Back to Journals » International Journal of Nanomedicine » Volume 12
Ravuconazole self-emulsifying delivery system: in vitro activity against Trypanosoma cruzi amastigotes and in vivo toxicity
Authors Spósito PA, Mazzeti AL, de Oliveira Faria C, Urbina JA, Pound-Lana G ![]() , Bahia MT, Mosqueira VF
, Bahia MT, Mosqueira VF ![]()
Received 2 February 2017
Accepted for publication 25 March 2017
Published 17 May 2017 Volume 2017:12 Pages 3785—3799
DOI https://doi.org/10.2147/IJN.S133708
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Pollyanna Álvaro Spósito,1 Ana Lia Mazzeti,1,2 Caroline de Oliveira Faria,1 Julio A Urbina,3 Gwenaelle Pound-Lana,1 Maria Terezinha Bahia,2 Vanessa Furtado Mosqueira1
1Laboratory of Pharmaceutics and Nanotechnology Research, Pharmacy Department, School of Pharmacy, Universidade Federal de Ouro Preto, Minas Gerais, Brazil; 2Parasite Diseases Research Laboratory, NUPEB, Medical School, Universidade Federal de Ouro Preto, MG, Brazil; 3Venezuelan Institute for Scientific Research, Apartado, Caracas, Venezuela
Abstract: Self-emulsifying drug delivery systems (SEDDSs) are lipid-based anhydrous formulations composed of an isotropic mixture of oil, surfactant, and cosurfactants usually presented in gelatin capsules. Ravuconazole (Biopharmaceutics Classification System [BCS] Class II) is a poorly water-soluble drug, and a SEDDS type IIIA was designed to deliver it in a predissolved state, improving dissolution in gastrointestinal fluids. After emulsification, the droplets had mean hydrodynamic diameters <250 nm, zeta potential values in the range of −45 mV to −57 mV, and showed no signs of ravuconazole precipitation. Asymmetric flow field-flow fractionation with dynamic and multiangle laser light scattering was used to characterize these formulations in terms of size distribution and homogeneity. The fractograms obtained at 37°C showed a polydisperse profile for all blank and ravuconazole–SEDDS formulations but no large aggregates. SEDDS increased ravuconazole in vitro dissolution extent and rate (20%) compared to free drug (3%) in 6 h. The in vivo toxicity of blank SEDDS comprising Labrasol® surfactant in different concentrations and preliminary safety tests in repeated-dose oral administration (20 days) showed a dose-dependent Labrasol toxicity in healthy mice. Ravuconazole–SEDDS at low surfactant content (10%, v/v) in Trypanosoma cruzi-infected mice was safe during the 20-day treatment. The anti-T. cruzi activity of free ravuconazole, ravuconazole–SEDDS and each excipient were evaluated in vitro at equivalent ravuconazole concentrations needed to inhibit 50% or 90% (IC50 and IC90), respectively of the intracellular amastigote form of the parasite in a cardiomyocyte cell line. The results showed a clear improvement of the ravuconazole anti-T. cruzi activity when associated with SEDDS. Based on our results, the repurposing of ravuconazole in SEDDS dosage form is a strategy that deserves further in vivo investigation in preclinical studies for the treatment of human T. cruzi infections.
Keywords: ravuconazole, self-emulsifying drug delivery, asymmetric flow field-flow fractionation, Trypanosoma cruzi, Chagas disease, in vitro activity
Introduction
Chagas disease, a parasitic infection caused by the protozoan Trypanosoma cruzi,1 is endemic in many countries in Latin America and is considered a major public health concern.2 Although Chagas disease was identified >100 years ago, current specific treatment options are limited to benznidazole and nifurtimox.3,4 Both drugs are effective in the acute phase; however, their effectiveness varies in the chronic phase depending on the genetic variability of the parasite strains, treatment schedules, the immune status, and age of the host.5–8 Unfortunately, both drugs induce serious adverse effects.6,9
Therefore, treatment alternatives that are more effective have been the target of concerted efforts, especially in recent decades.10,11 Few drug candidates are currently being tested as anti-T. cruzi drugs, and ergosterol biosynthesis inhibitors are among the most promising.12,13 Ravuconazole (RAV), a triazole antifungal drug (Figure 1), has demonstrated potent activity in vitro against extracellular epimastigotes and intracellular amastigotes of T. cruzi at concentrations as low as 1 nM.14 However, in vivo studies in murine and canine models have shown limitations in the effectiveness of RAV, probably due to inadequate pharmacokinetic profile, mainly related to relatively short terminal plasma half-life of RAV in these animal models.14,15 As a Biopharmaceutics Classification System (BCS) Class II drug, RAV possesses low aqueous solubility (<1 μg/mL in phosphate-buffered saline [PBS] pH 7.4 at 30°C),16 has high lipophilicity (calculated logarithmic partition coefficient [clogP]: 3.477; Advanced Chemistry Development, Inc. [ACD/Labs]), and can be associated with lipid excipients.17,18 Thus, the evaluation of RAV in new formulations that are able to improve its biopharmaceutical properties could turn it into a more promising alternative for the etiological treatment of experimental and human Chagas disease.
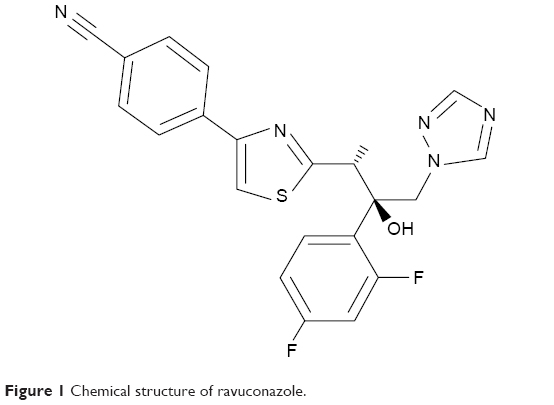 | Figure 1 Chemical structure of ravuconazole. |
Self-emulsifying drug delivery systems (SEDDSs) have often demonstrated the potential for improving oral bioavailability of lipophilic drugs, particularly BCS Class II drugs.19 SEDDSs are anhydrous isotropic mixtures of oil, surfactant, and drug, which, when placed in contact with an aqueous phase under gentle agitation, form fine oil-in-water (o/w) emulsions, usually with globules <250 nm in size.20,21 A SEDDS can also contain a cosurfactant and/or coemulsifier to facilitate emulsification or improve the incorporation of drug in the SEDDS. The ability of SEDDS to alter the drug permeability for different cell types and facilitate intestinal absorption via small emulsion droplets also influences its bioavailability. The small emulsion droplets, which form in the gastrointestinal tract (GIT), supply a large interfacial surface area utilizable for drug absorption and recruit lymphatic transport.22 We propose that these features may result in an increase in the anti-T. cruzi efficacy of RAV in both the acute and chronic phases of Chagas disease. In addition, our group recently reported that nanostructured formulation is a strategy that can make a marked difference in vivo with improved efficacy and reduced toxicity of active compounds, including in the treatment of experimental Chagas disease by the oral route.23–25
In this work, a RAV–SEDDS formulation for oral administration was developed and characterized. The safety of the formulation was evaluated in vivo in a 20-day treatment protocol in healthy mice and in T. cruzi-infected mice. The in vitro activity of this lipid-based formulation was determined on a cardiomyocyte cell line infected with T. cruzi amastigotes.
Materials and methods
Materials
RAV, ([R-(R*, R*)]-4-[2-[2-(2,4-difluorophenyl)-2-hydroxy-1-methyl-3-(1H-1,2,4-triazol-1-yl)propyl]-4-thiazolyl]benzonitrile), was purchased from Sigma-Aldrich (CAS Number: 182760-06-1; St Louis, MO, USA). Cremophor® EL, oleic acid, polyethylene glycol (PEG) 400, and Tween® 80 were also purchased from Sigma-Aldrich. Soy lecithin with ~75% phosphatidylcholine content (Epikuron® 170) was a gift from Cargill GmbH (Hamburg, Germany). Medium-chain triglyceride (Miglyol® 810N) was provided by Sasol GmbH (Bremen, Germany). Labrasol® was provided by Gattefossé (Cedex, France). Food-grade corn oil was from Mazola (São Paulo, Brazil). High-performance liquid chromatography (HPLC) grade acetonitrile was provided by Tedia (Rio de Janeiro, Brazil). Milli-Q water was obtained using a Simplicity® 185 System (Millipore, Bedford, MA, USA) and was used to prepare all aqueous systems used throughout the experiments. Cell culture medium Dulbecco’s Modified Eagle’s Medium (DMEM), fetal bovine serum (FBS), L-glutamine, and penicillin/streptomycin were purchased from Sigma-Aldrich Co (St Louis, MO, USA).
Parasites and animal ethics
Parasites of the Y strain of T. cruzi (DTU II),26 considered partially resistant to treatment with benznidazole and nifurtimox, were used.5 Bloodstream trypomastigotes were obtained from infected Swiss mice at the parasitemia peak day. The in vivo experimental protocol was approved by the ethics committee on Animal Experimentation of the Universidade Federal de Ouro Preto, Brazil (protocol number 2012/70) and was in agreement with the guidelines established by the National Council on Animal Experimentation Control (COBEA). Male and female Swiss mice weighing 25–30 g were used in this study and were maintained under the following environmental conditions: 12 h day/night cycles, temperature 22°C±2°C, standard diet, and water ad libitum.
Experimental methods
Selection of oil, surfactant, and cosurfactant
The excipients for the development of SEDDS were selected on the basis of RAV equilibrium solubility studies described below (Table 1). An amount of 20 mg of RAV was added to test tubes containing 1 mL of the different types of oil (Miglyol®, oleic acid, and corn oil), surfactants (Labrasol, Tween, and Cremophor), cosurfactants (Epikuron 170), and solubilizers (ethanol and PEG 400) (Table 1). The mixtures were vortexed (IKA® Vortex Genius 3) for 1 min, followed by stirring at 37°C for 48 h on a magnetic plate (cULus Listed® – 4D17). The samples were centrifuged at 5,000 rpm for 20 min to separate the nondissolved drug, and the supernatant was filtered through a 0.45 μm Millipore® syringe filter (Millipore). Aliquots of 200 μL of supernatant were diluted with 800 μL of acetonitrile, vortex-mixed, and centrifuged for 10 min (Eppendorf Centrifuge® 5415D). The supernatant was then filtered (0.45 μm) and analyzed by HPLC, according to the following RAV quantification method. All studies were conducted in triplicate.
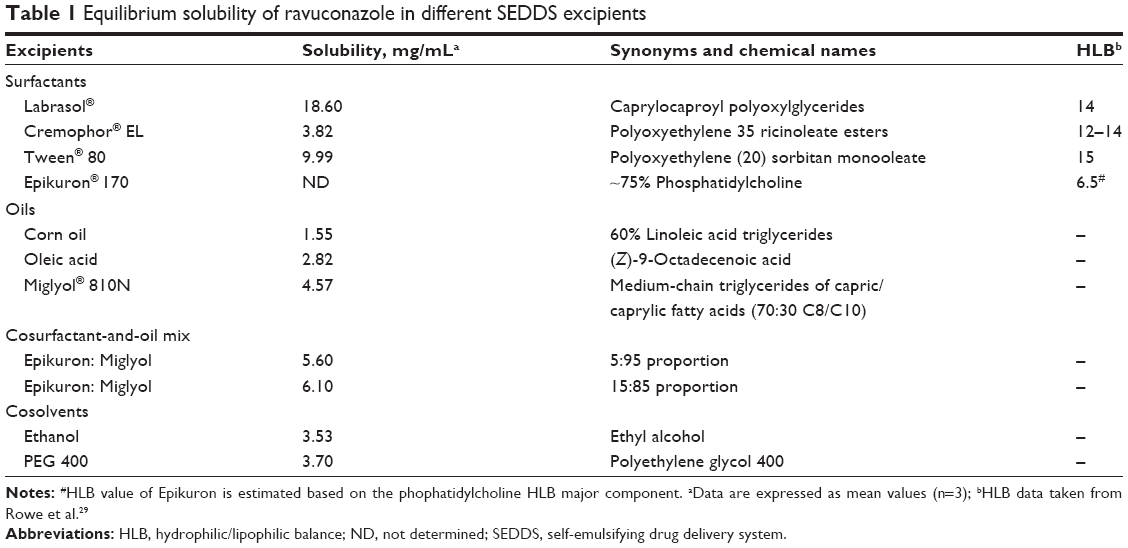 | Table 1 Equilibrium solubility of ravuconazole in different SEDDS excipients |
RAV quantification by HPLC
The HPLC system consisted of a Waters Alliance 2695 system with a Waters 2495 diode array detector. Samples were separated using a Phenomenex® C18 column (4.6×150 mm, 5 μm) with a Phenomenex C18 precolumn (4.6×5 mm, 5 μm). The mobile phase consisted of acetonitrile/water (70:30, v/v) pumped isocratically at 0.8 mL/min, following a method adapted from two methods previously reported.27,28 RAV was detected by monitoring the absorbance of the column eluent at 284 nm at 25°C. The injection volume was 25 μL, and the run time was 7 min, with a retention time of RAV of 4.3 min. The standard calibration curve was linear over a concentration range of 0.5 μg/mL–100.0 μg/mL, with a correlation coefficient of 0.9996 under our experimental conditions.
RAV–SEDDS development
Blank SEDDS (no drug) was prepared by mixing Epikuron and Miglyol in a mixture of ethanol and Labrasol or in Labrasol only. The proportions of oil, surfactant, cosurfactant, and coemulsifier varied from 60% to 70% (v/v), 10% to 40% (v/v), 0% to 15% (v/v), and 0% to 10% (v/v), respectively, as reported in Table 2. The RAV-loaded SEDDS (RAV–SEDDS) was prepared by dissolving RAV (6–12 mg/mL) in ethanol and Labrasol under magnetic stirring. In parallel, Miglyol and Epikuron were dissolved together under magnetic stirring at 40°C and added to the RAV solution. After mixing all excipients, the SEDDS was kept under magnetic stirring at 40°C for 20 min to guarantee complete drug dissolution.
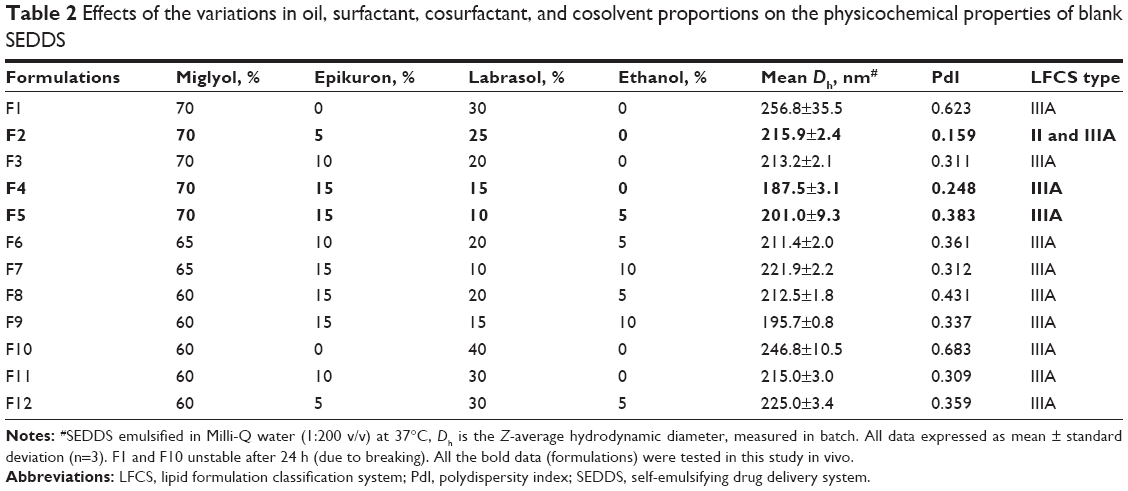 | Table 2 Effects of the variations in oil, surfactant, cosurfactant, and cosolvent proportions on the physicochemical properties of blank SEDDS |
Determination of globule size, polydispersity index (PdI), and zeta potential
The intensity-weighted (Z-average) hydrodynamic diameter (Dh) and PdI of the formulations were determined by dynamic light scattering (DLS) using a PN3702 Zetasizer Nano ZS (Malvern Instruments, Malvern, UK) equipped with a He–Ne Laser at 633 nm, set on 173° backscattering mode, at 25°C. Dh and PdI were calculated by the Zetasizer 7.11 software (Malvern Instruments) using the Stokes–Einstein equation with cumulant analysis, considering values of 0.8872 cP for water viscosity and 1.330 for water refractive index. To measure the mean emulsion droplet size and size dispersity (PdI), all formulations were previously emulsified in Milli-Q water at 37°C at 1:200 dilution and vortexed for 20 s. Values reported are the mean ± standard deviation of three readings of at least five formulations.
Zeta potential analysis was performed by laser Doppler anemometry (LDA) coupled to microelectrophoresis on the same equipment used for DLS analyses. The zeta potential values were determined on samples further diluted (100-fold) in Milli-Q water in disposable folded capillary zeta cells.
Size stability upon dilution
The RAV–SEDDS (10 mg/mL) was diluted 5, 100, 200, 500, and 1,000 times in Milli-Q water at 37°C and vortexed for 20 s. Droplet size, PdI, and zeta potential were determined. Signs of RAV precipitation were analyzed by light microscopy. All studies were conducted in triplicate.
Effect of pH
The RAV–SEDDS (10 mg/mL) was diluted (1:200) in compendial simulated gastric fluid without pepsin (pH 1.2) (SGF) and in simulated intestinal fluid without pancreatin (pH 6.8) (SIF) media following US Pharmacopeia (USP) XXIII.30 Subsequently, the emulsion droplet sizes, PdI, and zeta potential values were determined as described earlier. Signs of RAV precipitation in the different simulated media were analyzed by light microscopy (Olympus CX31, Olympus Corp, Tokyo, Japan). All the studies were conducted in triplicate.
Asymmetric flow field-flow fractionation
Emulsion droplet fractionation after emulsification was performed on a Postnova Analytics (Landberg, Germany) AF2000 MT AF4 system equipped with two PN1130 HPLC pumps (tip and focus pumps) and AF2000 module (cross flow pump), PN5300 autosampler, PN4020 channel oven, a separation channel fitted with a Postnova AF2000 MT Series NovaRC AQU 5 kDa cutoff regenerated cellulose membrane, and a 350 μm spacer. The fractionated sample was detected using a PN3211 ultraviolet (UV) detector with absorbance at 254 nm, the gyration diameter of the fractions were determined with a PN3621 multiangle laser light scattering (MALLS) detector with a 532 nm laser (7°–164°, 21 angles), and the hydrodynamic diameter with a Zetasizer Nano ZS (Malvern Instruments, as described earlier), in series. The carrier liquid was 10 mM NaCl in Milli-Q water filtered on a 0.1 μm polytetrafluoroethylene (PTFE) membrane filter (Millipore). The autosampler, channel, and detectors were kept at 37°C, and the detector flow rate was maintained at 0.5 mL/min. SEDDS formulations were emulsified in Milli-Q water at 37°C (1:200), and two successive dilutions were performed in the carrier liquid to prepare samples at 1:400 and 1:800 dilutions. The injection flow rate was set at 0.2 mL/min with injection time of 3 min, an injection volume of 20 μL, and a transition time of 1 min. An initial cross flow of 1.0 mL/min was applied during injection, maintained for 3 min, and set to decrease exponentially to 0.05 mL/min over a period of 10 min, followed by a linear decrease to zero over a period of 30 min, and further elution of the sample with a tip flow at 0.5 mL/min, accounting for a total analysis time of 65 min.
Diameter of gyration (Dg) was determined for each fraction at 7.8 s time intervals using the angular variation of the scattered light intensity at angles 20°–164° recorded on the MALLS detector using the Postnova AF2000 software calculation for a spherical shape model and the Dh at 5 s intervals.
In vitro RAV release studies
The RAV equilibrium solubility was determined in each dissolution medium to establish the sink conditions. For this purpose, 10 mg of RAV was added to test tubes containing 1 mL of SIF (pH 6.8) or SIF containing 2%, 5%, or 10% of Labrasol (pH 6.8), and the same methodology of RAV quantification was applied for solubility studies in oil, surfactant, and cosurfactant.
Then, the in vitro kinetic profiles of release of RAV from dispersed RAV–SEDDS (10 mg/mL) were determined using the direct dialysis method.31 The drug concentration was <8% (w/v) of the saturation solubility in the release medium of PBS with 5% Labrasol (medium II) to obey the sink conditions for both RAV powder and RAV–SEDDS.32 In the dissolution test in vitro, 200 μL of RAV–SEDDS was diluted up to 1 mL in the release medium and placed in a 12,000–14,000 molecular weight cutoff (MWCO) dialysis membrane tube (6.4 mm Spectra/Por®, Spectrum Labs) previously hydrated and equilibrated in release medium. The dialysis bag was suspended in 70 mL of medium (SIF 6.8 or SIF 6.8 plus Labrasol 5%) maintained at 37°C under continuous stirring in shaker bath (NovaEtica, Brazil). Aliquots (1 mL) were withdrawn at predetermined time intervals (0 min, 10 min, 30 min, 1 h, 2 h, 4 h, 6 h, 24 h, and 48 h) and replaced with an equivalent amount of fresh medium. The samples were vortex-mixed with same volume of acetonitrile and then centrifuged at 9,000 rpm for 10 min. The supernatant was collected, filtered on a Millipore 0.45 μm filter, and RAV concentration determined at 284 nm using reversed phase (RP)-HPLC method described earlier. All experiments were performed in triplicate. The same procedure was performed to evaluate the free RAV (powder) dissolution profile in the same media.
Stability studies
The anhydrous SEDDS was stored in sealed glass vials at 25°C and protected from light for a period of 6 months. The effects of storage time on macroscopic characteristics, RAV precipitation, droplet mean size, and zeta potential of the dispersed RAV–SEDDS and blank SEDDS were evaluated. All studies were conducted in triplicate.
Blank SEDDS safety study in healthy and infected mice
Swiss mice weighing 25–30 g were divided into nine experimental groups of seven animals each. Groups I–V were healthy animals and groups VI–IX were T. cruzi-infected ones. The infection with T. cruzi Y strain (5,000 trypomastigotes) was carried out as previously described33 and treatment started on Day 4, after confirmation of the infection.33
Healthy animals received 1.5 μL/g of blank SEDDS twice daily (groups II, III, and V) or once daily (group IV), or 1.5 μL/g of water twice daily (group I), for 20 consecutive days. The groups were as follows: I) water, II) blank SEDDS L25% (formulation F2, Table 2), III) blank SEDDS L15%-2 (formulation F4, Table 2), IV) blank SEDDS L15%-1 (formulation F4, Table 2), and V) blank SEDDS L10% with ethanol 5% (formulation F5, Table 2). T. cruzi-infected animals were treated with a daily dose of 30 mg/kg of RAV in the form of free RAV (group VI) or RAV–SEDDS (group VII, 1.5 μL/g of F5, administered twice daily) received by oral gavage for 20 consecutive days, in agreement with the recommended classical protocol of experimental treatment of T. cruzi-infected mice.5,34 The control groups received blank SEDDS (group VIII, 1.5 μL/g of F5 twice daily) or water (group IX, 2×1.5 μL/g).
The animals were inspected twice daily for clinical signs such as food and water intake, stool consistency, piloerection, dyspnea, and survival for 20 days during treatment. The animals were weighed every 5 days throughout the treatment period and the percentage weight variation was calculated relative to initial animal bodyweight on Day 0 using the same protocol as previously described.35
Evaluation of SEDDS anti-T. cruzi activity in vitro
The H9c2 cell line (American Type Culture Collection [ATCC] CRL 1446) derived from rat cardiomyoblasts was maintained in 75 cm2 bottles in DMEM supplemented with 10% fetal bovine serum (FBS), 1 mM L-glutamine, and 100 μg/mL penicillin/streptomycin, with weekly passages using trypsin (0.025%, w/v). H9c2 cells were seeded at 10,000 cells per well in 24-well plates covered with 13 mm cover slips, incubated at 37°C and 5% CO2 for 24 h. After 24 h, the cells were infected with trypomastigotes obtained from tissue culture at a ratio of 10 parasites per cell and incubated for 24 h. After interaction, the supernatant was removed, and each well was washed three times with PBS to remove noninternalized parasites. Then, RAV, RAV–SEDDS, blank SEDDS, and each isolated excipient of SEDDS at suitable proportions (Miglyol 60%, Epikuron 15%, Labrasol 10%, and ethanol 5%, v/v) were diluted in fresh culture medium and added at concentrations equivalent to that of the RAV-loaded SEDDS, referred to as the IC50 and IC90 of free RAV. The RAV IC50 and IC90 values of 0.1 nM and 0.5 nM, respectively, were previously determined in our experimental conditions using H9c2 cells infected with the Y strain of T. cruzi.14 The cells were then incubated for 72 h at 37°C. In all the experiments conducted in triplicate, the control wells were nontreated, infected H9c2 cells. After incubation, the cover slips were washed with PBS, fixed with methanol, stained with Giemsa solution, mounted on glass slides with Entellan (Merck, Darmstadt, Germany), and microscopically examined to determine the percentage of cells infected in treated and untreated controls. The inhibition percentage compared to controls was determined. All the experiments were conducted in triplicate with three repetitions.
Statistics
All experiments were performed in triplicate, and data were expressed as mean values ± standard deviations, unless otherwise stated. Mean size distribution, zeta potential, and drug release data at each time point were compared by analysis of variance (ANOVA) test. Survival data were analyzed by Kaplan–Meier non-parametric log rank test. In vitro release data were analyzed using one-way ANOVA followed by Dunnett’s multiple comparison test. In vitro anti-T. cruzi results were analyzed by ANOVA using the non-parametric version of Tukey’s multiple comparison test. All data were expressed as mean ± SD. A P-value of ≤0.05 was indicated as statistically significant. Statistical analyses of data were performed using GraphPad Prism® (Prism 6.01 for Windows).
Results and discussion
Selection of oil, surfactant, cosurfactant, and coemulsifier
The RAV equilibrium solubility was determined in different oils, surfactants, cosurfactants, and coemulsifiers generally used in SEDDS formulations. The solubility study is a prime tool to avoid drug precipitation after self-emulsification.21 The solubility of RAV was significantly higher (P<0.05) in Labrasol than in the other surfactants tested (Cremophor EL and Tween 80) (Table 1). The mixture of Epikuron and Miglyol, at a ratio of 15:85, significantly increased (P<0.05) the solubility of RAV in triglycerides. From the results presented in Table 1, it is clear that lecithin is essential to improve RAV solubility in Miglyol and that Labrasol surfactant is required to stabilize the nanodroplets. Labrasol presents a high hydrophilic/lipophilic balance (HLB) value. Furthermore, Labrasol and Miglyol paired with capric and caprylic fatty chains were chosen as the surfactant and oil, respectively (Table 1). The similarity in chemical structure between the fatty acid chains in the oily core triglycerides and the surfactant apolar backbone increases the emulsion stability following the rule of hydrocarbon chain length compatibility.36 The choice of surfactant is critical in the formulation of SEDDS. In particular, the HLB value and the critical micelle concentration (CMC) have considerable influence on the droplet sizes in the nanoemulsion formed.
Miglyol 810N, a medium-chain triglyceride, was the best oil to dissolve RAV and has many advantages because of its safety upon oral, parenteral, and topical administration.29 The oily phase directly influences the spontaneous formation of the emulsion, the emulsion droplet size, drug solubility in the GIT, intestinal transit, and the extent of lipolysis. The percentage of the oil phase also guides the classification of the lipid-based formulation (lipid formulation classification system [LFCS]) and must be chosen following its ability to dissolve the drug. At the same time, the polarity of the oil influences the production of a nanoemulsion with small globule size.19–21
The selected excipients for the development of optimized formulations of RAV–SEDDS were Labrasol, Epikuron, Miglyol 810N, and ethanol (Table 2). The concentrations of oils and surfactants in the SEDDS developed herein are in agreement with Pouton’s LFCS of type IIIA, as shown in Table 2.37,38 To improve the amount of RAV in the dissolved state in SEDDS, ethanol was chosen as a coemulsifier over PEG 400, which was found to be immiscible with the other constituents of the SEDDS formulation.
The zeta potential values of the droplets of all blank SEDDS formulations were around −45 mV and did not vary significantly between formulations (data not shown). It is known that high zeta potential values, >30 mV, either positive or negative, lead to emulsion droplets with increased colloidal stability due to electrostatic repulsion between same-charge particles, which prevents their aggregation.39 The phospholipids were essential to ensure small droplet sizes and low dispersity of particle size for blank SEDDS, as evidenced by F1 and F10 (Table 2), which were prepared without lecithin and did not remain stable (due to breaking) over a period of 24 h. Ethanol (up to 10%) and Labrasol variations from 10% to 30% did not significantly alter the nanoemulsion droplet sizes. Table 2 shows that all emulsions obtained from the SEDDS dispersion in water at 37°C produced relatively monodispersed (PdI =0.159–0.431) droplets in the 180–260 nm diameter range, in agreement with Type IIIA classification. Thus, they can be considered self-nanoemulsifying formulations and suitable for oral administration, as previously discussed.19 Formulations with the lowest content of surfactant and lowest size dispersity were chosen to load RAV.
The incorporation of drug in SEDDS can alter the size, polydispersity, and surface charge of emulsion droplets.40 Different ratios of surfactants were used to stabilize droplet surfaces with enhanced RAV incorporation capacity. RAV was loaded in F2 (5% Epikuron) at concentrations up to 6 mg/mL, with a significant increase in mean diameter from 215 nm to 247 nm (P<0.05). In F2, at higher RAV concentrations, the formulations were clearly unstable, and the drug precipitated upon dispersion in water (data not shown). The capacity to dissolve RAV was related to the increase in the percentage of Epikuron to 15%, whereby 8–10 mg/mL of RAV was efficiently loaded (F4 and F5 in Table 3). RAV loading in F4 did not influence any parameter of the formulation (P>0.05). F5, which had the highest RAV concentration and contained ethanol to ensure a higher RAV payload, was stable, monodispersed, and with mean droplet diameters <200 nm. The stability of the F5 formulation is attributed to the ability of the phosphatidylcholine from lecithin to form mixed micelles and stable monolayers in the presence of hydrophilic polyethylene oxide-based surfactants with improved solubilization capacity.41 The calculated HLB values of the final mixture of surfactants were 11.9, 9.75, and 9.1 for the F2, F4, and F5 formulations, respectively. This indicates that RAV is better associated with less-polar lipid/surfactant phases, in agreement with the use of LFCS type II and IIIA SEDDS. RAV–SEDDS type IIIA formulations are formed with good capacity to maintain RAV in the dispersed/solubilized state even after water dispersion (Table 3).
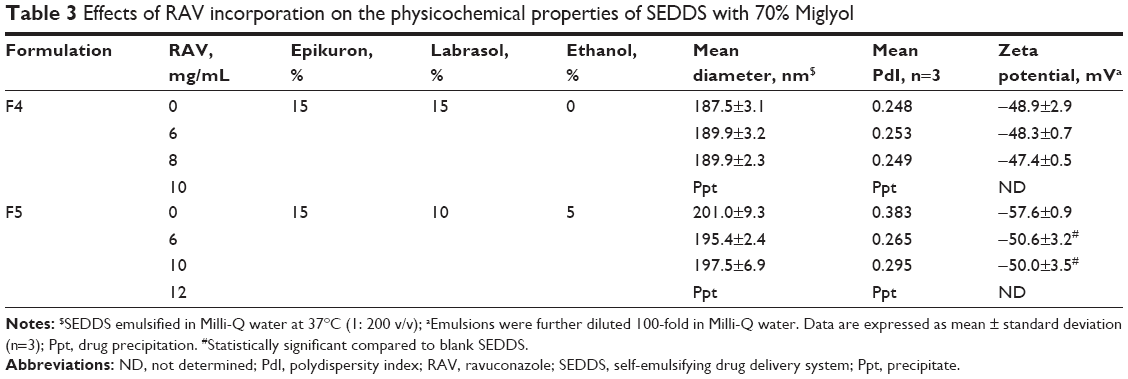 | Table 3 Effects of RAV incorporation on the physicochemical properties of SEDDS with 70% Miglyol |
The presence of RAV did not influence the droplet zeta potential when Labrasol was used at 15% (v/v), probably because its nonionic character better shields the polarizable groups at the oil/water droplet interface. However, due to the basic character of RAV, the positively charged RAV molecules influenced the surface charge when lower amounts of Labrasol were used (10%, v/v), indicating a potential location of the drug molecules near the droplet surface in F5. In the latter case, RAV influenced the interface and therefore may be involved in polar interactions with the phosphate groups of the phospholipids.
To examine the homogeneity of the formulations upon emulsification and the stability of the droplets to dilution, blank SEDDS and RAV–SEDDS F5 formulations were fractionated by AF4 at various dilutions (Figure 2). Both formulations showed two main populations with retention times (at peak maximum) of 16.3 min and 38.0 min, covering a broad range of diameters (Dg) between 100 nm and 600 nm, as determined by MALLS. The blank sample showed similar fractionation profiles at all three tested dilutions, whereas in RAV–SEDDS, the intensity of the UV signal for the first population to elute (16.3 min) decreased as the sample dilution increased. In our AF4 setup, the UV signal with absorbance at 254 nm was used to determine the relative proportion of each eluting fraction. It is a measure of the turbidity of the fraction and is unlikely to be affected by the chemical composition of the eluting structure. The almost linear decrease in intensity of the peak eluting at 16.3 min in the RAV–SEDDS emulsified formulation with increasing dilution from 1:200 to 1:400 and 1:800 indicates that this size population is sensitive to concentration effects. Micellar types of structures are known to disintegrate upon dilution. It is therefore likely that the lower-diameter population is a micellar type of structure, such as RAV-mixed micelles, or complex micellar structures, probably containing Labrasol and phospholipids. As the fractionation profiles indicate that this species is more abundant in the RAV–SEDDS samples than in the blank SEDDS samples, we can hypothesize that RAV plays an important role in stabilizing such structures. The shape factor, which is calculated as the ratio of the gyration radius (by MALLS) by the hydrodynamic radius (by DLS), Rg/Rh, gives an insight into the mass distribution within the volume of each structure and the shape of the structure formed. Spherical structures with a uniform distribution of mass have Rg/Rh close to 0.775,42 whereas values closer to 1.0 are typically obtained with spherical structures that have a nonuniform distribution of mass.43 Approximate values of 0.88 for blank SEDDS and 0.72 for RAV–SEDDS at retention time (tR) 16.25 min were obtained. The lower values obtained for RAV–SEDDS may indicate a more homogeneous distribution of mass than in blank SEDDS. The peak at higher retention times (~38 min) corresponds to structures of 500–600 nm (Dg), and this size does not vary upon dilution. These larger structures may correspond to oily droplets stabilized by surfactants at the interface. They are less sensitive to dilution, in agreement with the behavior of nanoemulsion droplets. It is interesting to note that DLS in batch mode was unable to distinguish between the two populations of similar sizes. To the best of our knowledge, this is the first report of SEDDS fractionation by AF4. AF4 provided additional information on the size distribution and was able to probe the heterogeneity of the structures formed after dispersion of the SEDDS in water.
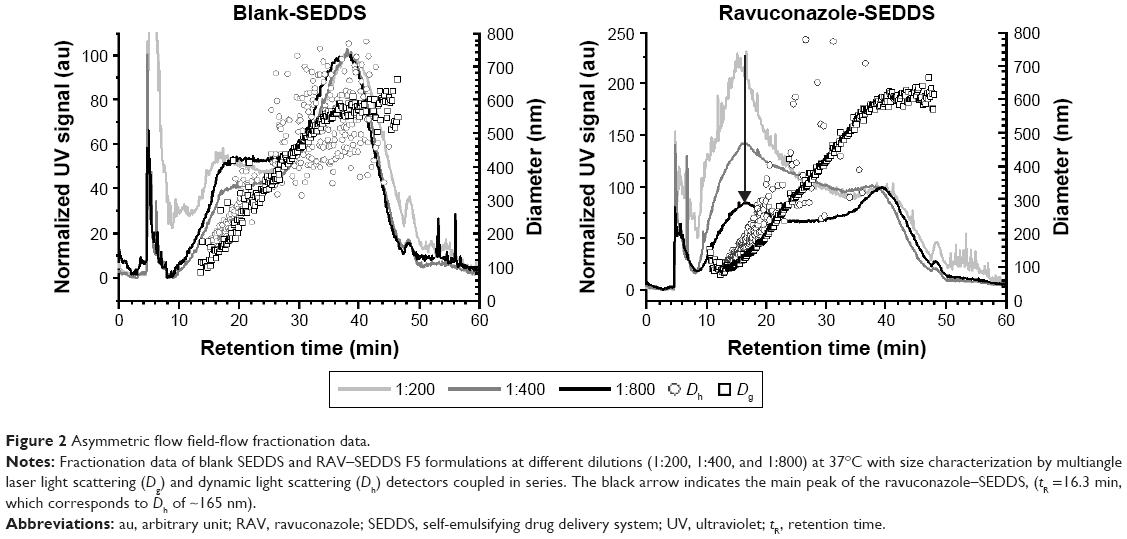 | Figure 2 Asymmetric flow field-flow fractionation data. |
The F5 formulation with RAV was characterized by batch DLS analysis in terms of robustness to dilution, effect of pH, in vitro drug release, and stability because of its high RAV payload (10 mg/mL) and low content of surfactant (Labrasol). Globule size and PdI are important parameters in characterizing the droplets in the LFCS. These parameters may not be affected upon dilution, and the diameter should be within the 100–250 nm range.37 Under conditions simulating physiological dilution through GIT transit from the stomach to the intestinal media, no significant variation in the droplet size was observed upon dilution (Table 4). RAV–SEDDS (10 mg/mL) in F5 was robust at different dilutions, without any macro or microscopic signs of drug precipitation. Even at Labrasol concentrations lower than its CMC in water (0.0123%, v/v),44 no RAV precipitation was observed. In agreement with the AF4 results, RAV may be forming mixed micelles with Labrasol, which may have a lower apparent CMC than the CMC of pure Labrasol in water. Consequently, these mixed micelles would still be present, even at Labrasol concentrations lower than its CMC in water. The solubilizing capacity of the SEDDS was not lost upon dilution, which may also indicate that a fraction of RAV was associated with the oily core of the emulsion droplets. This behavior is expected, due to the lipophilic nature of RAV (clogP close to 4.0) as well as its good solubility in the selected oil (Miglyol) and in the apolar region of the micelle core. Taken together, our results indicate that after dilution, RAV–SEDDS forms stable structures, probably related to mixed micelles and emulsion nanodroplets that maintain RAV molecules in a dispersed state, with no signs of crystal growth or amorphous precipitates.
 | Table 4 RAV–SEDDS robustness to dilution in water at 37°C (10 mg/mL) and influence of pH of different media |
Spontaneously emulsifying systems may suffer from considerable influence of the pH of the medium.45,46 In this context, the effect of pH on the characteristics of the nanoemulsions obtained by emulsification of the RAV–SEDDS (10 mg/mL) was investigated. The size, zeta potential, and PdI obtained in buffers with different pH values are shown in Table 4. Significant changes in droplet size, along with changes in the zeta potential, were observed at simulated stomach pH and intestinal pH. The pH of the release medium influences the drug solubility and association with surfactants at the emulsion droplet surface. RAV is a weak base, and at neutral pH, it is little ionized (pKa =2.73; ACD/Labs). At pH 1.2, >50% is ionized and positively charged.16 In aqueous HCl (pH 1.2), there was a reduction of negative surface charges (Table 4). This could be explained by the ionization of RAV in the acidic medium, producing positively charged groups. Lecithin is also mostly positively charged at low pH (1.2), which could contribute to the less-negative zeta potential values obtained, as shown in Table 4. Some structural phase changes seem to occur, which induce an increase in size and correspond to a reduction in the emulsion surface charge at low pH.
In phosphate buffer (pH 6.8), there was an increase in negative surface charge compared with 1:200 dilution in water at pH 6.3 (Table 4). At higher pH, ionization of acidic phospholipids (eg, phosphatidic acid) present in Epikuron may occur,47 rendering emulsion droplets more negative at duodenal pH, with no drug precipitation (Table 4). The presence of salts and electrolytes in the aqueous medium (buffer) reduces the solvation of nanodroplets and reduces the thickness of the electric double layer at the droplet surface, which usually reduces the colloidal stability of the nanoemulsion, leading to flocculation. These factors may contribute to an increase in the average size of the droplets and influence the dispersion system, resulting in flocculation and/or coalescence.
RAV equilibrium solubility in the dissolution medium was evaluated in order to avoid interference of the RAV solubility in the in vitro release study. As discussed herein, at intestinal pH, the solubility of RAV is expected to be <1 μg/mL.16 A surfactant concentration-dependent increase in RAV solubility in PBS (pH 6.8) was observed (Figure 3). Labrasol produced a significant (P<0.05) and linear (r2=0.9818) increase in RAV solubility in aqueous medium. This linearity indicates that RAV underwent micellar solubilization in the presence of Labrasol.
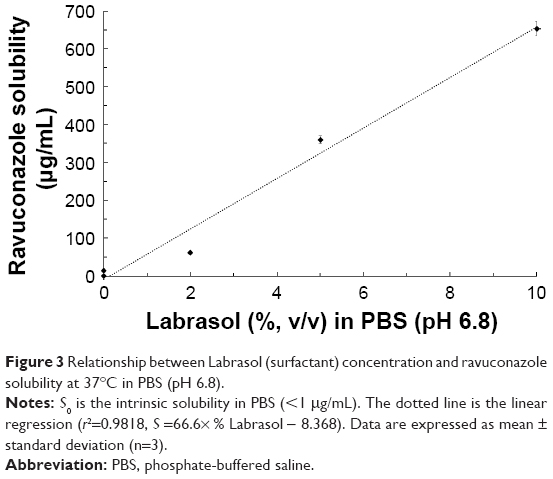 | Figure 3 Relationship between Labrasol (surfactant) concentration and ravuconazole solubility at 37°C in PBS (pH 6.8). |
In vitro RAV dissolution/release studies
A drug is classified as “poorly soluble” when its dissolution rate is so slow that it takes longer for it to dissolve than the transit time spent in the absorptive region in the gut lumen.48 From the RAV release profile (Figure 4), it is clear that RAV is a poorly soluble drug with rate-limiting dissolution characteristics. SEDDS clearly improved the rate and extent of RAV dissolution in the first 6 h. However, SEDDS in the presence of SIF, comprising agents that favor micellar solubilization, had a profound effect on the rate and extent of RAV dissolution. SEDDS significantly increased the amount of RAV dissolved (~20%) after 6 h, against 3% as free RAV in medium II (Figure 4). This characteristic suggests a probable uniform release of RAV–SEDDS in vivo, since Labrasol and lecithin will probably contribute to the formation of mixed micelles with bile salts in the intestinal medium, which simulates intestinal surfactant effects.49 In the absence of surfactant in the release medium, SEDDS also improved RAV dissolution, but to a smaller extent under nonsink conditions (Figure 4, inset). In the latter case, dissolution of free RAV is negligible, since no dissolution was observed from the RAV powder in the absence of a surfactant (medium I) for up to 48 h. Therefore, the small droplet sizes of the dispersed SEDDS also contribute to increasing RAV dissolution compared to free RAV, showing a strong effect of SEDDS on the rate and extent of dissolution. In this study, we used pharmacopeia (USPXXIII) medium and adapted the USP medium with synthetic surfactants (Figure 3) to simulate dissolution. Large differences in dissolution profiles with SEDDS formulations were observed depending on the release medium, and RAV release/dissolution was probably underestimated in these artificial media.
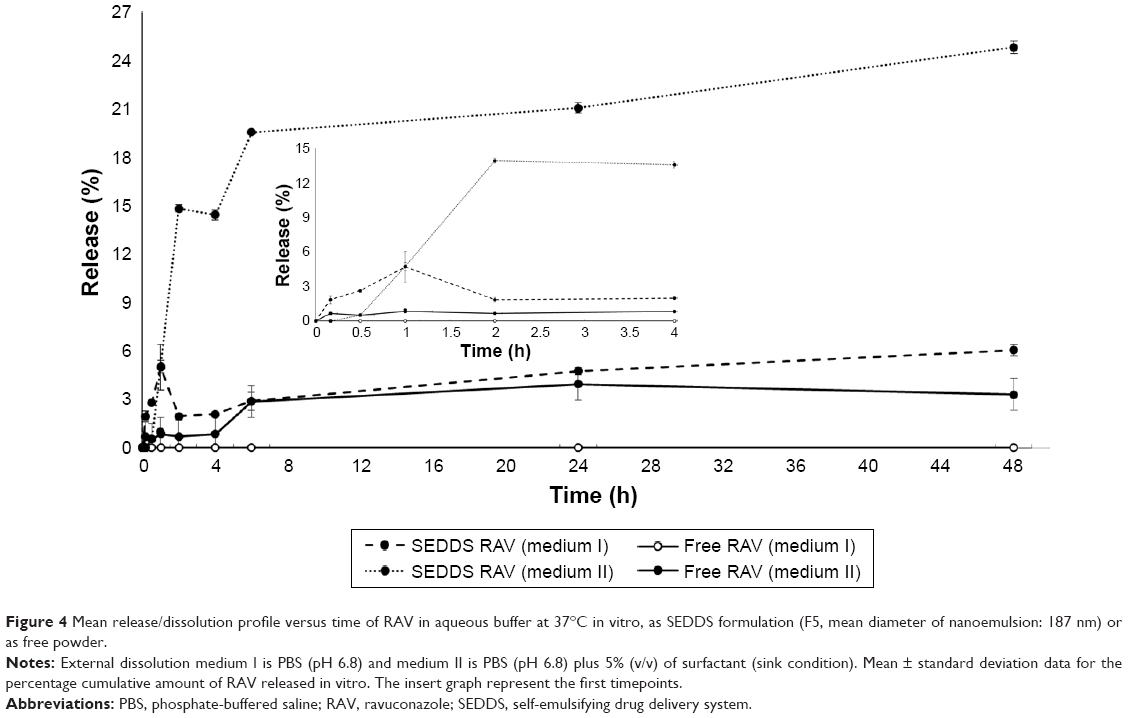 | Figure 4 Mean release/dissolution profile versus time of RAV in aqueous buffer at 37°C in vitro, as SEDDS formulation (F5, mean diameter of nanoemulsion: 187 nm) or as free powder. |
Several factors influence the stability of colloidal nanoemulsions, such as the adsorption of active molecules on the surface of the droplets and the presence of charged or nonionic surfactants. Table 5 shows the evaluation of the stability of SEDDS isotropic solutions (anhydrous) for a period of 6 months at 25°C. No precipitate was formed in the anhydrous mixture within the evaluation period and after emulsification. All dispersions remained homogeneous and translucent, and no drug crystallization was observed. Furthermore, there were no significant changes in the average size, PdI, and zeta potential of the blank SEDDS or RAV–SEDDS (10 mg/mL) during the 6 months. The formulations were considered stable under our storage conditions.
 | Table 5 Stability of blank SEDDS and RAV–SEDDS (10 mg/mL of RAV) after 6 months at room temperature (25°C) |
The overall physicochemical data indicate that SEDDSs are a promising option for oral delivery of RAV. Formulations F4 and F5 (Table 3) were tested in vivo with respect to their overall oral toxicity at excipient doses equivalent to 30 mg/kg/day of RAV, as previously reported for the treatment of experimental Chagas disease in mice.14 While all RAV–SEDDS formulations exhibited similar physicochemical characteristics, an important criterion for the selection of SEDDS formulations was the lesser amount of surfactant used, the smaller size, and the lower PdI (Tables 2 and 3).
In vivo toxicity of SEDDS in healthy and T. cruzi-infected mice
The toxicity of different formulations was tested in vivo in healthy mice with respect to the amount of Labrasol used and the treatment regimen during 20 days of repeated administration by oral gavage. Table 6 shows that Labrasol has a profound influence on the toxicity of blank SEDDS in mice under our experimental conditions. Higher toxicity is clearly related to a higher Labrasol content in the blank SEDDS. The highest concentration of Labrasol (equivalent to 0.75 g/kg), induced respiratory dysfunction, general toxicity, and mice death, even though this concentration was lower than the Labrasol DL50 (22 g/kg) previously reported in rats.29 From our data, mice were more sensitive to Labrasol content than rats.
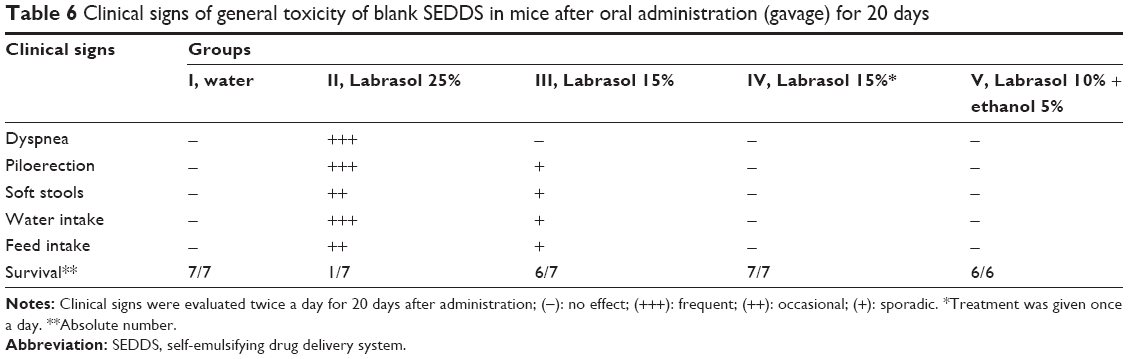 | Table 6 Clinical signs of general toxicity of blank SEDDS in mice after oral administration (gavage) for 20 days |
Figure 5 shows that the animals in groups I and V had a weight gain of 18.5% and 14%, respectively, until the end of the treatment (20 days). The mice in group II had a significant weight loss of 11% up to the 10th day posttreatment, followed by weight gain, however remaining significantly below the weight of control group I on Day 20. The mice in group III had a significant weight loss of 4.5% up to the fifth day posttreatment, followed by weight gain (12%), however significantly below that of control group I at the end of treatment. No body weight loss was observed in group IV during treatment (Table 6). Similarly, the survival was significantly reduced in group II, which received the highest amount of Labrasol, compared to that in all other groups (P<0.05). This experiment indicates that body weight loss is related to the percentage of Labrasol in blank SEDDS and shows the low tolerance of mice to Labrasol in such formulations.
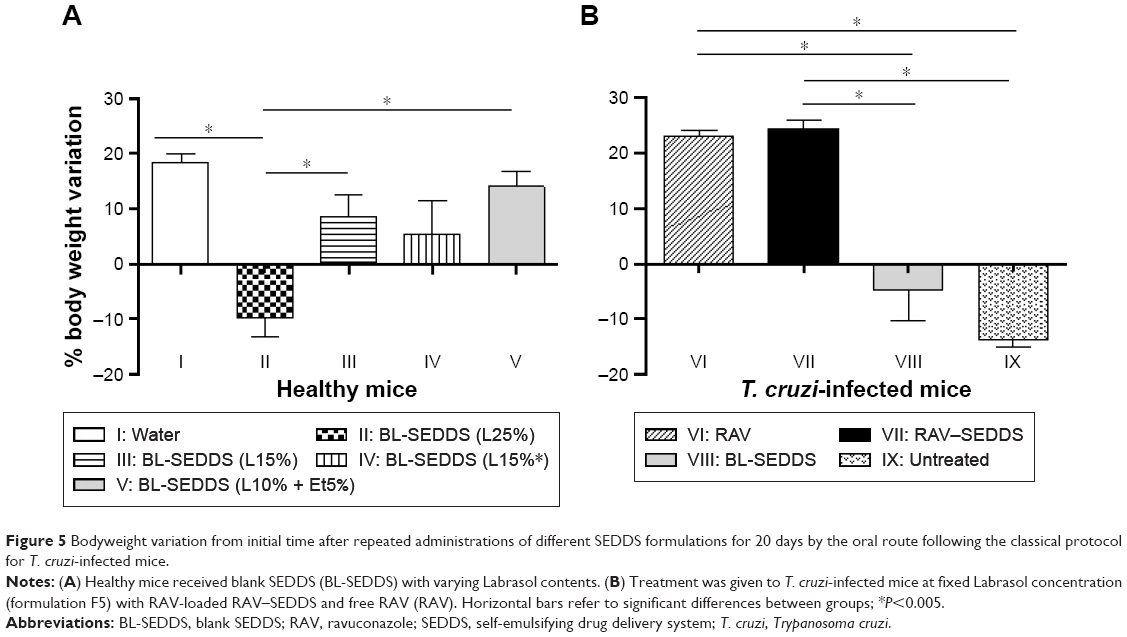 | Figure 5 Bodyweight variation from initial time after repeated administrations of different SEDDS formulations for 20 days by the oral route following the classical protocol for T. cruzi-infected mice. |
Lower doses of Labrasol (0.2–0.3 g/kg) divided in two daily doses induced no observable toxic effects, nor significant body weight loss, as shown in Figure 5A and Table 6. Sha et al50 evaluated the cytotoxicity and mannitol permeability of Labrasol in Caco-2 cells. The authors found that 0.1% and 1% (w/v) of Labrasol increased the permeability 4.6-fold and 33.8-fold and reduced the Caco-2 viability by ~3% and 43%, respectively, indicating dose-dependent effects on cell viability and permeability. It was shown that Labrasol alters the tight junction integrity of cells.50 Cornaire et al51 reported that 0.5% (w/v) of Labrasol increased lactate dehydrogenase release, indicating intestinal cell membrane damage in experiments on intestinal transport of digoxin.
The reduction in food consumption observed for groups II and III may be related to gastric stasis due to lipid consumption, in addition to GIT irritation induced by high amounts of surfactants.35,50 The latter effect contributed to the presence of more fluid feces, in particular in group II. The overall clinical observations indicate that even a single dose of 25% (w/v) of Labrasol in SEDDS (equivalent to 0.75 g/kg) is toxic for mice. It is important to note that a vast majority of studies concerning SEDDS involve surfactant percentages usually in the range of 25%–60% and, particularly, Labrasol concentrations of 5%–30%.52 In these studies, no systematic investigation of animal toxicity was reported. In general, surfactants cannot be considered pharmacologically or toxicologically inert excipients.53 Labrasol is widely used as a pharmaceutical excipient to dissolve lipophilic drugs.54 However, in this study, Labrasol was found to be toxic when used as a SEDDS excipient at proportions greater than 25%.
Due to the observed mortality, the formulation containing 25% of Labrasol was excluded from further in vivo experiments. The formulation containing 15% of Labrasol administered twice a day was also discarded because of the observed clinical alterations. The formulation containing the lowest amount of Labrasol (10%) and 5% of ethanol was selected for further studies. The use of phosphatidylcholine mixed with polyethylene oxide-based surfactants, such as Labrasol, dramatically reduces the toxicity of the latter, as already observed and discussed.55
Though we used low percentages of nonionic surfactant, F5 meets the criteria to be classified as SEDDS type IIIA, with good capability to solubilize RAV, high stability upon dilution, and safe characteristics in vivo. Although the promising physicochemical properties of lipid-based drug delivery systems, particularly of SEDDS, have generated enthusiasm in the pharmaceutical industry, there is already some reticence in developing and commercializing such formulations due to the absence of information regarding toxic effects of the vehicle, particularly in vivo. In particular, the field of SEDDS would benefit from detailed pathological analysis related to intestinal toxicity. F5 was further tested in mice experimentally infected with the T. cruzi Y strain. No weight loss was observed in animals treated with RAV–SEDDS or free RAV, indicating no additional toxicity due to the drug. The weight loss in group VIII, treated with the blank SEDDS, is related, in this case, to the damage caused by the acute infection, similarly to the nontreated control group (IX). Our results indicate that RAV–SEDDS can be used in long-term treatment schemes with minimal toxicity and highlight the importance of further in vivo investigation in nonclinical studies for the treatment of T. cruzi infection.
Anti-T. cruzi activity in vitro against intracellular amastigotes
The activity of each excipient used in our SEDDS was tested against the amastigote form of T. cruzi in H9c2 cells. The excipients were incubated at the concentrations corresponding to those in RAV–SEDDS required to provide RAV at its IC50 and IC90. From the data in Figure 6, at 0.1 nM, RAV produced >70% of inhibition of the infection in vitro when loaded in SEDDS (Figure 6). RAV–SEDDS increased the inhibition activity against amastigotes ~1.8-fold compared to free RAV at doses equivalent to IC50 (0.1 nM), showing a clear improvement of RAV potency against the intracellular form of the parasite (Figure 6). The IC50 was reduced to values <0.1 nM with no cytotoxicity to H9c2 cells. At 0.5 nM (IC90), no significant differences in activity were observed, probably because at higher doses, complete inhibition of the infection was already achieved with free RAV. No significant differences were observed between excipients and blank SEDDS in terms of infection inhibition.
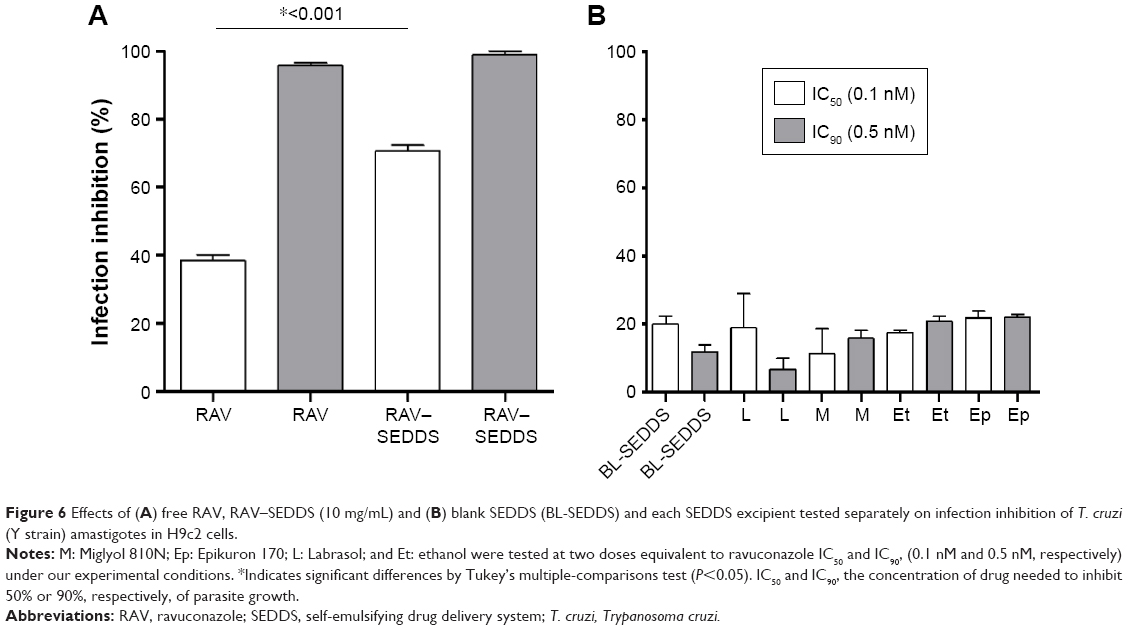 | Figure 6 Effects of (A) free RAV, RAV–SEDDS (10 mg/mL) and (B) blank SEDDS (BL-SEDDS) and each SEDDS excipient tested separately on infection inhibition of T. cruzi (Y strain) amastigotes in H9c2 cells. |
Conclusion
The differences in the activity of SEDDS formulations may be related to the mechanism of drug penetration into cells via micellar nanostructures, efflux transporter inhibition, and possible permeabilization of cell membranes. Our data suggest that using RAV loaded in SEDDS formulation to treat in vivo experimental T. cruzi infection could reduce the required dose. Consistent with this notion, a subsequent study from our group found that oral treatment with a daily dose of 20 mg/kg of RAV–SEDDS for 30 days was significantly more effective than treatment with free RAV at the same dose or benznidazole at 100 mg/kg in a murine model of acute Chagas disease caused by the Y strain of T. cruzi (Spósito et al. In vitro activity and oral in vivo efficacy of a new formulation of ravuconazole in a self-emulsifying delivery system against Trypanosoma cruzi, the etiological agent of Chagas disease, manuscript in preparation). Furthermore, the RAV–SEDDS formulation was simple to prepare, cost-effective, stable under storage, safe in mice, and easy to administer orally. Thus, the formulation developed here is a promising delivery form for RAV and other lipophilic drugs used for the etiological treatment of experimental and human Chagas disease and fungal infections.
Acknowledgments
The authors thank Coordination for the Improvement of Higher Level and Education Personnel-Brazil (CAPES)-Brazil and CNPq for scholarships. VCF Mosqueira and MT Bahia are recipients of researcher productivity grants from CNPq, Brazil. This work was also supported by NANOBIOMG-Network (# 7–14 and 40/11), PPM grant (#432-13) from FAPEMIG and UFOP, Minas Gerais – Brazil, and the bilateral Brazil-France CAPES-COFECUB 768/13 project.
Disclosure
The authors report no conflicts of interest in this work.
References
Chagas C. Nova tripanozomiase humana. Estudo sobre a morfologia e o ciclo evolutivo do Schizotrypanum cruzi. n. gen., n. sp. agente etiológico de nova entidade mórbida do homem. Mem Inst Oswaldo Cruz. 1909;1:159–218. | ||
World Health Organization [webpage on the Internet]. Chagas disease (American trypanosomiasis). Fact sheet No 340 [updated March, 2016]. Available from: http://www.who.int/mediacentre/factsheets/fs340/en/. Accessed April 10, 2017. | ||
Coura JR, Castro SL. A critical review on Chagas disease chemotherapy. Mem Inst Oswaldo Cruz. 2002;97(1):3–24. | ||
Pinto Dias JC. The treatment of Chagas disease (South American trypanosomiasis). Ann Int Med. 2006;144(10):772–774. | ||
Filardi LS, Brener Z. Susceptibility and natural resistance of Trypanosoma cruzi strains to drugs used clinically in Chagas disease. Trans R Soc Trop Med Hyg. 1987;81(5):755–759. | ||
Cançado JR. Long term evaluation of etiological treatment of Chagas disease with benznidazole. Rev Inst Med Trop São Paulo. 2002;44(1):29–37. | ||
Guedes PMM, Fietto JLR, Lana M, Bahia MT. Advances in Chagas disease chemotherapy. Anti Infect Agents Med Chem. 2006;5:175–186. | ||
Urbina JA. Specific chemotherapy of Chagas disease: relevance, current limitations and new approaches. Acta Trop. 2010;115(1–2):55–68. | ||
Castro JA, de Mecca MM, Bartel LC. Toxic side effects of drugs used to treat Chagas disease (American trypanosomiasis). Hum Exp Toxicol. 2006;25(8):471–479. | ||
Muñoz MJ, Murcia L, Segovia M. The urgent need to develop new drugs and tools for the treatment of Chagas disease. Expert Rev Anti Infect Ther. 2011;9(1):5–7. | ||
Bilbe G. Overcoming neglect of kinetoplastid diseases. Science. 2015;348(6238):974–976. | ||
Choi JY, Podust LM, Roush WR. Drug strategies targeting CYP51 in neglected tropical diseases. Chem Rev. 2014;114(22):11242–11271. | ||
Urbina JA, Docampo R. Specific chemotherapy of Chagas disease: controversies and advances. Trends Parasitol. 2003;19(11):495–501. | ||
Urbina JA, Payares G, Sanoja C, Lira R, Romanha AJ. In vitro and in vivo activities of ravuconazole on Trypanosoma cruzi, the causative agent of Chagas disease. Int J Antimicrob Agents. 2003;21(1):27–38. | ||
Diniz LF, Caldas IS, Guedes PMM, et al. Effects of ravuconazole treatment on parasite load and immune response in dogs experimentally infected with Trypanosoma cruzi. Antimicrob Agents Chemother. 2010;54(7):2979–2986. | ||
Cao X, Sun Z, Cao Y, et al. Design, synthesis, and structure-activity relationship studies of novel fused heterocycles-linked triazoles with good activity and water solubility. J Med Chem. 2014;57(9):3687–3706. | ||
Lentz KA, Quitko M, Morgan DG, Grace JE Jr, Gleason C, Marathe PH. Development and validation of a preclinical food effect model. J Pharm Sci. 2007;96(2):459–472. | ||
Ueda Y, Matiskella JD, Golik J, et al. Phosphonooxymethyl prodrugs of the broad spectrum antifungal azole, ravuconazole: synthesis and biological properties. Bioorg Med Chem Lett. 2003;13(21):3669–3672. | ||
Müllertz A, Ogbonna A, Rena S, Rades T. New perspectives on lipid and surfactant based drug delivery systems for oral delivery of poorly soluble drugs. J Pharm Pharmacol. 2010;62(11):1622–1636. | ||
Pouton CW, Porter CJ. Formulation of lipid-based delivery systems for oral administration: materials, methods and strategies. Adv Drug Deliv Rev. 2008;60(6):625–637. | ||
Date AA, Desai N, Dixit R, Nagarsenker M. Self-nanoemulsifying drug delivery systems. Nanomedicine. 2010;5(10):1595–1616. | ||
Driscoll CM. Lipid-based formulations for intestinal lymphatic delivery. Eur J Pharm Sci. 2002;15(5):405–415. | ||
De Mello CGC, Branquinho RT, Oliveira MT, et al. Efficacy of lychnopholide polymeric nanocapsules after oral and intravenous administration in murine experimental Chagas disease. Antimicrob Agents Chemother. 2016;60(9):5215–5222. | ||
Branquinho RT, Mosqueira VCF, Oliveira-Silva JCVD, Simões-Silva MR, Saúde-Guimarães DA, De Lana M. Sesquiterpene lactone in nanostructured parenteral dosage form is efficacious in experimental Chagas disease. Antimicrob Agents Chemother. 2014;58(4):2067–2075. | ||
Branquinho RT, Roy J, Farah C, et al. Biodegradable polymeric nanocapsules prevent cardiotoxicity of anti-trypanosomal lychnopholide. Sci Rep. 2017;7:44998. | ||
Zingales B, Andrade SG, Briones MRS, et al; Second Satellite Meeting. A new consensus for Trypanosoma cruzi intraspecific nomenclature: second revision meeting recommends TcI to TcVI. Mem Inst Oswaldo Cruz. 2009;104(7):1051–1054. | ||
Groll AH, Mickiene D, Petraitis V, et al. Compartmental pharmacokinetics and tissue distribution of the antifungal triazole ravuconazole following intravenous administration of its di-lysine phosphoester prodrug (BMS-379224) in rabbits. J Antimicrob Chemother. 2005;56(5):899–907. | ||
Cendejas-Bueno E, Rodríguez-Tudela JL, Cuenca-Estrella M, Gómez-López A. Development and validation of a fast HPLC/photodiode array detection method for the measurement of voriconazole in human serum samples. A reference laboratory experience. Enferm Infecc Microbiol Clin. 2013;31(1):23–28. | ||
Rowe RC, Sheskey PJ, Quinn ME. Handbook of Pharmaceutical Excipients. 6th ed. London: Royal Pharmaceutical Society of Great Britain; 2009. | ||
Galia E, Nicolaides E, Hörter D, Löbenberg R, Reppas C, Dressman JB. Evaluation of various dissolution media for predicting in vivo performance of Class I and Class II drugs. Pharm Res. 1998;15(5):698–705. | ||
Soundararajan R, Sasaki K, Godfrey L, et al. Direct in vivo evidence on the mechanism by which nanoparticles facilitate the absorption of a water insoluble, P-gp substrate. Int J Pharm. 2016;514(1):121–132. | ||
Oliveira LT, Garcia GM, Kano EK, Tedesco AC, Mosqueira VCF. HPLC-FLD methods to quantify chloroaluminum phthalocyanine in nanoparticles, plasma and tissue: application in pharmacokinetic and biodistribution studies. J Pharm Biomed Anal. 2011;56(1):70–77. | ||
Diniz LF, Urbina JA, Andrade IM, et al. Benznidazole and posaconazole in experimental Chagas disease: positive interaction in concomitant and sequential treatments. PLoS Negl Trop Dis. 2013;7(8):e2367. | ||
Romanha AJ, Castro SL, Soeiro MN, et al. In vitro and in vivo experimental models for drug screening and development for Chagas disease. Mem Inst Oswaldo Cruz. 2010;105(2):233–238. | ||
Delongeas JL, Conchard GV, Beamonte A, et al. Assessment of labrasol/labrafil/transcutol (4/4/2, v/v/v) as a non-clinical vehicle for poorly water-soluble compounds after 4-week oral toxicity study in Wistar rats. Regul Toxicol Pharmacol. 2010;57(2–3):284–290. | ||
Ghaicha L, Leblanc RM, Villamagna F, Chattopadhyay AK. Monolayers of mixed surfactants at the oil-water interface, hydrophobic interactions, and stability of water-in-oil emulsions. Langmuir. 1995;11(2):585–590. | ||
Pouton CW. Formulation of poorly water-soluble drugs for oral administration: physicochemical and physiological issues and the lipid formulation classification system. Eur J Pharm Sci. 2006;29(3–4):278–287. | ||
Porter CJ, Pouton CW, Cuine JF, Charman WN. Enhancing intestinal drug solubilisation using lipid-based delivery systems. Adv Drug Deliv Rev. 2008;60(6):673–691. | ||
O’Brien RD. Fats and Oils: Formulating and Processing for Applications. 3th ed. Boca Raton, FL: CRC Press; 2008. | ||
Chudasama A, Patel V, Nivsarkar M, Vasu K, Shishoo C. Role of lipid-based excipients and their composition on the bioavailability of antirretroviral self-emulsifying formulations. Drug Deliv. 2015;22(4):531–540. | ||
Jumma M, Müller BW. Influence of the non-ionic surfactant PEG-660-12-hydroxy stearate on the surface properties of phospholipid monolayers and their effect on lipid emulsion stability. Colloid Polym Sci. 1999;277(4):347–353. | ||
Brewer AK, Striegel AM. Particle size characterization by quadruple-detector hydrodynamic chromatography. Anal Bioanal Chem. 2009;393(1):295–302. | ||
Stauch O, Schubert R, Savin G, Burchard W. Structure of artificial cytoskeleton containing liposomes in aqueous solution studied by static and dynamic light scattering. Biomacromolecules. 2002;3(3):565–578. | ||
Ujhelyia Z, Fenyvesia F, Váradia J, et al. Evaluation of cytotoxicity of surfactants used in self-micro emulsifying drug delivery systems and their effects on paracellular transport in Caco-2 cell monolayer. Eur J Pharm Sci. 2012;47(3):564–573. | ||
Kawakami K, Yoshikawa T, Moroto Y, et al. Microemulsion formulation for enhanced absorption of poorly soluble drugs. I. Prescription design. J Control Release. 2002;81(1–2):65–74. | ||
Kim HJ, Yoon KA, Hahn M, Park ES, Chi SC. Preparation and in vitro evaluation of self-microemulsifying drug delivery systems containing idebenone. Drug Dev Ind Pharm. 2000;26(5):523–529. | ||
Mosqueira VCF, Legrand P, Pinto-Alphandary H, Puisieux F, Barratt G. Poly (D,L-lactide) nanocapsules prepared by solvent displacement process: influence of composition on physico-chemical and structural properties. J Pharm Sci. 2000;89(5):614–626. | ||
Hörter D, Dressman JB. Influence of physicochemical properties on dissolution of drugs in the gastrointestinal tract. Adv Drug Deliv Rev. 1997;25:3–14. | ||
Elnaggar YSR, El-Massik MA, Abdallah OY. Self-nanoemulsifying drug delivery systems of tamoxifen citrate: design and optimization. Int J Pharm. 2009;380(1–2):133–141. | ||
Sha X, Yan G, Wu Y, Li J, Fang X. Effect of self-microemulsifying drug delivery systems containing Labrasol on tight junctions in Caco-2 cells. Eur J Pharm Sci. 2005;24(5):477–486. | ||
Cornaire G, Woodley J, Hermann P, Cloarec A, Arellano C, Houin G. Impact of excipients on the absorption of P-glycoprotein substrates in vitro and in vivo. Int J Pharm. 2004;278(1):119–131. | ||
Gursoy RN, Benita S. Self-emulsifying drug delivery systems (SEDDS) for improved oral delivery of lipophilic drugs. Biomed Pharmacother. 2004;58(3):173–182. | ||
Budden R, Kühl G, Bahlsen J. Experiments on the toxic, sedative and muscle relaxant potency of various drug solvents in mice. Pharmacol Ther B. 1979;5(1–3):467–474. | ||
Strickley RG. Solubilizing excipients in oral and injectable formulations. Pharm Res. 2004;21(2):201–230. | ||
Jumaa M, Müller BW. Lipid emulsions as a novel system to reduce the hemolytic activity of lytic agents: mechanism of the protective effect. Eur J Pharm Sci. 2000;9(3):285–290. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.