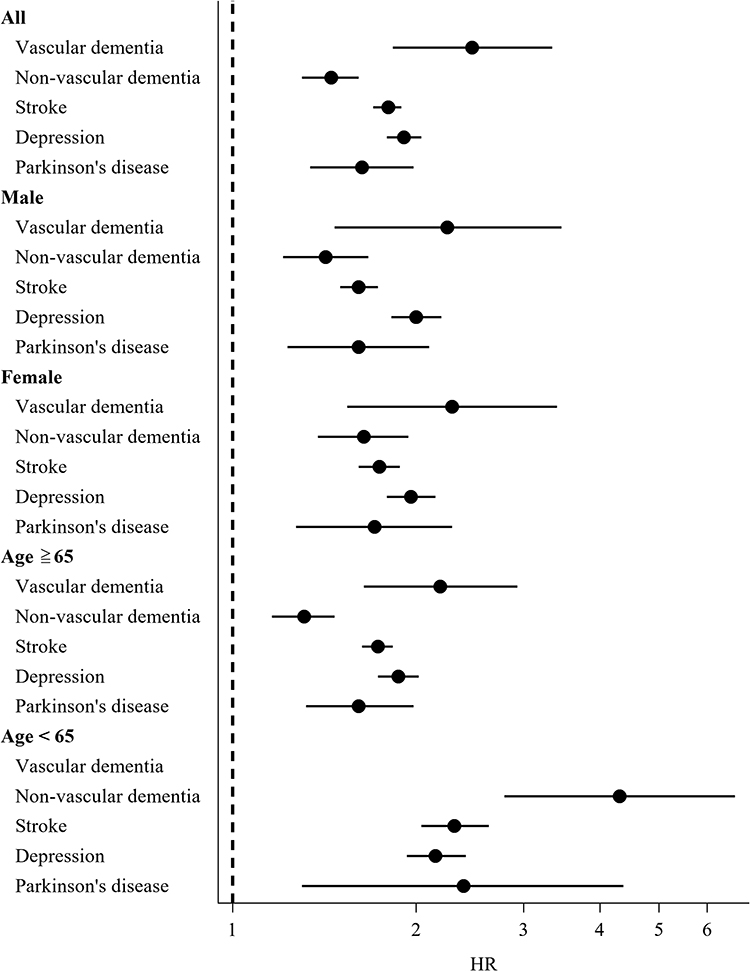 |
Figure 2 Forest plot illustrates subgroup analyses of statin use and neurological outcomes. Forest plot demonstrated that pre-injury statin uses significantly increased post-TBI neurological outcomes across prespecified subgroups (male, female, age ≥65 years, age <65 years). |
In Figure 3a–e, across all five panels, the Kaplan–Meier curves consistently demonstrated higher cumulative incidence rates in statin users compared with matched non-users. The early and persistent divergence of curves indicated that the increased risks, including dementia (vascular and non-vascular), stroke, depression, and Parkinson’s disease, were not transient but remain evident throughout the 5-year follow-up.
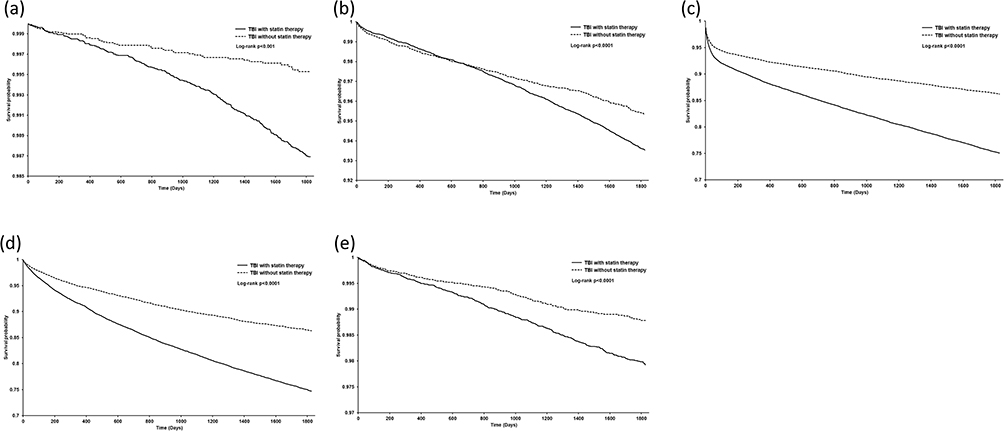 |
Figure 3 Cumulative incidence of neurological outcomes after traumatic brain injury. Across all five panels, the Kaplan–Meier curves consistently demonstrated higher cumulative incidence rates in statin users compared with matched non-users. The early and persistent divergence of curves indicated that the increased risks, including (a) vascular dementia, (b) non-vascular dementia, (c) stroke, (d) depression, and (e) Parkinson’s disease, were not transient but remain evident throughout the 5-year follow-up. Hazard ratios were estimated using Cox proportional hazards models. |
Discussion
In this large, global real-world cohort study using the TriNetX Research Network, we examined the association between sustained statin use and long-term neurological outcomes following TBI. Our propensity score–matched analyses demonstrated that patients receiving continuous statin therapy before and after TBI exhibited higher risks of vascular dementia, non-vascular dementia, stroke, depression, and Parkinson’s disease during a five-year follow-up period. These associations were consistent across sex and age strata, underscoring the robustness of the observed findings in a heterogeneous real-world population. Contrary to several preclinical and selected clinical reports suggesting neuroprotective effects of statins,11,22 however, such studies often focused on older populations, short-term outcomes, or less stringent exposure definitions, which may partially explain the discrepancy with our findings.
Several plausible explanations for the observed increased risk are confounding by indication, which is given the strict continuous exposure definition extending 5 years after TBI and the potential for immortal time bias; patients prescribed statins are more likely to have underlying vascular risk factors such as diabetes, hypertension, and atherosclerotic cardiovascular diseases27 conditions independently associated with both cerebrovascular events and neurodegenerative disorders. Although we applied rigorous propensity score matching to balance measured covariates, residual confounding from unmeasured factors, including subclinical vascular disease burden, lifestyle variables, selection of a particularly adherent or high-risk cohort of statin users, cannot be fully excluded.
Another important consideration is the heterogeneity of statin effects in the central nervous system, which may differ by statin class, dose, timing, and duration. While lipophilic statins such as simvastatin readily cross the blood–brain barrier and exert anti-inflammatory effects in experimental settings, chronic inhibition of cholesterol synthesis within the brain may adversely affect synaptic integrity and neuronal repair, particularly in the setting of injury.15,16 Recent evidence indicates that rosuvastatin-loaded nanoemulsions exert neuroprotective effects by attenuating neuroinflammation and oxidative stress in experimental models, supporting the potential of nanoemulsion-based statin delivery strategies for neuroinflammatory disorders in post-TBI patients.28 Cholesterol is essential for synaptogenesis and membrane remodeling, processes that are critical for recovery after TBI; long-term suppression may therefore impair adaptive neuroplasticity.29
Our findings also align with large population-based studies in non-TBI cohorts that failed to demonstrate cognitive protection from statins. A systematic review by Richardson et al found no consistent association between statin use and reduced cognitive decline or dementia incidence in older adults.22 Similarly, the PROSPER randomized controlled trial reported no significant benefit of pravastatin on cognitive outcomes despite vascular risk reduction.25
The increased risk of stroke observed among statin-treated TBI patients in our cohort deserves particular attention. Although statins are well established for primary and secondary prevention of ischemic stroke, TBI-related cerebrovascular vulnerability is multifactorial and includes microvascular injury, endothelial dysfunction, and blood–brain barrier disruption.30,31 It is conceivable that patients requiring statins represent a subgroup with heightened baseline cerebrovascular fragility, in whom the cumulative burden of vascular pathology outweighs any potential statin-mediated benefit.27
Similarly, the association between statin use and depression following TBI may reflect complex neuropsychiatric interactions. While statins have been proposed to exert antidepressant effects via anti-inflammatory pathways,32 clinical evidence remains inconsistent, and some observational studies have suggested increased depressive symptoms among long-term statin users.33 Our findings support the need for cautious interpretation of statin effects on neuropsychiatric outcomes in vulnerable populations. TBI is a recognized risk factor for Parkinsonian syndromes, potentially through α-synuclein aggregation, mitochondrial dysfunction, and chronic microglial activation.34,35 While statins have been hypothesized to reduce α-synuclein pathology, epidemiologic data remain conflicting.36 Our results suggest that in the context of TBI, statins may not mitigate and may even exacerbate neurodegenerative vulnerability.
This study has several important strengths. The use of a large, multinational real-world database enhances generalizability, while the strict definition of continuous statin exposure minimizes misclassification. The application of propensity score matching across a wide range of comorbidities and concomitant medications strengthens causal inference.
Limitations
Several limitations should be considered when interpreting the findings of this study.
First, the TriNetX platform does not provide detailed information regarding traumatic brain injury severity. Important clinical variables such as Glasgow Coma Scale scores, neuroimaging findings, mechanism of injury, and standardized TBI severity classifications were unavailable. Because TBI severity is a strong determinant of long-term neurological outcomes, the inability to stratify or adjust for injury severity may have influenced the observed associations.
Second, several potentially important genetic, environmental, and socioeconomic factors could not be captured in the database. These include race-specific genetic predisposition, family history of neurodegenerative disease, dietary habits, sleep quality, income level, social support, herbal medicine use, immune function markers, and out-of-network or unrecorded medical treatments.
Third, medication exposure was determined based on prescription records rather than confirmed adherence. Therefore, actual patient compliance with statin therapy cannot be guaranteed.
Fourth, a potential methodological limitation is the risk of immortal time bias, as patients were classified as continuous statin users based on prescriptions extending up to 5 years after the index TBI, while follow-up began at the index date. In theory, this approach could introduce a period during which patients must survive to be classified as exposed, potentially leading to an underestimation of early post-TBI events in the statin group. However, in our cohort, most patients in the statin-treated group initiated therapy within the first year after TBI, and the requirement for continuous use mainly affects later follow-up; therefore, the magnitude of this bias is expected to be limited.
Finally, death was treated as a censoring event rather than being modeled as a competing risk. Given that mortality is substantial in post-TBI populations, the occurrence of death may preclude the development of the neurological outcomes of interest, which could potentially bias incidence estimates. Future studies using time-varying exposure definitions and competing risk models, such as Fine–Gray subdistribution hazard models, may provide more accurate risk estimates. Accordingly, these results should be interpreted with caution and validated in prospective TBI-focused studies.
In conclusion, this large real-world cohort study suggests that continuous statin use before and after TBI was associated with higher observed risk of multiple adverse neurological outcomes, including dementia, stroke, depression, and Parkinson’s disease. These statins remain essential for cardiovascular indications; the study does not argue against their use for established cardiovascular prevention, but rather against assuming added neuroprotective benefit in the TBI context. Careful prospective validation is warranted before statins can be recommended as disease-modifying agents for TBI-associated neurodegeneration.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
Yu-Hui Lin and I-Ju Tsai are co-first authors for this study. Ching-Hui Huang and Chia-Chu Chang are co-correspondence authors for this study. The authors report no conflicts of interest in this work.
References
1. Sultan W, Sapkota A, Khurshid H, et al. Effect on cognitive outcome after traumatic brain injury: a systematic review. Cureus. 2021;13(8):e16953. PMID: 34405076; PMCID: PMC8352842. doi:10.7759/cureus.16953
2. Taylor CA, Bell JM, Breiding MJ, Xu L. Traumatic brain injury-related emergency department visits, hospitalizations, and deaths - United States, 2007 and 2013. MMWR Surveill Summ. 2017;66(9):1–12. PMID: 28301451; PMCID: PMC5829835. doi:10.15585/mmwr.ss6609a1
3. Cortel-LeBlanc A, Cortel-LeBlanc M, Webster RJ, et al. TRANSCENDENT Concussion Research Team. Post-traumatic headache phenotypes and clinical characteristics. Cephalalgia. 2025;45(12):3331024251404912. PMID: 41370085. doi:10.1177/03331024251404912
4. Smith DH, Johnson VE, Stewart W. Chronic neuropathologies of single and repetitive TBI: substrates of dementia? Nat Rev Neurol. 2013;9(4):211–221. PMID: 23458973; PMCID: PMC4513655. doi:10.1038/nrneurol.2013.29
5. Fann JR, Ribe AR, Pedersen HS, et al. Long-term risk of dementia among people with traumatic brain injury in Denmark: a population-based observational cohort study. Lancet Psychiatry. 2018;5(5):424–431. PMID: 29653873. doi:10.1016/S2215-0366(18)30065-8
6. Gardner RC, Yaffe K. Epidemiology of mild traumatic brain injury and neurodegenerative disease. Mol Cell Neurosci. 2015;66(Pt B):75–80. PMID: 25748121; PMCID: PMC4461453. doi:10.1016/j.mcn.2015.03.001
7. Werner C, Engelhard K. Pathophysiology of traumatic brain injury. Br J Anaesth. 2007;99(1):4–9. PMID: 17573392. doi:10.1093/bja/aem131
8. El Baassiri MG, Raouf Z, Badin S, Escobosa A, Sodhi CP, Nasr IW. Dysregulated brain-gut axis in the setting of traumatic brain injury: review of mechanisms and anti-inflammatory pharmacotherapies. J Neuroinflammation. 2024;21(1):124. PMID: 38730498; PMCID: PMC11083845. doi:10.1186/s12974-024-03118-3
9. Loane DJ, Kumar A. Microglia in the TBI brain: the good, the bad, and the dysregulated. Exp Neurol. 2016;275 Pt 3(0 3):316–327. PMID: 26342753; PMCID: PMC4689601. doi:10.1016/j.expneurol.2015.08.018
10. Delcy SAS, Farrugia A, Diaz Nieves IA, O’Brien CA, Bennett FC, Cohen AS. Microglia depletion improves hippocampal circuit function after mild traumatic brain injury in male mice. Brain Behav Immun. 2026;131:106178. PMID: 41232618. doi:10.1016/j.bbi.2025.106178
11. Khokhar B, Simoni-Wastila L, Slejko JF, Perfetto E, Zhan M, Smith GS. Mortality and associated morbidities following traumatic brain injury in older medicare statin users. J Head Trauma Rehabil. 2018;33(6):E68–E76. PMID: 29385012; PMCID: PMC6066463. doi:10.1097/HTR.0000000000000369
12. Zhang F, Lv T, Xu T, et al. Acetylation of fatty acid synthase regulates microglial lipid droplets accumulation and pro-inflammatory activity following traumatic brain injury. Redox Biol. 2025;89:103978. PMID: 41412037. doi:10.1016/j.redox.2025.103978
13. Huo J, Feng L, Cheng Y, et al. Delayed simvastatin treatment improves neurological recovery after cryogenic traumatic brain injury through downregulation of ELOVL1 by inhibiting mTOR signaling. Brain Res Bull. 2024;217:111072. PMID: 39243948. doi:10.1016/j.brainresbull.2024.111072
14. Premnath SM, Nanda SK, Ray L, Arokiaraj MC. Effect of statins on the inflammatory markers in patients with coronary artery disease. J Lab Physicians. 2023;15(4):498–502. PMID: 37780883; PMCID: PMC10539054. doi:10.1055/s-0043-1768167
15. Choudhary A, Rawat U, Kumar P, Mittal P. Pleotropic effects of statins: the dilemma of wider utilization of statin. Egypt Heart J. 2023;75(1):1. PMID: 36602642; PMCID: PMC9816367. doi:10.1186/s43044-023-00327-8
16. Jain MK, Ridker PM. Anti-inflammatory effects of statins: clinical evidence and basic mechanisms. Nat Rev Drug Discov. 2005;4(12):977–987. PMID: 16341063. doi:10.1038/nrd1901
17. Gorabi AM, Kiaie N, Hajighasemi S, et al. Statin-induced nitric oxide signaling: mechanisms and therapeutic implications. J Clin Med. 2019;8(12):2051. PMID: 31766595; PMCID: PMC6947613. doi:10.3390/jcm8122051
18. Xu X, Gao W, Cheng S, et al. Anti-inflammatory and immunomodulatory mechanisms of atorvastatin in a murine model of traumatic brain injury. J Neuroinflammation. 2017;14(1):167. PMID: 28835272; PMCID: PMC5569493. doi:10.1186/s12974-017-0934-2
19. Wijaya A, Wang Y, Tang D, et al. A study of lovastatin and L-arginine co-loaded PLGA nanomedicine for enhancing nitric oxide production and eNOS expression. J Mater Chem B. 2022;10(4):607–624. PMID: 34994373. doi:10.1039/d1tb01455b
20. Vandresen-Filho S, Martins WC, Bertoldo DB, et al. Atorvastatin prevents glutamate uptake reduction induced by quinolinic acid via MAPKs signaling. Neurochem Res. 2016;41(8):2017–2028. PMID: 27084771. doi:10.1007/s11064-016-1913-1
21. Pordel S, McCloskey AP, Almahmeed W, Sahebkar A. The protective effects of statins in traumatic brain injury. Pharmacol Rep. 2024;76(2):235–250. PMID: 38448729. doi:10.1007/s43440-024-00582-9
22. Westphal Filho FL, Moss Lopes PR, de Almeida A M, et al. Statin use and dementia risk: a systematic review and updated meta-analysis. Alzheimers Dement. 2025;11(1):e70039. PMID: 39822593; PMCID: PMC11736423. doi:10.1002/trc2.70039
23. Richardson K, Schoen M, French B, et al. Statins and cognitive function: a systematic review. Ann Intern Med. 2013;159(10):688–697. PMID: 24247674. doi:10.7326/0003-4819-159-10-201311190-00007
24. Zissimopoulos JM, Barthold D, Brinton RD, Joyce G. Sex and race differences in the association between statin use and the incidence of Alzheimer disease. JAMA Neurol. 2017;74(2):225–232. PMID: 27942728; PMCID: PMC5646357. doi:10.1001/jamaneurol.2016.3783
25. Rea TD, Breitner JC, Psaty BM, et al. Statin use and the risk of incident dementia: the Cardiovascular Health Study. Arch Neurol. 2005;62(7):1047–1051. PMID: 16009757. doi:10.1001/archneur.62.7.1047
26. Shepherd J, Blauw GJ, Murphy MB, et al. PROSPER study group. PROspective study of pravastatin in the elderly at risk. Pravastatin in elderly individuals at risk of vascular disease (PROSPER): a randomised controlled trial. Lancet. 2002;360(9346):1623–1630. PMID: 12457784. doi:10.1016/s0140-6736(02)11600-x
27. Khaliq MA, Nofal S, Israr S, et al. A retrospective audit of statin prescription practices in diabetic patients with cardiovascular risk. Cureus. 2025;17(8):e90152. PMID: 40955263; PMCID: PMC12433618. doi:10.7759/cureus.90152
28. Saberi Z, Rostamkhani N, Saghatchi Zanjani M, et al. Neuroprotective effect of rosuvastatin-loaded nanoemulsions against lipopolysaccharide-induced neuroinflammation and oxidative stress. J Drug Target. 2026;34(1):57–67. PMID: 40751388. doi:10.1080/1061186X.2025.2538037
29. Cheon SY. Impaired cholesterol metabolism, neurons, and neuropsychiatric disorders. Exp Neurobiol. 2023;32(2):57–67. PMID: 37164646; PMCID: PMC10175956. doi:10.5607/en23010
30. Serban NL, Ungureanu G, Florian IS, Ionescu D. Cerebral vascular disturbances following traumatic brain injury: pathophysiology, diagnosis, and therapeutic perspectives–a narrative review. Life. 2025;15(9):1470. PMID: 41010412; PMCID: PMC12471948. doi:10.3390/life15091470
31. Sun B, Li L, Harris OA, Luo J. Blood-brain barrier disruption: a pervasive driver and mechanistic link between traumatic brain injury and Alzheimer’s disease. Transl Neurodegener. 2025;14(1):16. PMID: 40140960; PMCID: PMC11938631. doi:10.1186/s40035-025-00478-5
32. Tsai PY, Chen SM, Lin CY, et al. Possible association of statin use with the risk of depression: an up-to-date systematic review and meta-analysis. Gen Hosp Psychiatry. 2025;97:118–125. PMID: 41075578. doi:10.1016/j.genhosppsych.2025.10.001
33. Agustini B, Mohebbi M, Woods RL, et al; ASPREE Investigator Group. Association between statin use and depressive symptoms in a large community-dwelling older population living in Australia and the USA: a cross-sectional study. CNS Drugs. 2019;33(7):685–694. PMID: 31062260; PMCID: PMC6719539. doi:10.1007/s40263-019-00633-3
34. Delic V, Beck KD, Pang KCH, Citron BA. Biological links between traumatic brain injury and Parkinson’s disease. Acta Neuropathol Commun. 2020;8(1):45. PMID: 32264976; PMCID: PMC7137235. doi:10.1186/s40478-020-00924-7
35. Yildirim-Balatan C, Fenyi A, Besnault P, et al. Parkinson’s disease-derived α-synuclein assemblies combined with chronic-type inflammatory cues promote a neurotoxic microglial phenotype. J Neuroinflammation. 2024;21(1):54. PMID: 38383421; PMCID: PMC10882738. doi:10.1186/s12974-024-03043-5
36. Dai L, Wang J, He M, et al. Lovastatin alleviates α-synuclein aggregation and phosphorylation in cellular models of synucleinopathy. Front Mol Neurosci. 2021;14:682320. PMID: 34381332; PMCID: PMC8350347. doi:10.3389/fnmol.2021.682320
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.