Back to Journals » OncoTargets and Therapy » Volume 9
Ras-related associated with diabetes gene acts as a suppressor and inhibits Warburg effect in hepatocellular carcinoma
Authors Yan Y, Xie M, Zhang L ![]() , Zhou X, Xie H, Zhou L, Zheng S
, Zhou X, Xie H, Zhou L, Zheng S ![]() , Wang W
, Wang W ![]()
Received 19 February 2016
Accepted for publication 31 March 2016
Published 30 June 2016 Volume 2016:9 Pages 3925—3937
DOI https://doi.org/10.2147/OTT.S106703
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr William C. Cho
Yingcai Yan,1,* Minjie Xie,1,* Linshi Zhang,1 Xiaohu Zhou,1 Haiyang Xie,1 Lin Zhou,1 Shusen Zheng,2,3 Weilin Wang2,3
1Key Laboratory of Combined Multi-Organ Transplantation, Ministry of Public Health, The First Affiliated Hospital, College of Medicine, Zhejiang University, 2Division of Hepatobiliary and Pancreatic Surgery, Department of Surgery, 3Collaborative Innovation Center for Diagnosis and Treatment of Infectious Diseases, The first Affiliated Hospital, College of Medicine, Zhejiang University, Hangzhou, People’s Republic of China
*These authors contributed equally to this work
Abstract: Hepatocellular carcinoma (HCC) is rapidly becoming one of the most prevalent cancers worldwide and is a prominent source of mortality. Ras-related associated with diabetes (RRAD), one of the first members of the 35–39 kDa class of novel Ras-related GTPases, is linked to several types of cancer, although its function in HCC remains unclear. In this study, we observed that RRAD was downregulated in HCC compared with adjacent normal tissues. This change was associated with a poor prognosis. Furthermore, knockdown of RRAD in SK-Hep-1 cells facilitated cell proliferation, accelerated the G1/S transition during the cell cycle, induced cell migration, and reduced apoptosis. In contrast, overexpression of RRAD in Huh7 cells had the opposite effects. Moreover, we demonstrated that RRAD induced cell proliferation through regulation of the cell cycle by downregulating cyclins and cyclin-dependent kinases. RRAD induced tumor cell apoptosis through the mitochondrial apoptosis pathway. In addition, we confirmed that knockdown of RRAD promoted aerobic glycolysis by upregulating glucose transporter 1, whereas overexpression of RRAD inhibited aerobic glycolysis. In conclusion, RRAD plays a pivotal role as a potential tumor suppressor in HCC. An improved understanding of the roles of RRAD in tumor metabolism may provide insights into its potential as a novel molecular target in HCC therapy.
Keywords: hepatocellular carcinoma, Ras-related associated with diabetes, aerobic glycolysis, tumorigenicity, glucose transporter 1
Introduction
Hepatocellular carcinoma (HCC) is the most common primary hepatic malignancy and the third most common cause of cancer-associated mortality worldwide. The average survival time of patients with HCC is between 6 months and 20 months.1,2 There is a pressing need for a better understanding of the mechanisms involved in HCC, with the goal of developing better therapies for individuals with this disease.
In contrast to normal cells that depend on mitochondrial oxidative phosphorylation to obtain energy, most cancer cells prefer aerobic glycolysis (the Warburg effect), even though it is an inefficient way to generate adenosine triphosphate (ATP).3 Thus, metabolic reprogramming plays a critical role in biobehavioral mechanisms of cancer. Recent studies have verified that a higher rate of glucose uptake and lactate production is associated with tumor progression and survival.4,5 Therefore, a better understanding of the molecular events associated with glycolysis can be of great significance.
Ras-related associated with diabetes (RRAD), a prototypic member of a subfamily of Ras-related GTPases, has a unique sequence and is overexpressed in the skeletal muscle of humans with type II diabetes.6 The Ras-related GTPases have been implicated in a wide spectrum of cellular functions, including cell proliferation/differentiation,7 intracellular vesicular trafficking,8 and cytoskeletal control.9 Recent studies have suggested that RRAD may act as a tumor suppressor in some tumor tissues. For example, the expression of RRAD is lower in nasopharyngeal,10 lung,11 and ovarian12 cancer tissues. Ectopic expression of RRAD suppresses tumor cell proliferation and migration, with the concomitant inhibition of aerobic glycolysis. In addition, RRAD shows significantly higher methylation in cancer than in nonmalignant tissues.13 Overall, the decreased expression of RRAD in tumor tissues is linked to a poorer prognosis. However, the role that RRAD plays in HCC remains unknown.
In the current study, we demonstrate that RRAD expression is downregulated in HCC cells. Knockdown of RRAD in an HCC cell line with high basal levels of this protein promotes cell proliferation and migration, and reduces apoptosis, while overexpression in a line with low basal levels has the opposite effects. Furthermore, RRAD appears to inhibit the Warburg effect in HCC cells.
Materials and methods
Tissue microarrays and immunohistochemistry
Tissue microarray chips that contained 90 cases of paired HCC tumor and peritumor specimens (HLiv-HCC180Sur-03) were purchased from Shanghai Outdo Biotech Company (Shanghai, People’s Republic of China). Ethical approval for this study was given by the Medical Ethics Committee of Shanghai, People’s Republic of China. All specimens spotted on the tissue microarray chips included complete postoperative follow-up information for 2–3.7 years. Tumor staging was evaluated according to the tumor node metastasis classification of malignant tumors. A rabbit polyclonal antibody against human RRAD (ab75100; Abcam, Cambridge, MA, USA) was used for immunohistochemistry (IHC) that was performed as described previously.14 All slides were examined and scored by two pathologists who were blinded to clinical patient data. The IHC score was calculated by combining the quantity score (percentage of positively stained cells) with the staining intensity score. The quantity scores ranged from 0 to 3 (0, 0%; 1, ≤5%; 2, 5%–50%; 3, ≥50% immunoreactive cells). Scores of 0 or 1 were considered to indicate “low,” while those of 2 or 3 indicated “high” expression of RRAD.
IHC was also used to detect expression of several proteins in tumor tissues removed from nude mice. The following antibodies were used: RRAD (ab75100; Abcam), glucose transporter 1 (GLUT1, ab652; Abcam), cleaved-PARP (ab32064; Abcam), and cleaved-caspase-3 (ab32150; Abcam).
Cell culture
The HCC cell lines 97H, 97L, HepG2, LM3, Huh7, SK-Hep-1, and L02 were maintained at our institute. All HCC cell lines, except L02, were cultured in Dulbecco’s Modified Eagle’s Medium (Thermo Fisher Scientific, Waltham, MA, USA). The L02 line was cultured in Medium 1640 (Thermo Fisher Scientific). Both media were supplemented with 10% heat-inactivated fetal bovine serum (FBS; Sigma-Aldrich Co., St Louis, MO, USA). All cells were maintained in a humidified atmosphere containing 95% O2 and 5% CO2 at 37°C.
Cell transfection
siRNAs for RRAD and a negative control (NC) were synthesized by GenePharma (Shanghai, People’s Republic of China) and used to transfect SK-Hep-1 cells at a concentration of 10 nM. The sequences of siRNAs were as follows: NC 5′-UUCUCCGAACGUGUCACGUTT-3′ and antisense 5′-ACGUGACACGUUCGGAGAATT-3′; si-RRAD 5′-GACUCAGACGAGAGCGUUUTT-3′, and antisense 5′-AAACGCUCUCGUCUGAGUCTT-3′. Oligonucleotide transfection was performed using Lipofectamine 2000 (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s protocol. Cells were collected 48 hours after transfection. Adenoviruses overexpressing RRAD or a NC were constructed by Hanbio (Shanghai, People’s Republic of China). Huh7 cells were treated with the adenovirus at a multiplicity of infection of 50 plaque forming units per cell in Dulbecco’s Modified Eagle’s Medium with 10% FBS. Cells were collected 48 hours after transfection. A replication-defective lentivirus encoding the RRAD or NC was constructed by Hanbio. Huh7 cells were treated with the lentivirus at a multiplicity of infection of 50 pfu/cell in 2% FBS with 8 μg/mL Polybrene (Sigma-Aldrich Co). Forty-eight hours after infection, puromycin (Thermo Fisher Scientific) was added to the medium to select stable transformants.
Western blot analyses
Western blotting was performed as described previously.14 Briefly, cells were first washed twice with cold phosphate-buffered saline (PBS). Total proteins were extracted from the cells after incubating in radioimmunoprecipitation assay lysis buffer (Cell Signaling Technology, Danvers, MA, USA) supplemented with complete protease inhibitor cocktail (Hoffman-La Roche Ltd., Basel, Switzerland) for 1 hour on ice. The supernatants were collected after centrifugation at 15,000× g at 4°C for 20 minutes. Protein concentrations were measured using a bicinchoninic acid protein assay kit (Thermo Fisher Scientific). Equal amounts of denatured proteins were separated by electrophoresis on SDS-PAGE gels and transferred onto polyvinylidene difluoride membranes. After blocking nonspecific binding for 1 hour using 5% nonfat milk, the membranes were incubated overnight on ice with primary antibodies against β-actin (Sigma-Aldrich Co.) and RRAD, cyclin A2, cyclin B1, cyclin D1, cyclin E1, cyclin-dependent kinase (CDK) 2, CDK 4, CDK 6, Bax, PARP, caspase-3, GLTU1, GLUT4, LDHA, LDHB, FBP1, PKM2, and PGK1 (Abcam).
Cell proliferation analyses
Cell proliferation assays were performed using Cell Counting Kit-8 (Dojindo Laboratories, Kumamoto, Japan). Absorbance was read at 450 nm. Cell proliferation was also evaluated using a colorimetric immunoassay (Cell-Light™ EdU Apollo567 In Vitro Imaging Kit; Ribobio, Guangzhou, People’s Republic of China) according to the manufacturer’s instructions. Cells were cultured in a confocal dish at a density of 1×105 cells per dish for 24 hours before transfection. The immunoassay was performed 48 hours after transfection. At least five independent experiments with three replicates for each condition were performed.
Cell cycle analysis
Cell cycle distribution was determined by flow cytometry. Briefly, 5×105 cells were fixed overnight at 4°C with 70% ethanol. After washing with cold PBS, cells were resuspended in a cell cycle staining kit (Multisciences, Hangzhou, People’s Republic of China) and incubated for 30 minutes at room temperature. All cells were then measured on a flow cytometer (CYTOMICS FC 500; Beckman Coulter, Miami, FL, USA) and analyzed using Modfit LT software (Verity Software House, Topsham, ME, USA).
Cell migration assay
The ability of cells to migrate into a defect in a monolayer culture was determined using the wound healing assay. Cells were grown to confluence in six-well plates and scratches were created using a 100 μL pipette tip. The medium was removed and cells were washed with PBS. The medium was then replaced and scratch closure was analyzed by capturing images under a microscope at different time points. Calculations were carried out according to the formula, (STime zero - STime point/STime zero) ×100, where S is the distance in μm.
Cell migration assays were also performed using transwells from Costar (Sigma-Aldrich Co.) with a 6.5 mm diameter and 8.0 μm pore size. Cells (1×104) were suspended in 300 μL serum-free medium, placed into the upper chamber of the transwell, and incubated for 24 hours. The lower chamber contained medium with 10% FBS. After incubation, membranes were isolated and stained with Diff-Quik (Polysciences, Inc., Warrington, PA, USA) according to the manufacturer’s protocol. The cells were then counted in five fields using an inverted microscope. The percentage of migrating cells was calculated relative to that of the control cells.
Apoptosis assay
Cells were cultured in six-well plates. Forty-eight hours after transfection, the cells were collected and resuspended in staining solution containing Annexin V-fluorescein isothiocyanate and propidium iodide. The stained cells (1×105) were analyzed using a flow cytometer.
Glucose uptake and lactate production assays
Glucose uptake was measured using a fluorescent D-glucose analog, 2-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino]-2-deoxy-D-glucose (Thermo Fisher Scientific), as described previously.15 For measuring lactate production, 1×106 cells were cultured in 10 cm dishes and analyzed using assay kits from BioVision (Milpitas, CA, USA).
Subcutaneous tumor growth
All experimental animal procedures were performed in compliance with the Animal Experiments Ethical Committee of Zhejiang University and were approved by the Zhejiang Medical Experimental Animal Care Commission. Huh7 cells were suspended in 0.2 mL of PBS and subcutaneously injected into the left flank of mice (5-week-old female BALB/c nude mice, 5×106 cells per mouse). The developed subcutaneous xenografts were extracted and cut into 1 mm3 pieces, which were then inoculated into the liver parenchyma of nude mice under anesthesia with ketamine after opening up the abdomen. The mice were monitored once every 2 days. The animals were sacrificed 5 weeks later, and relative data (including the weight, length, and width of the tumor) were recorded. Tumor volume was calculated using the following formula
|
|
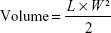
where L and W are the longest and shortest diameters of the tumors, respectively.
Statistical analysis
Data were analyzed using SPSS, Version 18 (SPSS Inc., Chicago, IL, USA). The results are presented as mean ± standard error of the mean. All experiments were performed in triplicate. Differences between groups were determined using Student’s t-test. P<0.05 was considered to indicate statistical significance.
Results
Expression of RRAD in HCC cell lines
Recently, a study showed that low expression of RRAD was associated with a poor prognosis in HCC.16 Here, we drew a similar conclusion that the expression of RRAD in tumor tissues was lower than matched peritumor tissues of a 90-patient cohort. Additionally, patients with relatively high RRAD expression had a better prognosis compared with those with low RRAD expression (P=0.044; Figure S1). Furthermore, investigation of the correlation between the expression of RRAD and clinicopathological features in the 90-patient cohort showed that low levels of RRAD were significantly associated with large tumor size and high tumor node metastasis stage (Table S1). Next, we speculated that RRAD affects HCC cells. The expression of RRAD was high in SK-Hep-1 cells and low in Huh7 cells (Figure 1A). Therefore, we transfected SK-Hep-1 cells with siRNAs of RRAD while Huh7 cells were transfected with an adenovirus for RRAD (Figure 1B and C). Si-1, Si-2, and Si-3 are three different siRNAs of RRAD affecting different truncations. The results showed that Si-1 was the most effective and was used for subsequent experiments.
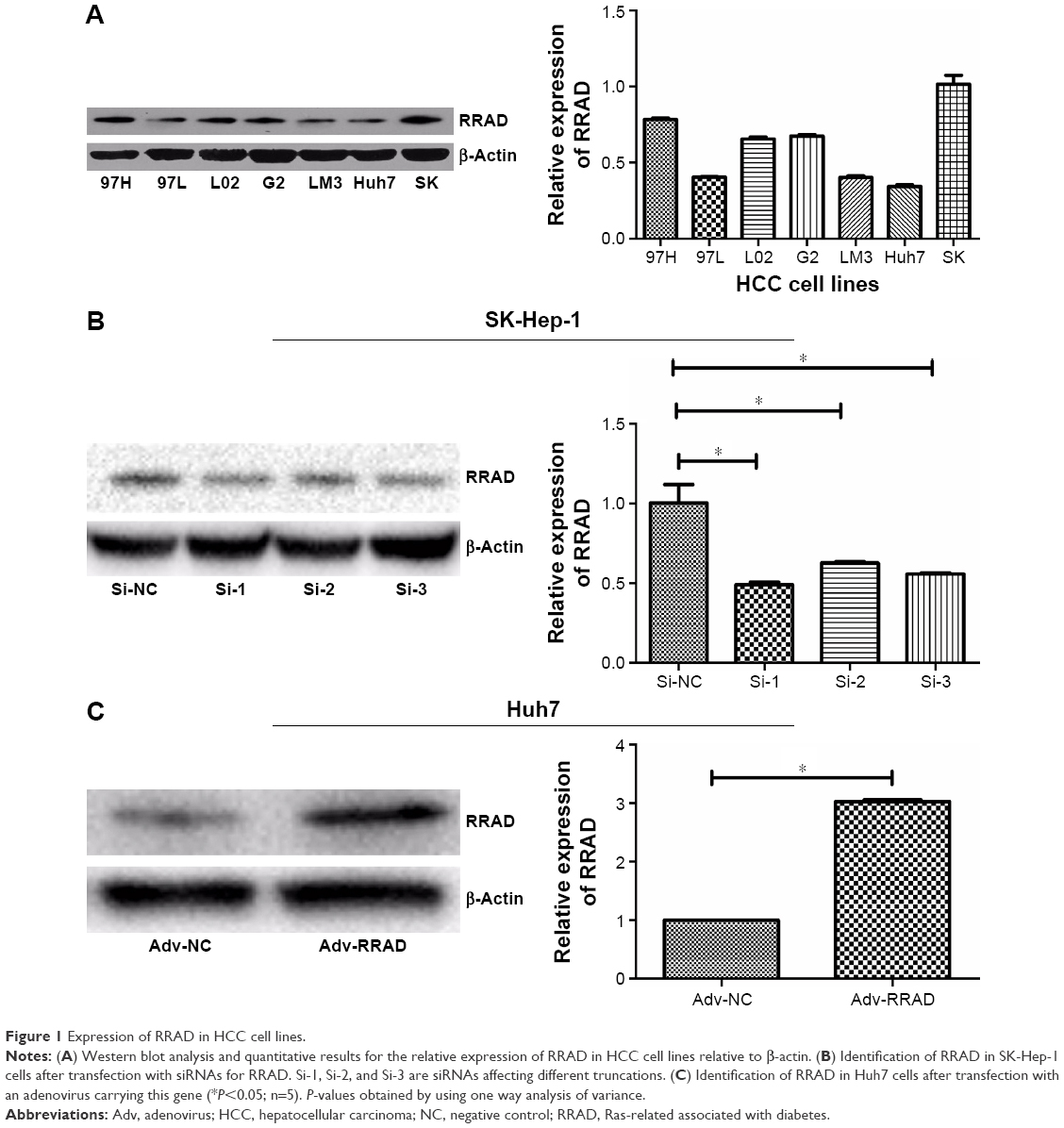 | Figure 1 Expression of RRAD in HCC cell lines. |
RRAD influences HCC cell proliferation and the cell cycle
The Cell Counting Kit-8 assay showed that knockdown of RRAD increased, while overexpression of RRAD decreased, the viability of SK-Hep-1 and Huh7 cells, respectively (Figure 2A and B). The 5-ethynyl-2′-deoxyuridine results showed that silencing RRAD promoted the proliferation of SK-Hep-1 cells while overexpression of RRAD inhibited the proliferation of Huh7 cells (Figure 2C and D).
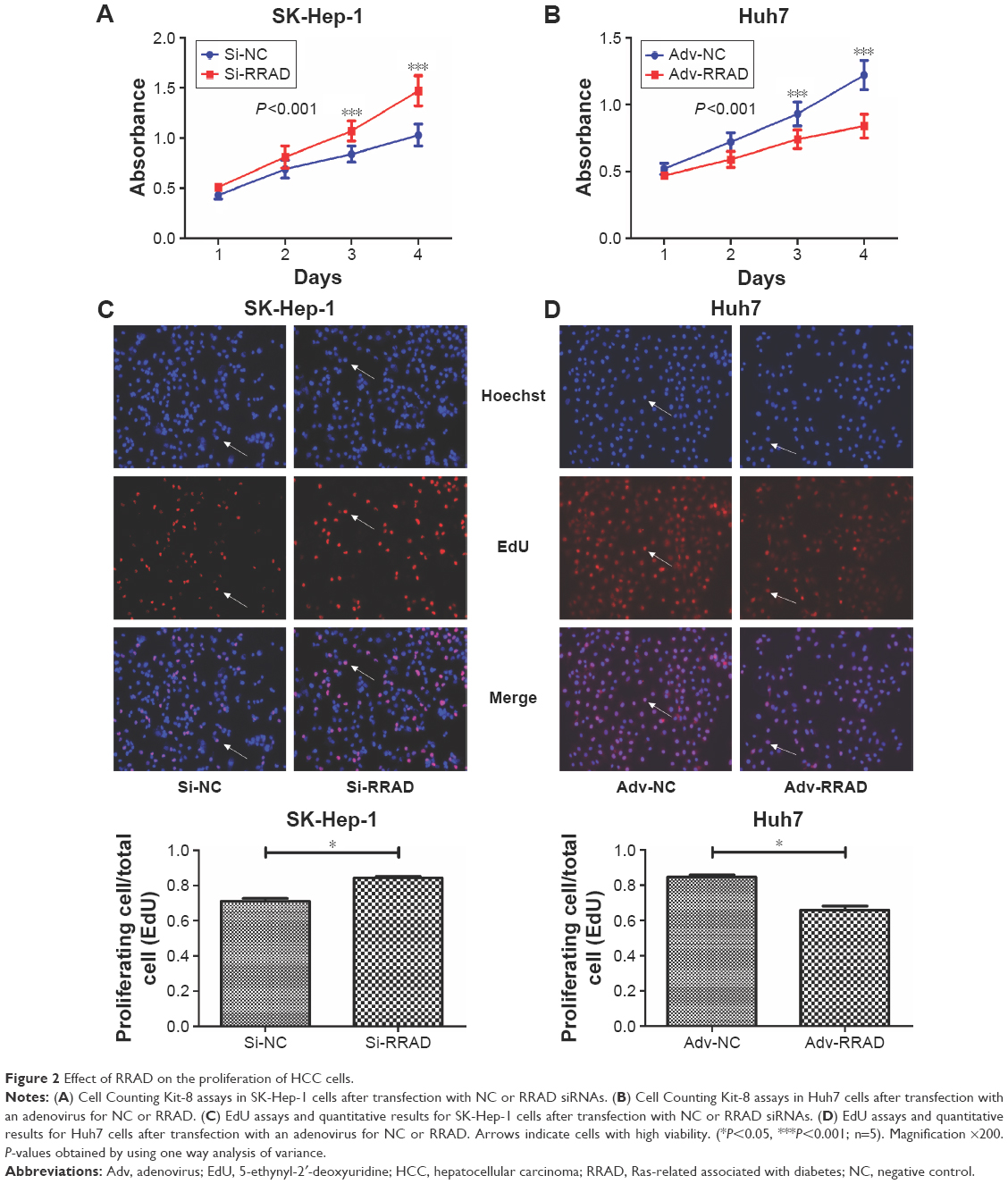 | Figure 2 Effect of RRAD on the proliferation of HCC cells. |
Because RRAD affected cell proliferation significantly, we next assessed changes to the cell cycle. Knockdown of RRAD significantly influenced the cell cycle in SK-Hep-1 cells. More cells transited to S phase so that cells in G1 phase were fewer compared with cells transfected with the NC. Cells in G2 phase showed no significant change (Figure 3A). Overexpression of RRAD in Huh7 cells prevented premature entry into S phase while cells in G2 phase were unchanged (Figure 3B).
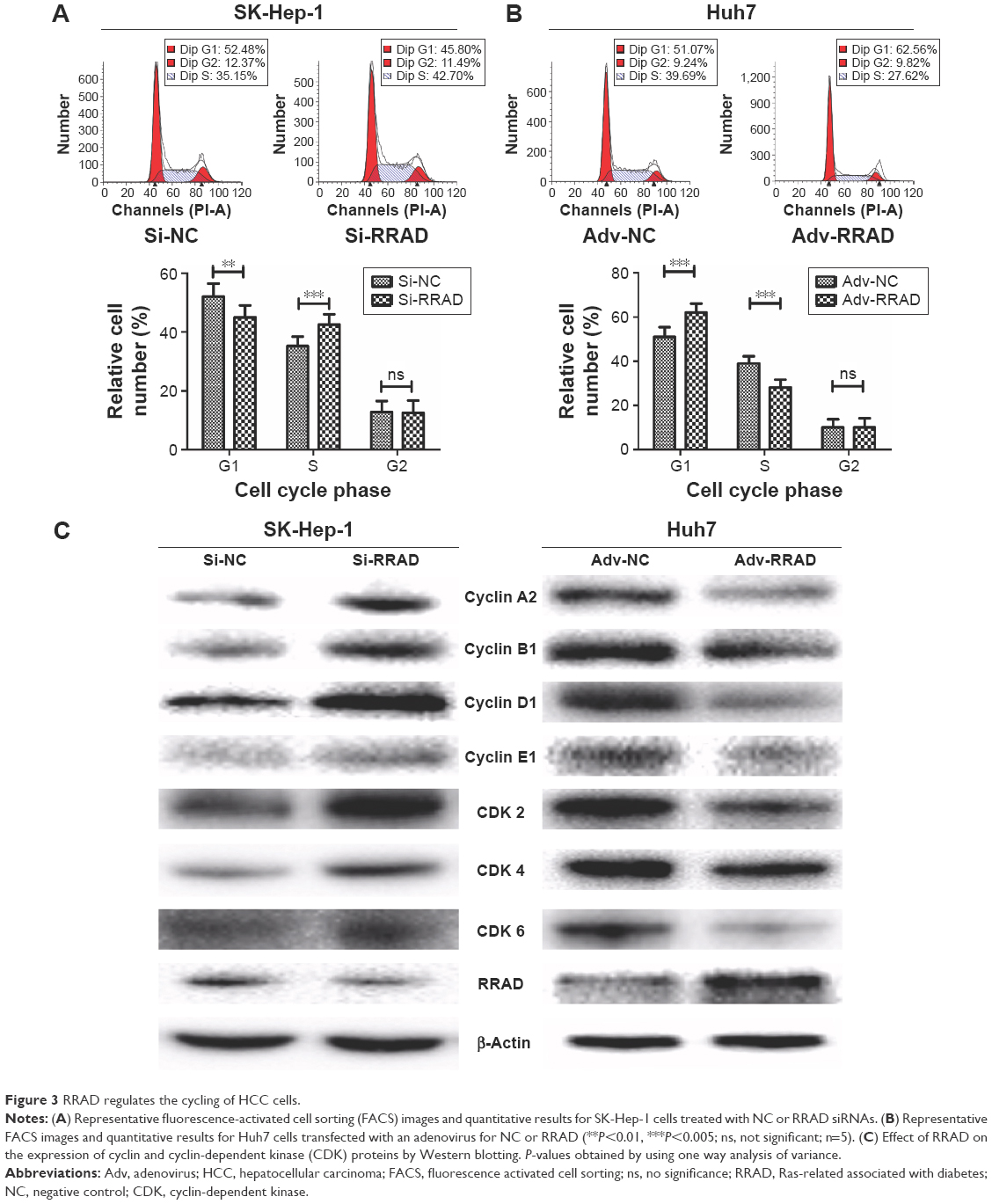 | Figure 3 RRAD regulates the cycling of HCC cells. |
We next determined whether the proteins that play important roles in regulating the cell cycle were changed in response to RRAD. Knockdown of RRAD upregulated the levels of cyclin A2, B1, D1, and E1 proteins, as well as CDK 2, CDK 4, and CDK 6 in SK-Hep-1 cells. Conversely, overexpression of RRAD in Huh7 cells downregulated levels of these proteins and protein kinases (Figure 3C). Consistent with these results, silencing RRAD significantly decreased the number of cells in G0/G1 phase with a sharp increase of those in S phase so that cell proliferation was promoted. Overexpression of RRAD had the opposite effect.
RRAD is associated with the migration of HCC cells
In order to investigate the function of RRAD in cell migration, wound healing and transwell assays were performed. The results indicated that knockdown of RRAD increased the migration ability of SK-Hep-1 cells (Figure 4A and B). In contrast, a significant decrease in the rate of migration was observed when RRAD was overexpressed in Huh7 cells (Figure 4C and D).
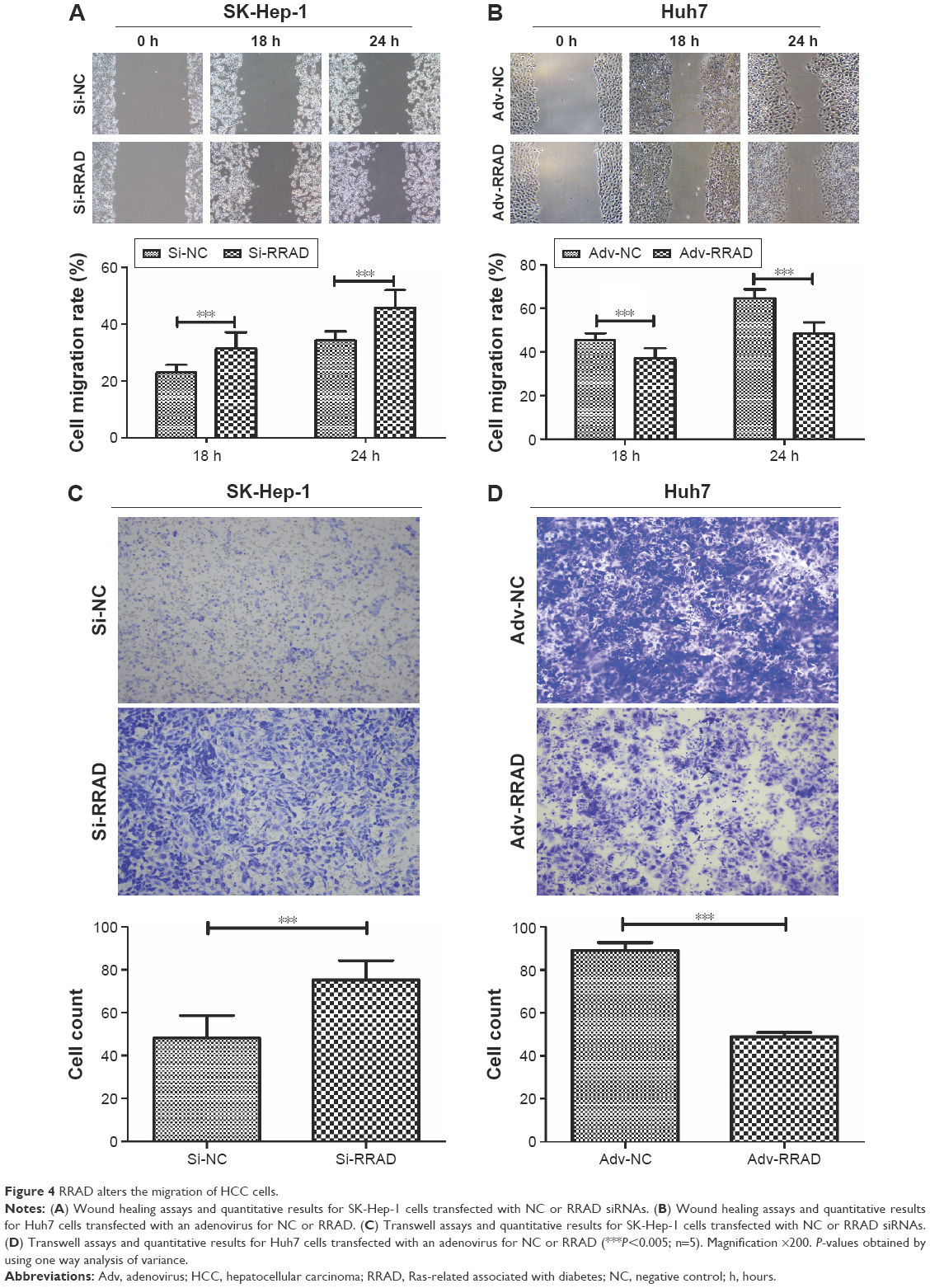 | Figure 4 RRAD alters the migration of HCC cells. |
RRAD affects apoptosis in HCC cells
Knockdown of RRAD reduced apoptosis in SK-Hep-1 cells (Figure 5A) while its overexpression increased apoptosis in Huh7 cells (Figure 5B). In addition, we found that proteins related to apoptosis including Bax, PARP, and caspase-3 were altered in response to the knockdown/overexpression of RRAD (Figure 5C).
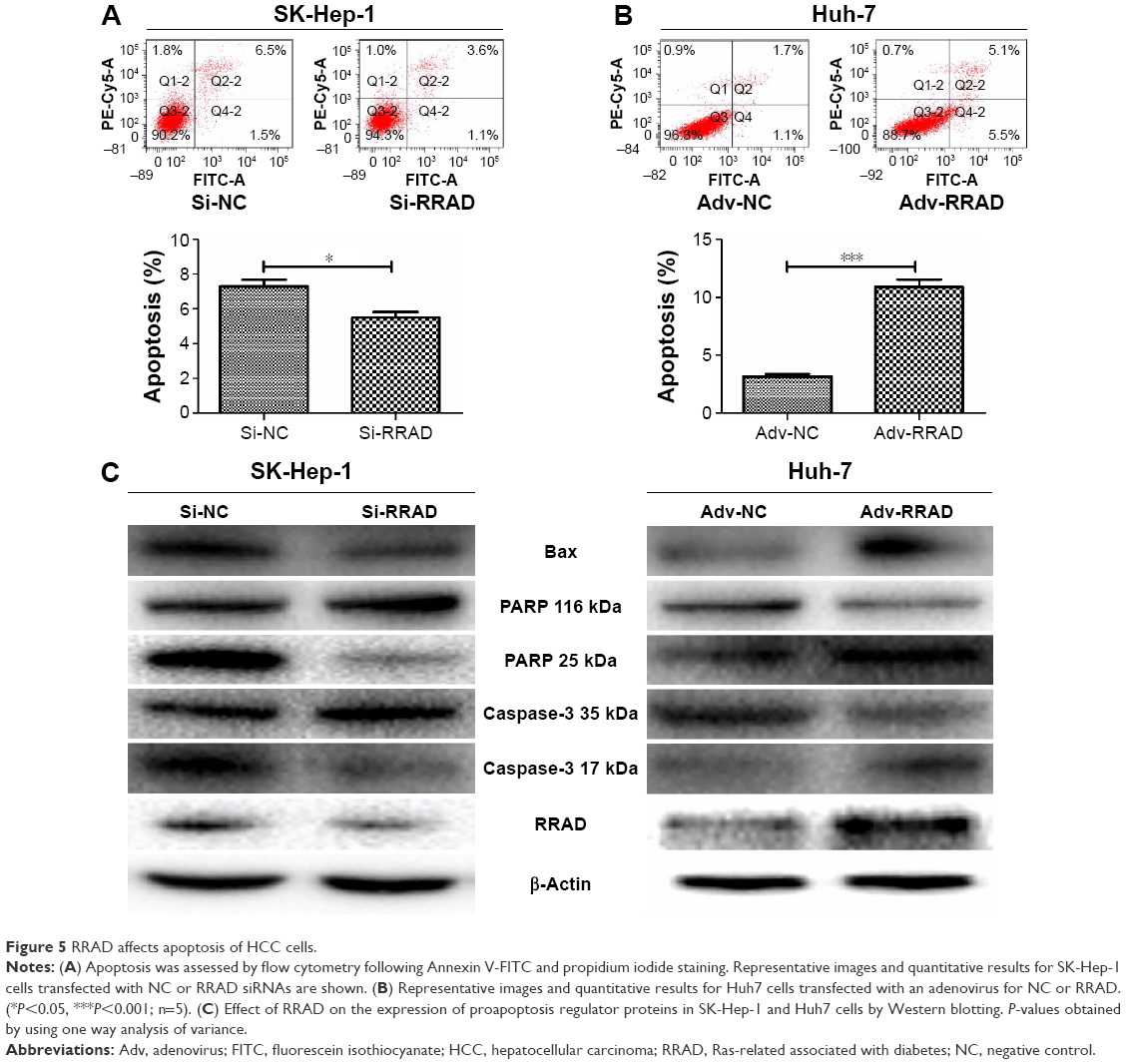 | Figure 5 RRAD affects apoptosis of HCC cells. |
RRAD suppresses glucose uptake and lactate production in HCC cells
The motility of cancer cells is associated with glucose and lactate levels in the extracellular milieu.17 Because RRAD expression was related to cell migration, we analyzed glucose uptake and lactate production. Knockdown of RRAD significantly enhanced glucose uptake and lactate production in SK-Hep-1 cells (Figure 6A and B). In contrast, overexpression of RRAD decreased glucose uptake and lactate production in Huh7 cells (Figure 6C and D). To better understand the role that RRAD plays in glucose metabolism, the levels of proteins related to aerobic glycolysis were determined. GLUT1 was upregulated with knockdown of RRAD while overexpression of RRAD led to a lower GLUT1 level. However, other proteins associated with glucose metabolism showed no significant alteration (Figure 6E).
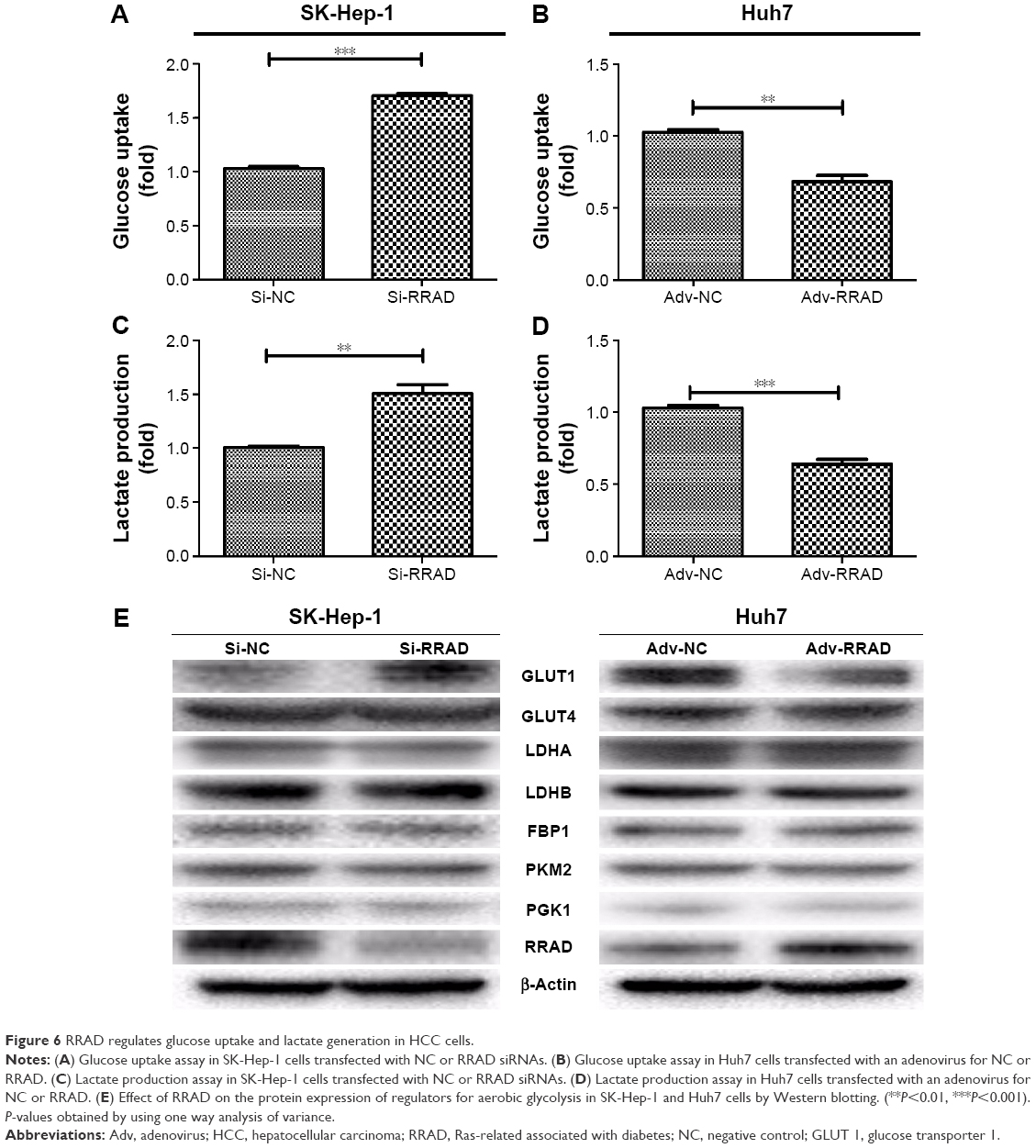 | Figure 6 RRAD regulates glucose uptake and lactate generation in HCC cells. |
RRAD inhibits tumor growth of HCC cells in nude mice
To further determine the effect of RRAD on HCC tumor growth in vivo, we constructed a lentiviral vector expressing RRAD and used it to establish stable Huh7 cell lines. The RRAD (n=6) and NC (n=6) groups showed successfully formed orthotropic liver tumors. The tumor size of the NC group was significantly larger than that of the RRAD group (Figure 7A–C).
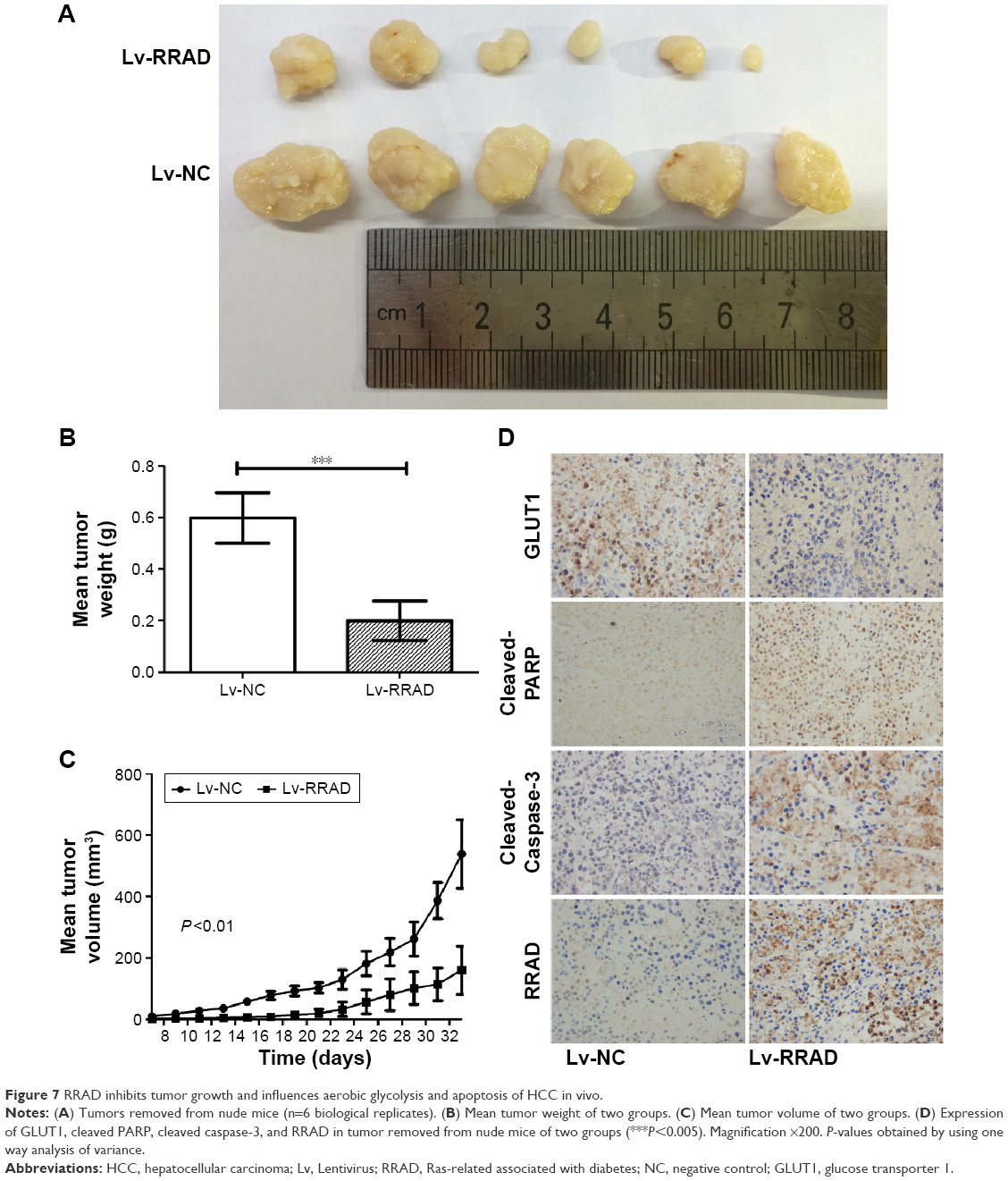 | Figure 7 RRAD inhibits tumor growth and influences aerobic glycolysis and apoptosis of HCC in vivo. |
RRAD suppresses aerobic glycolysis and promotes apoptosis in vivo
To further investigate the effect of RRAD on HCC in vivo, we performed an IHC assay on the tumor removed from nude mice. The expression of GLUT1 was higher in the NC group, which implied that RRAD suppresses aerobic glycolysis in vivo. Yet, the expression of cleaved PARP and cleaved caspase-3 was lower in the NC group, which implied that RRAD promotes apoptosis in vivo (Figure 7D).
Discussion
The Ras superfamily of small GTPases is composed of over 150 members. These GTPases play multiple roles, being versatile and key regulators of virtually all fundamental cellular processes. Conversely, their dysfunction plays a critical role in the pathogenesis of many human diseases including cancer.18 RRAD was one of the first members of the 35–39 kDa class of novel Ras-related GTPases, along with Gem, Kir, and other uncharacterized enzymes.19 In addition to its interaction with skeletal muscle β-tropomyosin, RRAD is associated with tumorigenicity. Expression of RRAD is downregulated in human lung cancer, which is related to a poor prognosis.6,20 RRAD is also downregulated in nasopharyngeal carcinoma cell lines by aberrant methylation.10
RRAD acts as a tumor suppressor in both solid and nonsolid tumors.21 The antitumor effect of RRAD in HCC was reported recently. This study showed that RRAD inhibited HCC cell migration, invasion, and aerobic glycolysis, and was associated with a poor prognosis.16 However, there remain questions about the mechanisms by which RRAD inhibits tumorigenicity of HCC.
In the current study, we focused on the impact that RRAD had on HCC to provide more information about its inhibitory effects. We showed, for the first time, that RRAD was negatively associated with HCC. RRAD was downregulated in HCC specimens and a lower expression was associated with a poorer prognosis. Furthermore, small tumor size and low tumor node metastasis stage were related to higher expression of RRAD.
The presumptive tumor suppressor function of RRAD in human HCC was further investigated by demonstrating that RRAD inhibited cell proliferation and the cell cycle was arrested in G0/G1 phase. Proteins and protein kinases that regulate the cell cycle (cyclins A2, B1, D1, and E1 and CDKs 2, 4, and 6) were downregulated by RRAD. CDK 2 may accelerate the G1/S transition when binding to cyclin E/A.22 Cyclin D combined with CDK 4/6 is necessary to phosphorylate and inactivate retinoblastoma family members, which dissociates these members from E2Fs, allowing E2F-dependent transcription. This may support DNA replication and upregulate cyclin B, promoting several early mitosis events that are essential for cell proliferation.23,24 Thus, the downregulation of cyclins and CDKs by RRAD indicates its antitumorigenic property.
Apoptosis is a type of cell death that can be initiated by extrinsic or intrinsic pathways. The extrinsic (death receptor) pathway provides a connection to the extracellular surroundings and links signals coming from the microenvironment to the intracellular signaling networks. The death receptor family of proteins is composed of cell surface receptors including CD95 (Fas/Apo1) and tumor necrosis factor.25 The intrinsic (mitochondrial) apoptosis pathway is initiated by the Bcl-2/Bax system.26 Specifically, when Bax is overexpressed, cell death signaling is enhanced, while Bcl-2 inhibits apoptosis. Caspase-3 acts as a central executioner in the caspase apoptotic cascade pathway and cleaves poly(ADP-ribose) polymerase to process apoptosis when activated.27 In the current study, apoptosis was increased, and Bax and cleaved poly(ADP-ribose) polymerase/caspase-3 were increased, by overexpression of RRAD, while silencing RRAD had the opposite effect. Collectively, the antitumor growth effect of RRAD may be related to the induction of apoptosis via various pathways mediated by RRAD.
Metabolic reprogramming is a hallmark of cancer.28 Cancer cells rely on aerobic glycolysis to meet their requirements for maintaining continuous growth and proliferation.29 However, this results in a lower efficiency of ATP production relative to mitochondrial oxidative phosphorylation. To compensate, cancer cells upregulate glucose transporters, notably GLUT1, that substantially increase glucose import into the cytoplasm.30 Recent studies have demonstrated the ectopic expression of GLUT1 in several types of malignancies,31 and GLUT1 may be a new target for tumor therapy.32,33 RRAD is linked to glucose metabolism in several types of cancers.11,12 Therefore, we analyzed RRAD-induced glucose consumption and lactate generation in HCC cells. The results indicated that RRAD decreased glucose uptake and lactate production, suggesting that RRAD acts as a suppressor of the Warburg effect. Knockdown of RRAD enhanced GLUT1 but had no effect on the levels of other proteins related to aerobic glycolysis. Taken together, these data indicated that RRAD may reduce the Warburg effect by downregulating GLUT1 in HCC cells.
Recently, a study revealed the inhibitory effect of RRAD in HCC21 by showing that RRAD inhibited migration, invasion, and aerobic glycolysis, and low expression was related to a poor prognosis. In our study, we reached a similar conclusion and showed that the negative effect of RRAD on HCC cell proliferation involved downregulation of cyclins and CDKs, resulting in cell cycle arrest in G0/G1. We also verified that RRAD facilitated apoptosis through the mitochondrial apoptosis pathways. Additionally, we found alterations of the GLUT1 protein level related to the expression of RRAD, whereas other molecules involved in the Warburg effect were unchanged.
Conclusion
We have identified a novel functional recessive oncogene, RRAD, in HCC. Our findings indicate that RRAD regulates aerobic glycolysis and that metabolic reprogramming induced by RRAD inhibits cell proliferation and arrests HCC cells in G0/G1. Moreover, cellular migration is impeded while apoptosis is promoted. These effects are consistent with a role for RRAD as a tumor suppressor in HCC. Importantly, in addition to contributing to our understanding about tumor metabolism, RRAD may serve as a potential epigenetic biomarker and provide a novel molecular target in cancer therapy.
Acknowledgments
This work was supported by The National Natural Science Foundation of China (no 81172315/H1617) and National S&T Major Project (no 2012ZX10002017).
Disclosure
The authors report no conflicts of interest in this work.
References
Byam J, Renz J, Millis JM. Liver transplantation for hepatocellular carcinoma. Hepatobiliary Surg Nutr. 2013;2(1):22–30. | ||
Bravi F, Bosetti C, Tavani A, Gallus S, La Vecchia C. Coffee reduces risk for hepatocellular carcinoma: an updated metaanalysis. Clin Gastroenterol Hepatol. 2013;11(11):1413–1421. | ||
Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–1033. | ||
Hitosugi T, Kang S, Vander Heiden MG, et al. Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Sci Signal. 2009;2(97):ra73. | ||
Zhu Y, Shi M, Chen H, et al. NPM1 activates metabolic changes by inhibiting FBP1 while promoting the tumorigenicity of pancreatic cancer cells. Oncotarget. 2015;6(25):21443–21451. | ||
Zhu J, Bilan PJ, Moyers JS, Antonetti DA, Kahn CR. Rad, a novel Ras-related GTPase, interacts with skeletal muscle β-tropomyosin. J Biol Chem. 1996;271(2):768–773. | ||
Downward J. Regulatory mechanisms for ras proteins. Bioessays. 1992;14(3):177–184. | ||
Tisdale EJ, Bourne JR, Khosravi-Far R, Der CJ, Balch WE. GTP- bindingmutants of rab1 and rab2 are potent inhibitors of vesicular transport from the endoplasmic reticulum to the Golgi complex. J Cell Biol. 1992;119(4):749–761. | ||
Ridley AJ, Hall A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell. 1992;70(3):389–399. | ||
Mo Y, Midorikawa K, Zhang Z, et al. Promoter hypermethylation of Ras related GTPase gene RRAD inactivates a tumor suppressor function in nasopharyngeal carcinoma. Cancer Lett. 2012;323(2):147–154. | ||
Zhang C, Liu J, Wu R, et al. Tumor suppressor p53 negatively regulates glycolysis stimulated by hypoxia through its target RRAD. Oncotarget. 2014;5(14):5535–5546. | ||
Wang Y, Li G, Mao F, et al. Ras-induced epigenetic inactivation of the RRAD (Ras-related associated with diabetes) gene promotes glucose uptake in a human ovarian cancer model. J Biol Chem. 2014;289(20): 14225–14238. | ||
Sova P, Feng Q, Geiss G, et al. Discovery of novel methylation biomarkers in cervical carcinoma by global demethylation and microarray analysis. Cancer Epidemiol Biomarkers Prev. 2006;15(1):114–123. | ||
Wang W, Xie Q, Zhou X, et al. Mitofusin-2 triggers mitochondria Ca2+ influx from the endoplasmic reticulum to induce apoptosis in hepatocellular carcinoma cells. Cancer Lett. 2015;358(1):47–58. | ||
Zou C, Wang Y, Shen Z. 2-NBDG as a fluorescent indicatorfor direct glucose uptake measurement. J Biochem Biophys Methods. 2005;64(3):207–215. | ||
Shang R, Wang J, Sun W, et al. RRAD inhibits aerobic glycolysis, invasion, and migration and is associated with poor prognosis in hepatocellular carcinoma. Tumour Biol. Epub 2015 Nov 6. | ||
Dhup S, Dadhich RK, Porporato PE, Sonveaux P. Multiple biological activities of lactic acid in cancer: influences on tumor growth, angiogenesis and metastasis. Curr Pharm Des. 2012;18(10):1319–1330. | ||
Goitre L, Trapani E, Trabalzini L, Retta SF. The Ras superfamily of small GTPases: the unlocked secrets. Methods Mol Biol. 2014;1120:1–18. | ||
Hsiao BY, Chen CC, Hsieh PC, et al. Rad is a p53 direct transcriptional target that inhibits cell migration and is frequently silenced in lung carcinoma cells. J Mol Med. 2011;89(5):481–492. | ||
Suzuki M, Shigematsu H, Shames DS, et al. Methylation and gene silencing of the Ras-related GTPase gene in lung and breast cancers. Ann Surg Oncol. 2007;14(4):1397–1404. | ||
Yeom SY, Lee SJ, Kim WS, Park C. Rad knockdown induces mitochondrial apoptosis in bortezomib resistant leukemia and lymphoma cells. Leuk Res. 2012;36(9):1172–1178. | ||
Tsai LH, Harlow E, Meyerson M. Isolation of the human cdk2 gene that encodes the cyclin A- and adenovirus E1A-associated p33 kinase. Nature. 1991;353(6340):174–177. | ||
Kimura K, Hirano M, Kobayashi R, Hirano T. Phosphorylation and activation of 13S condensin by Cdc2 in vitro. Science. 1998;282(5388):487–490. | ||
Joan M. G1 cell-cycle control and cancer. Nature. 2004;432(7015): 298–306. | ||
Ashkenazi A. Targeting the extrinsic apoptosis pathway in cancer. Cytokine Growth Factor Rev. 2008;19(3–4):325–331. | ||
Gu W, Li C, Yin W, Guo Z, Hou X, Zhang D. Shen-Fu injection reduces postresuscitation myocardial dysfunction in a porcine model of cardiac arrest by modulating apoptosis. Shock. 2012;38(3):301–306. | ||
Lazebnik YA, Kaufmann SH, Desnoyers S, Poirier GG, Earnshaw WC. Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature. 1994;371(6495):346–347. | ||
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. | ||
Dang CV. Links between metabolism and cancer. Genes Dev. 2012;26(9):877–890. | ||
Jones RG, Thompson CB. Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev. 2009;23(5):537–548. | ||
Li S, Yang X, Wang P, Ran X. The effects of GLUT1 on the survival of head and neck squamous cell carcinoma. Cell Physiol Biochem. 2013;32(3):624–634. | ||
Shibuya K, Okada M, Suzuki S, et al. Targeting the facilitative glucose transporter GLUT1 inhibits the self-renewal and tumor-initiating capacity of cancer stem cells. Oncotarget. 2015;6(2):651–661. | ||
Abouzeid AH, Patel NR, Rachman IM, Senn S, Torchilin VP. Anti-cancer activity of anti-GLUT1 antibody-targeted polymeric micelles co-loaded with curcumin and doxorubicin. J Drug Target. 2013;21(10): 994–1000. |
Supplementary materials
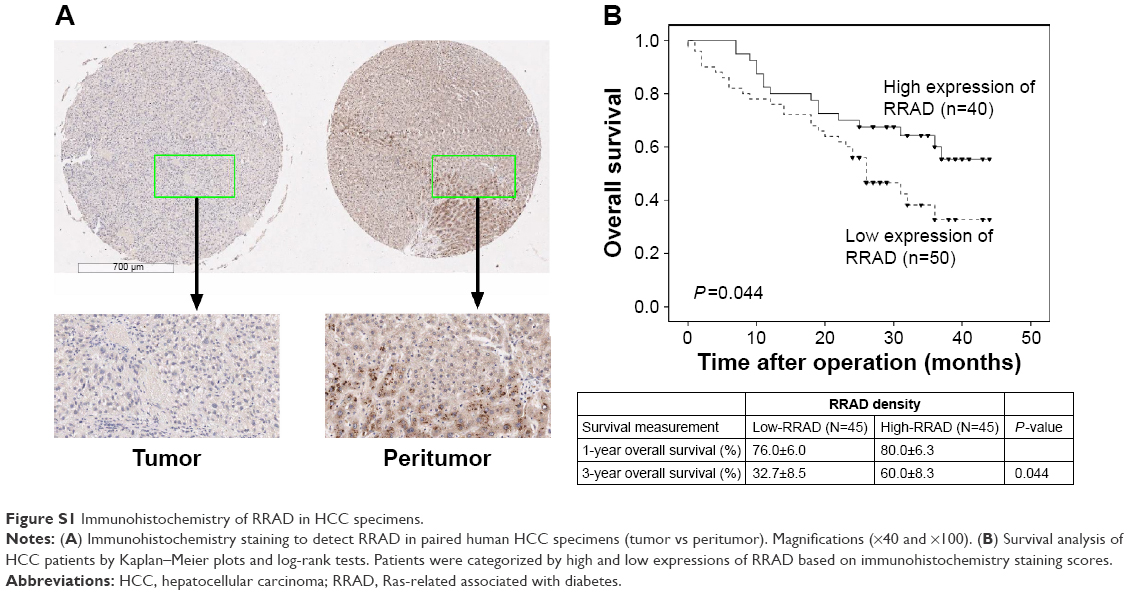 | Figure S1 Immunohistochemistry of RRAD in HCC specimens. |
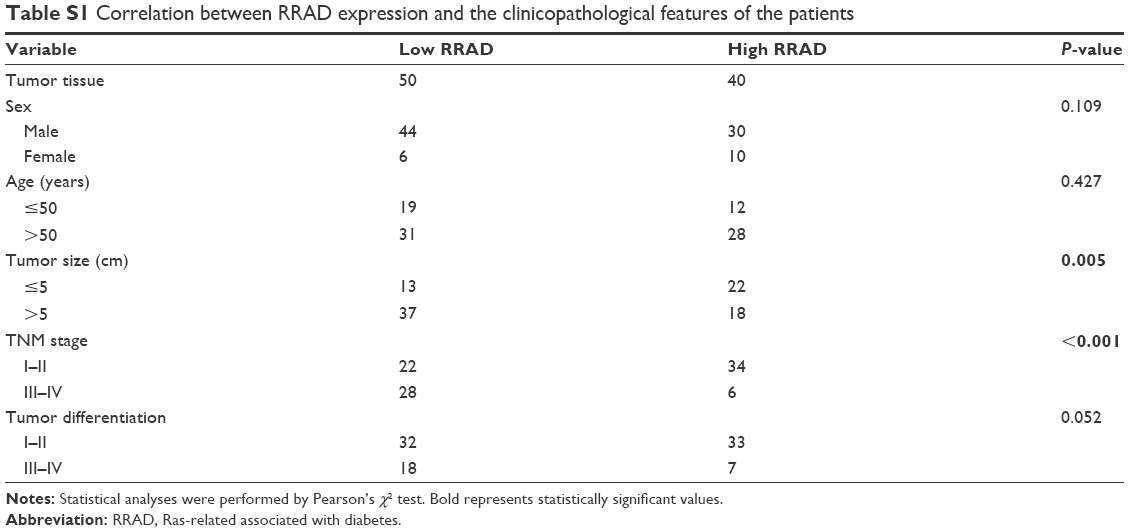 | Table S1 Correlation between RRAD expression and the clinicopathological features of the patients |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.