Back to Journals » Clinical Ophthalmology » Volume 15
Randomised, Double-blind, Comparative Clinical Study of New Ranibizumab Biosimilar in Neovascular (Wet) Age-Related Macular Degeneration
Authors Apsangikar P, Ghadge P, Naik M, Nair S, Payghan R
Received 25 March 2021
Accepted for publication 25 May 2021
Published 16 July 2021 Volume 2021:15 Pages 3087—3095
DOI https://doi.org/10.2147/OPTH.S307746
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Scott Fraser
Prasad Apsangikar, Pravin Ghadge, Manoj Naik, Santosh Nair, Ravikiran Payghan
Reliance Life Sciences Pvt. Ltd, Mumbai, India
Correspondence: Prasad Apsangikar
Reliance Life Sciences Pvt. Ltd., 282 MIDC, Thane Belapur Road, Rabale, Navi Mumbai, 400701 Tel +919867610090
Fax +912235338099
Email [email protected]
Purpose: The study was undertaken for regulatory purposes to establish clinical biosimilarity and interchangeability of a ranibizumab biosimilar with reference product.
Patients and Methods: A total of 159 subjects with neovascular (wet) age-related macular degeneration (AMD) were dosed with ranibizumab. Initial double blind period of 16 weeks was followed by open-label phase till week 24. Efficacy assessment was performed at weeks 1, 4, 8, 12, 16, 20 and 24 based on best corrected visual acuity. Change in central macular thickness was assessed by optical coherence tomography from baseline to week 24. Immunogenicity assessment was done in both arms at baseline, week 16 and week 24. Safety evaluation included clinical and ophthalmic examination, adverse events, vital signs, laboratory parameters and immunogenicity in both treatment arms.
Results: In the biosimilar test arm, 104 (98.11%) and 105 (99.06%) patients lost fewer than 15 letters in visual acuity at week 16 and week 24, respectively, compared with 53 (100%) at both follow-ups in reference arm. In the test arm, 27 (25.47%) and 34 (32.08%) patients gained at least 15 letters in visual acuity till week 16 and week 24 respectively, compared with 17 (32.08%) and 23 (43.30%) in the reference arm. In the test arm, mean change in central macular thickness at 24 weeks was − 89.93 μm against − 64.42 μm in the reference arm. Difference was statistically not significant for any endpoint at 16 and 24 weeks for the primary and secondary endpoints.
Conclusion: The evaluation of efficacy, safety and immunogenicity was concluded to show no meaningful clinical difference for biosimilar ranibizumab with the reference product.
Keywords: ranibizumab, biosimilar, AMD, intravitreal, visual acuity
Introduction
Vascular endothelial growth factor-A (VEGF-A) is the principal target for treatment of neovascular age-related macular degeneration (nAMD) and VEGF-A inhibitors are currently the standard of care for most cases of newly occurring, symptomatic nAMD. Ranibizumab, a recombinant, humanized, monoclonal antibody fragment that binds to and neutralizes active isoforms of VEGF-A, has been approved for the treatment of nAMD1. The relatively high cost of original biological agents likely limits some patient access to these treatments. Biosimilar products are highly similar to an approved reference biological product; that is, there are no clinically meaningful differences in terms of efficacy, safety, and immunogenicity. Similar biologics are developed through a sequential process to demonstrate the similarity by extensive characterization studies revealing the molecular and quality attributes with regard to the reference biologic. For certain attributes it is customary to use multiple, orthogonal methods for characterization. Characterization studies for similar biologics include physicochemical properties, biological activity, immunological properties, functional assays, purity (process and product-related impurities etc.), contamination, strength and content. Principles outlined in the ICH Q6B guidelines should be followed2.
RanizuRelTM (Ranibizumab) is produced as a biosimilar to the innovator product following global standard guidelines. Following the establishment of comparative physicochemical and biological characterization, the present study was done as a regulatory requirement for establishing non-inferiority to the reference innovator product in terms of efficacy, safety and immunogenicity thereby to establish absence of any clinically meaningful difference and interchangeability.
Materials and Methods
The study was conducted in compliance with the approved protocol by Drug Control General of India (DCGI) and New Drugs and Clinical Trial Rules (NDCT) and in accordance with the Declaration of Helsinki, International Conference on Harmonization-Good clinical practices (ICH-GCP). This was an active-controlled, parallel-group, comparative randomized, double blind, clinical study to evaluate efficacy, pharmacokinetics, safety and immunogenicity of test biosimilar ranibizumab with reference ranibizumab in patients with neovascular (wet) age-related macular degeneration. This study’s essential documents were also approved by the Institutional Ethics Committee (EC) and registered with Clinical Trial Registry of India (CTRI) registration number CTRI/2018/ 05/ 014065 before initiation of the study.
This was a prospective, multi-centre, double blind, two-arm, parallel group, active control, randomized comparative clinical study conducted at 16 hospitals, nursing homes and ophthalmology clinics across India. The randomized controlled trial is the most preferred clinical study design; hence, this design was adopted for this study. Random allocation minimized the heterogeneity between the test and control arms. Male or female patients of age ≥ 50 years satisfying the inclusion and exclusion criteria were included in the study. Subjects included in the study were having active primary or recurrent subfoveal lesions with classic or occult choroidal neovascularization (CNV) secondary to age-related macular degeneration (AMD) with corrected visual acuity, using Early Treatment of Diabetic Retinopathy Study (ETDRS) charts, of 20/40 to 20/320 (Snellen equivalent) in the study eye. Subjects with CNV in either eye due to other causes, such as ocular histoplasmosis, trauma, or pathologic myopia were excluded. Subjects with previous intravitreal drug delivery or vitrectomy surgery in the study eye or on verteporfin photodynamic therapy in the study eye within 6 months, previous external-beam radiation therapy, transpupillary thermotherapy or subfoveal focal laser photocoagulation in the study eye and with laser photocoagulation (juxtafoveal or extrafoveal) one month prior were also excluded. Exclusion was also done for subjects with history of submacular surgery or other surgical intervention for AMD, previous intravitreal drug delivery or vitrectomy surgery in the study eye, patients with current vitreous hemorrhage, sub retinal hemorrhage that involved the fovea, subfoveal fibrosis or atrophy, active intraocular inflammation or infection in the study eye. Subjects with history of retinal pigment epithelial tear, aphakia or absence of the posterior capsule in the study eye, infectious conjunctivitis, keratitis or scleritis in either eye and uncontrolled glaucoma, were also not included in the study. Uncontrolled glaucoma (defined as intraocular pressure of 30 mmHg or more despite treatment with antiglaucoma medications) or history of glaucoma filtering surgery in the study eye was also included in the exclusion criteria.
The randomization schedule was generated by a statistician at sponsor site. Once a subject was found to be eligible for randomization, the site would request a randomization code for the subject. Randomization was managed centrally. Patients were allocated (2:1) to one of the two treatment arms using a computer-generated randomization schedule. Treatment assignment for individual subjects remained double-blind until after the study data had been cleaned and the database locked as per the statistical analysis plan. In order to maintain blinding, an unblinded person was employed in the study during the blinding activities who maintained the blinding records and codes for medications and was responsible for release of medications and maintaining the logs.
Eligible subjects as per the randomization and double blind code received either biosimilar or reference ranibizumab 0.5 mg (0.05 mL of 10 mg/mL solution) as intravitreal injection once every 4 weeks for the treatment period of 24 weeks. The treatment period consisted of initial double blind-period of 16 weeks followed by open-label phase till week 24. Primary Objective was to compare the efficacy of intravitreal injections of the biosimilar ranibizumab against the reference product in preventing vision loss, as determined by the proportion of patients who lost fewer than 15 letters in visual acuity at week 16 compared with baseline. As per the available literature, the beneficial effect of intravitreal injection of ranibizumab peaks at 3 months (injections administered at month 0, 1 and 2) whereafter a plateau is reached. Hence, in accordance with pooled data from previous studies, the decision for primary efficacy assessment at week 16 following initiation of therapy was made. The assessments of the study were conducted as a double-blind study up to week 16. All patients in both treatment arms continued treatment in the open-label phase up to week 24. This allowed this study to conform to the studies conducted on the Reference Medicinal Product (RMP), as well as address the scientific question of how the biosimilar version is tolerated, thereby establishing the safety and therapeutic profile over a total period of approximately 24 weeks. Secondary efficacy endpoints were, proportion of patients who lost fewer than 15 letters and who gained at least 15 letters in visual acuity from baseline to week 24, mean change in best corrected visual acuity (no. of letters) from baseline to week 24, proportion of patients with a visual acuity Snellen equivalent of 20/40 or better from baseline to week 24, proportion of patients with a visual acuity Snellen equivalent of 20/200 or worse from baseline to week 24 and change in central macular thickness assessed by Optical Coherence Tomography from baseline to week 24. Ocular examination, tonometry and slit lamp examination were done at every visit for recording of any abnormality. Optical Coherence Tomography were performed at screening, week 4, 8, 12, 16, 20 and 24. The other secondary objectives were to evaluate immunogenicity, safety and tolerability in both ranibizumab arms up to week 24. Efficacy assessment was performed at week 1, 4, 8, 12, 16, 20 and 24 based on the best corrected visual acuity as assessed with the Early Treatment Diabetic Retinopathy Study (ETDRS) chart, with the use of standardized refraction and testing protocol. Change in central macular thickness was assessed by Optical Coherence Tomography from baseline to week 24. Pharmacokinetic assessment in 24 subjects (12 from each group i.e., 1:1 ratio of test and reference product) was planned to be done to evaluate the systemic exposure to ranibizumab. Hence, PK data were not analysed. Safety was evaluated based on clinical and ophthalmic examination, adverse events, vital signs and laboratory parameters in both the treatment arms. All adverse events (AE) and serious adverse events (SAE) from the screening visit till the end of study were recorded for safety analysis. Treatment emergent adverse events (TEAE) were followed till resolution, death or loss to follow-up. Immunogenicity assessment was done in both the treatment arms at baseline, week 16 and week 24. Clinically significant abnormal laboratory values were recorded and were considered in the safety analysis. Study flow chart is given below in Figure 1.
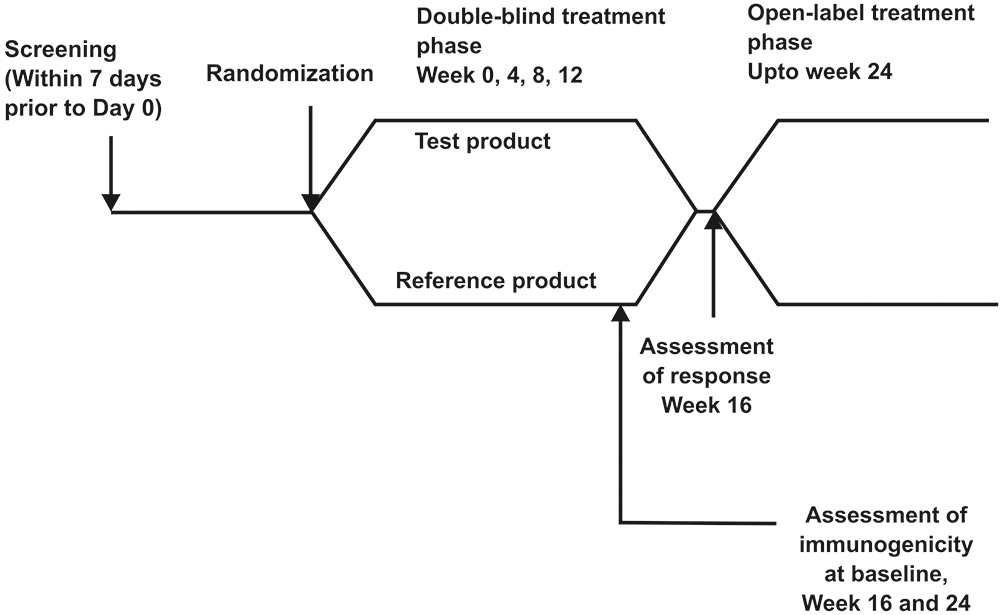 |
Figure 1 Study flow chart. |
Statistical Analysis
Statistical analysis plan (SAP) was prepared to describe the statistical methods to be employed in the study and the data presentations required for this study. Statistical analyses were performed using the R version 3.6.1. Null hypothesis was that a proportional difference less than 9% yields a statistical significance for proportion of patients who lost fewer than 15 letters in visual acuity from baseline to week 16. Alternate hypothesis (H1) was that a proportional difference greater than 9% yields a statistically significant difference with power at 80% and alpha at 0.05. Hence, it was assumed that the lower and upper boundaries can be 9% and 15% respectively, for the proportion of patients who lost fewer than 15 letters in visual acuity from baseline to week 16, to show a statistical significance. With the above assumptions, a total of 150 patients could provide the study with a statistical power of more than 80% with alpha at 0.05. The details of efficacy equivalence margins and statistical aspect of efficacy analysis was detailed in a separate statistical analysis plan.
Results
In this study, a total of 160 subjects (107 subjects in the biosimilar test arm and 53 subjects in the reference ranibizumab arm) were randomized across 16 centers across India and were included in intent to treat (ITT) population. A total of 159 randomized subjects received at least one dose of the study medication (106 subjects in the test arm and 53 subjects in the reference arm) and were included in the Safety/modified ITT population (mITT) population. Out of 159 dosed subjects, no subject had a major protocol deviation. Therefore, per protocol (PP) population for this study included 159 subjects i.e. 106 subjects in the test or study arm and 53 subjects in the reference arm. Out of 106 subjects included in PP population from biosimilar arm, 43 (40.57%) were females and 63 (59.43%) were males. The mean age of these subjects was 67.04 years, mean height was 160.19 cm, mean weight was 61.39 kg and mean BMI was 24.00 kg/m2. In the reference product arm, out of 53 subjects in PP population, 16 (30.19%) were females and 37 (69.81%) were males with mean age of 67.06 years, mean height of 160.30 cm, mean weight of 69.84 kg and mean BMI of 23.99 kg/m2. The demographic characteristics of the subjects enrolled in both arms were comparable for age, height and weight.
A total of 141 subjects completed the study, which included 91 (85.85%) subjects in the biosimilar test arm and 50 (94.34%) subjects in the reference arm. A total of 18 subjects discontinued from the study before completion, which included 15 (14.15%) subjects in the test arm and 3 (5.66%) subjects in the reference arm. The details of patient disposition are presented in Table 1.
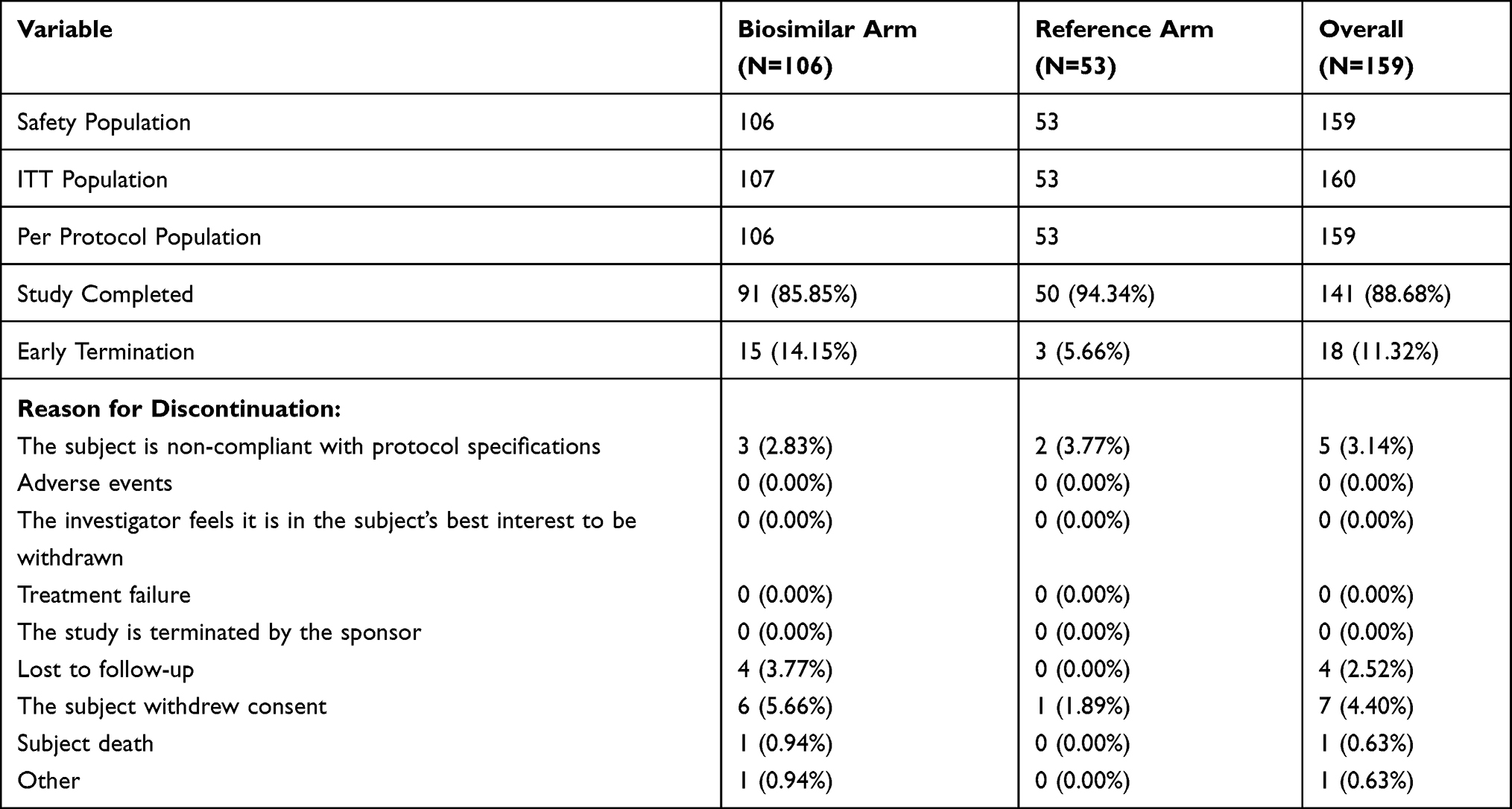 |
Table 1 Subject Disposition |
Efficacy Assessment
In the primary efficacy analysis, in test arm, 104 (98.11%) patients lost fewer than 15 letters in visual acuity from baseline to week 16 compared with 53 (100%) patients in reference arm. At week 24, 105 (99.06%) patients in the test arm lost fewer than 15 letters in visual acuity from baseline as compared with 53 (100%) patients in the reference arm. The difference in proportion of patients who lost fewer than 15 letters in visual acuity from baseline to week 16 (p = 0.314) and week 24 (p = 0.478) was statistically not significant between the two treatment arms (Table 2).
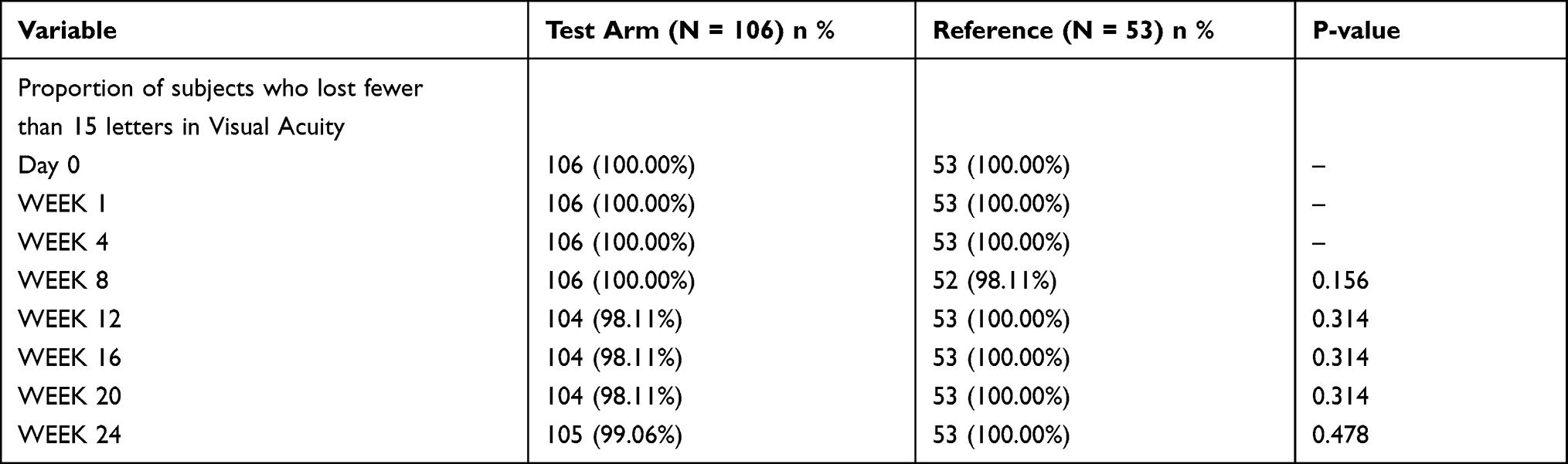 |
Table 2 Proportion of Patients Who Lost Fewer Than 15 Letters in Visual Acuity (PP Population) |
In the evaluation of secondary endpoints, in the test arm, 27 (25.47%) patients gained at least 15 letters in visual acuity from baseline to week 16 compared with 16 (30.19%) patients in the reference arm. At week 24, 34 (32.08%) patients in test arm and 23 (43.30%) patients in the reference arm gained at least 15 letters in visual acuity from baseline. The difference in proportion of patients who gained at least 15 letters in visual acuity from baseline to week 16 (p = 0.535) and week 24 (p = 0.161) was statistically not significant between the two treatment arms. In the test arm, the mean number of letter gain was 10.47 compared with 12.58 letters in the reference arm at week 16. At week 24, the mean number of letter gain was 12.11 and 15.66 in the test arm and the reference arm, respectively. The difference in mean number of letter gain from baseline to week 16 (p = 0.24234) and week 24 (p = 0.07534) was statistically not significant between the two treatment arms.
In secondary efficacy analysis, biosimilar ranibizumab arm, 14 (14.74%) patients had visual acuity Snellen equivalent of 20/40 or better or Snellen equivalent of 20/200 or worse, compared with 12 (23.53%) and 6 (11.76%) patients respectively in the reference arm at week 16. At week 24, 16 (17.58%) and 9 (9.89%) patients from the biosimilar arm and 13 (26.00%) and 4 (8.00%) patients from the reference arm had visual acuity Snellen equivalent of 20/40 or better and Snellen equivalent 20/200 or worse respectively. The difference between the two treatment arms for both Snellen equivalents was statistically not significant at week 16 (p = 0.129, 0.735) and week 24 (p = 0.146, 0.838).
In biosimilar test arm, the mean change in central macular thickness was −78.22 µm compared with −66.80 µm in the reference arm at week 16. At week 24, the mean change in central macular thickness was −89.93 µm in the test arm and −64.42 µm in the reference arm. The difference between the two treatment arms was statistically not significant at week 16 (p = 0.5502) and week 24 (p = 0.2160) (Table 3).
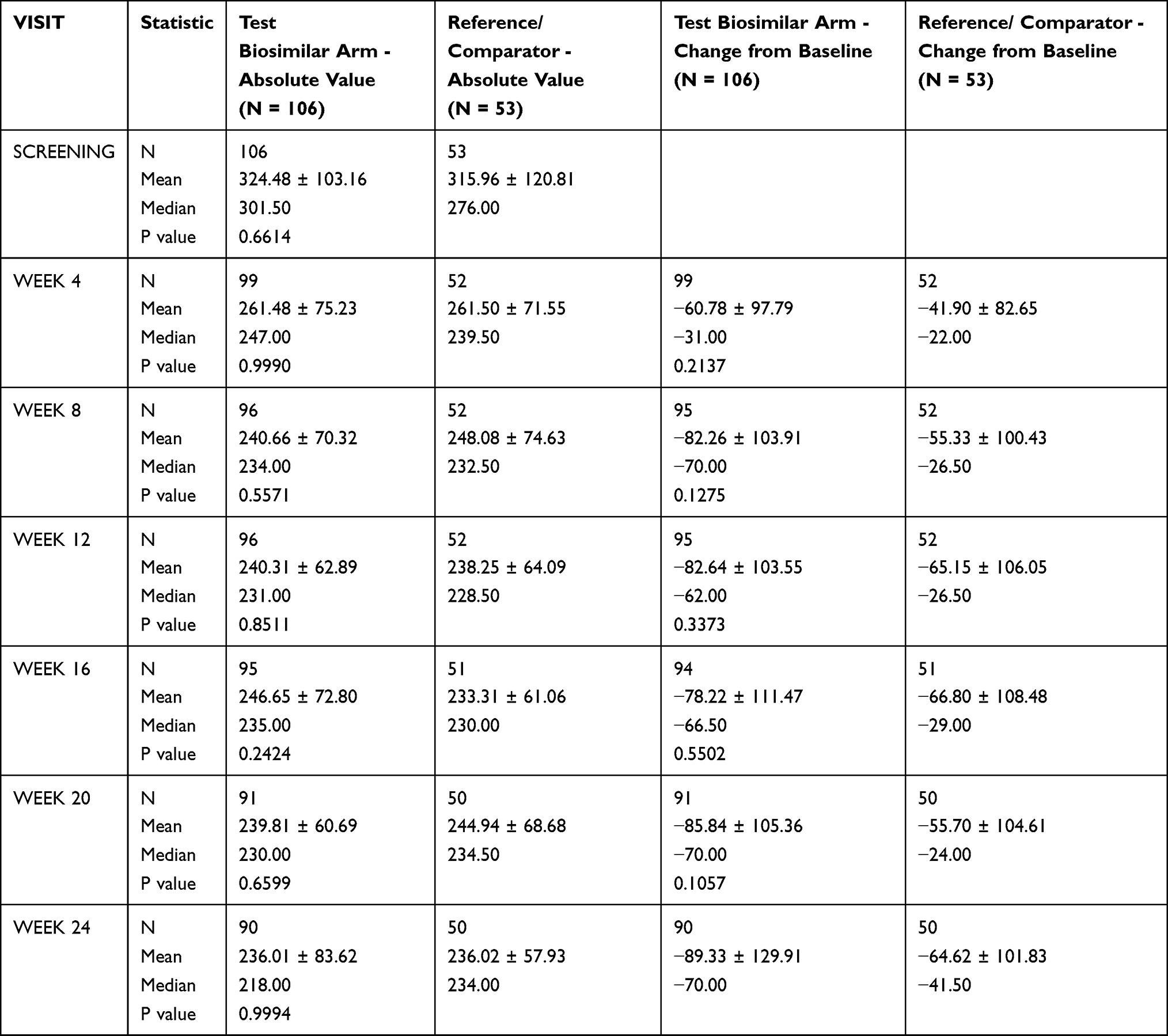 |
Table 3 Change in Central Macular Thickness Assessed by Optical Coherence Tomography (PP Population) |
In the pharmacokinetic analysis, the assessment of concentrations was tried with two different methods, however the methods were not sensitive enough to detect systemic concentrations.
Safety Assessment
In this study, a total of 38 adverse events were reported out of which 26 were reported in the test or biosimilar arm and 12 were reported in the reference arm. There were 16 (15.09%) subjects in the test arm and 5 (9.43%) subjects in the reference arm with at least one TEAE. There were 5 (4.72%) subjects in the test arm and 01 (1.89%) subjects in the reference arm with at least one TEAE related to study medication. In this study, 1 (0.94%) case of death was reported in the test arm which was unlikely related to the study drug and was attributed to the patient’s co-morbid condition of ischemic heart disease. No other serious adverse events were reported during this study.
According to SOC (System Organ Class) in the test biosimilar arm, the most commonly reported (incidence ≥ 5%) TEAEs were related to eye disorders (9; 8.49%). Other less common (≤ 5%) TEAEs were related to nervous system disorders, infections and infestation disorders, general disorders and administration site conditions, gastrointestinal disorders, immune system disorders and respiratory, thoracic and mediastinal disorders. In the reference or comparator arm, the most commonly reported (incidence ≥ 5%) TEAEs were related to eye disorders (4; 7.55%). In the test arm, adverse events related to eye disorders included conjunctival hyperemia, eye discharge, conjunctival hemorrhage, macular fibrosis, eye pain, vitreous floaters and vitreous haze. In the reference arm, adverse events related to eye disorders included conjunctival hyperemia, cystoid macular edema, macular edema and ocular hyperemia. In the test arm, the adverse events related to laboratory parameters (SOC-Investigations) including increased creatinine and increased blood urea nitrogen (BUN). In the reference arm, there were no adverse events related to laboratory parameters.
All the samples analyzed in this study were negative for anti-ranibizumab antibodies. There were no apparent immunologically mediated safety or efficacy concerns reported in this study.
Discussion
For biosimilar comparability, the comparative clinical experience with the reference product, including efficacy, safety and risk-benefit profile are to be considered. The potential differences between the biosimilar product and the reference product in the incidence and severity of human immune responses is the main objective for clinical immunogenicity assessment.3 Biosimilar ranibizumab in the present study4 has shown a comparable efficacy profile to that of reference ranibizumab as evident from the efficacy variables analysed between the treatment arms. In terms of the primary endpoint of this study the difference in proportion of patients with score less than 15 letters in visual acuity from baseline to week 16 (p = 0.314) and week 24 (p = 0.478) was statistically not significant between the two treatment arms. The results of other efficacy parameters were also similar which included proportion of patients who lost fewer than 15 letters and who gained at least 15 letters in visual acuity from baseline to week 24, mean change in best corrected visual acuity from baseline to week 24, proportion of patients with a visual acuity Snellen equivalent of 20/40 or better from baseline to week 24, proportion of patients with a visual acuity Snellen equivalent of 20/200 or worse from baseline to week 24 and change in central macular thickness assessed by Optical Coherence Tomography from baseline to week 24. No subjects in the study required any rescue treatment. The profile of adverse events was similar between two treatment arms supporting the similarity in safety profile. Overall, both drugs were well tolerated. The safety profile observed during this study is in line with known safety profile of ranibizumab. Immunogenicity samples from all subjects were negative for anti-drug antibodies (ADA) against ranibizumab. There were no apparent immunologically mediated safety or efficacy concerns reported in this study. The validated efficacy and safety endpoints evaluated during this study prove the clinical comparability of the ranibizumab biosimilar with the reference innovator product.
Conclusions
The biosimilar of ranibizumab studied, has comparable efficacy and safety profile to that of reference ranibizumab as evident from the efficacy variables analyzed between the treatment arms. Therefore, based on this comparability, it is proposed that the indigenous ranibizumab biosimilar may be considered as a viable alternative to the reference innovator product in patients with neovascular (wet) age-related macular degeneration.
Abbreviations
VEGF, Vascular endothelial growth factor; ICH-GCP, International Conference on Harmonization-Good clinical practices; DCGI, Drug control General of India; NDCT, New Drugs and Clinical Trial Rules; EC, Ethics Committee; ICF, Informed Consent Form; CNV, Choroidal neovascularization; AMD, Age-related macular degeneration; ETDRS, Early Treatment Diabetic Retinopathy Study; AE, adverse events; SAE, serious adverse events; SAP, statistical analysis plan; ITT, intent to treat; mITT, modified intent to treat; PP, Per protocol; TEAE, treatment emergent adverse event; SOC, System organ class; ADA, anti-drug antibodies.
Data Sharing Statement
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Ethics Approval and Informed Consent
The study was conducted in compliance with ethical principles and in accordance with the Declaration of Helsinki, ICH-GCP, protocol, Drug Controller General of India (DCGI) and New Drugs and Clinical Trial Rules (NDCT). Prior to initiation of study, approval was obtained from Ethics Committee (EC) at site institute and the EC also reviewed the Informed Consent Form (ICF) and endorsed it in writing.
List of Ethics Committee for Ranibizumab Clinical Trial
- Ethics Committee GSVM Medical College, Kanpur: Approved 22/06/2018
- Ethics Committee of King George Medical University, Lucknow: Approved 12/06/2018
- Ethics Committee, KLE University, Belgaum: Approved 20/04/2018
- Institutional Ethics Committee KIMS, Bangalore: Approved 06/10/2018
- Institutional Ethics Committee, Dr. B R Ambedkar Medical College, Bangalore: Approved 25/08/2018
- Institutional Ethics Committee, B J Medical College and Civil Hospital, Ahmedabad : Approved 17/12/2018
- Institutional Ethics Committee, D Y Patil University School of Medicine, Nerul: Approved 22/03/2019
- Institutional Ethics Committee, Datta Meghe Institute of Medical Sciences, Vardha: Approved 29/09/2018
- Institutional Ethics Committee, JPM Rotary Club of Cuttack Eye Hospital and Research Institute, Cuttack: Approved 28/02/2019
- Institutional Ethics Committee, King George Hospital, Vishakhapatnam: Approved 04/04/2019
- Institutional Ethics Committee Mysore Medical College and Research Institute and Associated Hospitals, Mysore: Approved 10/09/2018
- Institutional Ethics Committee, Regional Institute of Ophthalmology, Kolkata Approved 05/06/2018
- L. V. Prasad Eye Institute Ethics Committee, Hyderabad: Approved 19/07/2018
- Medilink Ethics Committee, Ahmedabad: Approved 17/12/2018
- Narayana Nethralaya Ethics Committee, Bangalore: Approved 30/01/2019
- Sanjivani Hospital Ethics Committee, Ahmedabad: Approved 27/02/201
Consent for Publication
The study was conducted as regulatory study by sponsor and has the consent for publication.
Acknowledgments
We acknowledge the investigators who contributed to the generation of the study data; Dr. Perwez Khan, Kanpur; Dr. Rekha Mudhol, Belagavi; Dr. Vivek Dave, Hyderabad; Dr. Shachi Desai, Ahmedabad; Dr. Satish K, Mysore; Dr. Lakshmi Kanta Mondal, Kolkata; Dr. Sachin Daigavane, Wardha; Dr. Rani Sujatha, Bengaluru; Dr. Somesh Aggarwal, Ahmedabad; Dr. Sandeep Saxena, Lucknow; Dr. Niveditha H, Bengaluru; Dr. Rohan Chauhan, Ahmedabad; Dr. Naresh Kumar Yadav, Bengaluru; Dr. Nita Shanbhag, Navi Mumbai; Dr. Santosh Kumar Mahapatra, Cuttack; Dr. VVL Rao, Vishakhapatnam.
Author Contributions
PA: Concept, analysis and interpretation, drafted or written, agreed on the journal, reviewed all versions of the article before submission, significant changes introduced at the proofing stage and agree to take responsibility and be accountable for the contents of the article.
MN: Concept, analysis and interpretation, drafted or written, agreed on the journal, reviewed all versions of the article before submission, significant changes introduced at the proofing stage and agree to take responsibility and be accountable for the contents of the article.
SN: Concept, analysis and interpretation, drafted or written, agreed on the journal, reviewed all versions of the article before submission, significant changes introduced at the proofing stage and agree to take responsibility and be accountable for the contents of the article.
PG: Concept, analysis and interpretation, drafted or written, agreed on the journal, reviewed all versions of the article before submission, significant changes introduced at the proofing stage and agree to take responsibility and be accountable for the contents of the article.
RP:, Concept, analysis and interpretation, drafted or written, agreed on the journal, reviewed all versions of the article before submission, significant changes introduced at the proofing stage and agree to take responsibility and be accountable for the contents of the article.
All authors read and approved the final manuscript and agree to take responsibility and be accountable for the contents of the article.
Funding
The study was funded by the sponsor, Reliance Life Sciences Pvt. Ltd. The study was a regulatory study with design of the study and collection, analysis and interpretation of data with writing the manuscript was done by the respective groups of the sponsor.
Disclosure
The authors declare that they have no competing interests in this work.
References
1. Woo SJ, Veith M, Hamouz J, et al. Efficacy and safety of a proposed ranibizumab biosimilar product vs a reference ranibizumab product for patients with neovascular age-related macular degeneration. JAMA Ophthalmol. 2021;139(1):68–76. doi:10.1001/jamaophthalmol.2020.5053
2. Guidelines on similar biologics: regulatory requirements for marketing authorization in India; August 15, 2016.
3. Scientific Considerations in Demonstrating Biosimilarity to a Reference Product. Guidance for Industry. U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER); April, 2015.
4. RanizuRel (ranibizumab) Clinical Study Report (Jan 2020). Reliance Life Sciences. Version 2.0.; January, 2020.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.