Back to Journals » Drug Design, Development and Therapy » Volume 9
Quinoline-based clioquinol and nitroxoline exhibit anticancer activity inducing FoxM1 inhibition in cholangiocarcinoma cells
Authors Chan-on W, Huyen NTB, Songtawee N, Suwanjang W, Prachayasittikul S, Prachayasittikul V
Received 15 December 2014
Accepted for publication 28 January 2015
Published 8 April 2015 Volume 2015:9 Pages 2033—2047
DOI https://doi.org/10.2147/DDDT.S79313
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 6
Editor who approved publication: Professor Shu-Feng Zhou
Waraporn Chan-on,1 Nguyen Thi Bich Huyen,2 Napat Songtawee,3 Wilasinee Suwanjang,1 Supaluk Prachayasittikul,3 Virapong Prachayasittikul2
1Center for Research and Innovation, 2Department of Clinical Microbiology and Applied Technology, 3Center of Data Mining and Biomedical Informatics, Faculty of Medical Technology, Mahidol University, Bangkok, Thailand
Purpose: Fork head box M1 (FoxM1) is an oncogenic transcription factor frequently elevated in numerous cancers, including cholangiocarcinoma (CCA). A growing body of evidence documents its diverse functions contributing to tumorigenesis and cancer progression. As such, discovery of agents that can target FoxM1 would be valuable for the treatment of CCA. The quinoline-based compounds, namely clioquinol (CQ) and nitroxoline (NQ), represent a new class of anticancer drug. However, their efficacy and underlying mechanisms have not been elucidated in CCA. In this study, anticancer activities and inhibitory effects of CQ and NQ on FoxM1 signaling were explored using CCA cells.
Methods: The effects of CQ and NQ on cell viability and proliferation were evaluated using the colorimetric 3-(4,5-dimethylthiazol-2yl)-5-(3-carboxymethoxyphenyl)-(4-sulfophenyl)-2H-tetrazolium (MTS assay). Colony formation and cell migration affected by CQ and NQ were investigated using a clonogenic and a wound healing assay, respectively. To demonstrate the agents’ effects on FoxM1 signaling, expression levels of the target genes were quantitatively determined using real-time polymerase chain reaction.
Results: CQ and NQ significantly inhibited cell survival of HuCCT1 and Huh28 in a dose- and a time-dependent fashion. Further investigations using the rapidly proliferating HuCCT1 cells revealed significant suppression of cell proliferation and colony formation induced by low doses of the compounds. Treatment of CQ and NQ repressed expression of cyclin D1 but enhanced expression of p21. Most importantly, upon CQ and NQ treatment, expression of oncogenic FoxM1 was markedly decreased concomitant with downregulation of various FoxM1’s downstream targets including cdc25b, CENP-B, and survivin. In addition, the compounds distinctly impaired HuCCT1 migration as well as inhibited expression of matrix metalloproteinase (MMP)-2 and MMP-9.
Conclusion: Collectively, this study reports for the first time the anticancer effects of CQ and NQ against CCA cells, and highlights new insights into the mechanism of actions of the quinoline-based compounds to disrupt FoxM1 signaling.
Keywords: FoxM1, cholangiocarcinoma, 8-hydroxyquinoline derivatives, clioquinol, nitroxoline, migration
Introduction
Cholangiocarcinoma (CCA) is an epithelial malignancy of the bile duct, representing the second most common hepatic malignancy.1 Incidence and mortality rates of CCA increase progressively with extremely poor prognosis.2 The majority of CCA cases have a short median survival time of less than 24 months, which could partially result from resistance of CCA to currently available treatment strategies.3 Presently, surgical resection remains the only curative option for CCA; however, a substantial proportion of CCA patients are diagnosed with advanced tumor that is not suitable for operation. The first-line chemotherapeutic treatment for inoperable patients is a combination of gemcitabine and cisplatin.4,5 However, the efficacy of this standard regimen is still limited, pointing to the need to develop alternative therapeutic options for this devastating malignancy.
Fork head box M1 (FoxM1) is an oncogenic transcription factor belonging to the fork head/winged–helix family of transcription factors.6 Its expression is silenced in terminally differentiated cells, but highly elevated in proliferating and tumor cells.7 Upregulation of FoxM1 has been observed in most human malignancies including CCA, cancer of prostate, lung, liver, and breast.8–11 In CCA, elevated expression of FoxM1 was ranked among the top genes differentially upregulated in tumor tissues.8,11 Besides its critical role regulating transcription of genes involved in G1/S and G2/M transitions of cell cycle,12–14 emerging evidence suggests that FoxM1 enhances cancer progression by increasing cancer cell invasion, metastasis, angiogenesis as well as drug resistance.15–20 A growing body of evidence suggests that FoxM1 represents a potential target for cancer treatment in which its downregulation results in inhibition of tumor growth, invasion, and angiogenesis as well as increase in chemosensitivity.18,21–23 It has been reported that expression and transcriptional activity of FoxM1 could be inhibited by several proteasome inhibitors including bortezomib, MG132, Siomycin A, and thiostrepton.24 Additionally, FoxM1 inhibition by Siomycin A resulted in downregulation of several FoxM1’s target genes such as cdc25B, CENP-B, and survivin.25 This information suggests that any other proteasome inhibitor may have FoxM1 inhibitory activity, and thus represents an option for the potential treatment of CCA that highly upregulates FoxM1.
Clioquinol (5-chloro-7-iodo-8-hydroxyquinoline; CQ) is an antibiotic agent with the proteasome inhibiting property.26 CQ has been recognized as a novel anticancer drug that is able to disrupt proteasome activity.27–29 In recent years, several lines of evidence have revealed its cytotoxicity in various cancer models including leukemia, multiple myeloma, and cancer of prostate, bladder, and breast.27–30 CQ has been demonstrated to induce cancer cell death via several mechanisms including inhibition of lysosome, NF-kappa B, histone deacetylases, and mTOR signaling pathway.30–34 Investigation of CQ has also been extended to clinical trial of Alzheimer’s disease, suggesting its broad therapeutic efficacies and safe administration in humans.35 Nitroxoline (5-nitro-8-hydroxyquinoline; NQ), a CQ analog, is a Food and Drug Administration (FDA)-approved antibiotic used for the treatment of urinary tract infection.36 Like CQ, NQ was recently reported to exhibit anticancer activities in cancer models of breast and bladder.37 In addition to its inhibitory action on tumor growth, the same study demonstrated that NQ inhibited angiogenesis associated with modulation of type 2 methionine aminopeptidase.37
In the present study, antitumor effects of CQ and NQ were explored in the human CCA model using in vitro assays. Molecular studies were also conducted to reveal their inhibitory effects on FoxM1 signaling. Herein, reduction of CCA cell survival, proliferation, colony formation, and migration exerted by CQ and NQ were demonstrated. This study also provides novel evidence that expression of FoxM1 and its downstream targets can be suppressed upon exposure of CQ and NQ to CCA cells, suggesting the potential application of CQ and NQ to the treatment of CCA.
Materials and methods
Cell lines and reagents
The human CCA cell lines, HuCCT1 and Huh28, were purchased from RIKEN (Saitama, Japan) and cultured in RPMI-1640 (Gibco, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS) (Gibco), penicillin (100 U/mL), and streptomycin (100 mg/mL) (Invitrogen, Waltham, MA, USA) in a humidified incubator at 37°C and 5% CO2. CQ, NQ, and dimethyl sulfoxide (DMSO) were procured from Sigma-Aldrich (St Louis, MO, USA). Thiostrepton was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Stock solutions of CQ, NQ, and thiostrepton were dissolved in DMSO and stored at −20°C in aliquots.
Cell viability and cell proliferation assay
Cells were seeded in triplicate wells of 96-well plates at a density of 4×103 cells per well in 10% FBS-supplemented medium. The next day, the medium was removed and replaced with 100 μL of 10% FBS-supplemented medium containing various concentrations of CQ (20, 25, and 30 μM), NQ (10, 20, and 40 μM) or DMSO (vehicle control). Controls were treated with DMSO at a concentration equal to that of agent-treated cells. Cells were then incubated at 37°C for 24, 48, and 72 hours. Viability of the cells was assessed using the colorimetric 3-(4,5-dimethylthiazol-2yl)-5-(3-carboxymethoxyphenyl)-(4-sulfophenyl)-2H-tetrazolium (MTS assay) according to the manufacturer’s protocol (Promega, Fitchburg, WI, USA). In brief, 20 μL of MTS solution was added to each well, and cells were incubated at 37°C for 2 hours. The optical density was measured using Infinite 200 PRO (Tecan Trading AG, Zurich, Switzerland) at 490 nm. The assay was performed in triplicate and repeated three times independently. The percentage of cell viability = (absorbance of compound-treated cells/absorbance of DMSO-treated control) ×100%.
To evaluate cell proliferation, cells were plated at a density of 2×103 cells per well in 96-well plates in 100 μL of medium. After treatment with CQ (5 and 10 μM), NQ (2 and 5 μM), or DMSO for 24, 48, and 72 hours, the number of viable cells was determined using MTS assay as described above. The assay was performed three times independently. The proliferation rate = (absorbance of cells at 24, 48, or 72 hours/absorbance of cells at 0 hour) ×100%.
Colony formation assay
Cells were seeded at 1×103 cells/well in 6-well plates in duplicate. After 24 hours, culture medium was removed and replaced with fresh medium containing CQ (5 and 10 μM), NQ (2 and 5 μM), or DMSO. Media containing compounds were changed every 3 days to maintain the compounds’ activity. After 2 weeks of culture, the colonies of cells were fixed with methanol and stained with 0.5% crystal violet. The colonies containing at least 50 cells were considered positive and were counted in each well. At least three independent experiments were performed. The percentage of colony formation = (colony number of compound-treated cells/colony number of DMSO-treated control) ×100%.
Quantitative real-time polymerase chain reaction
Total RNA was isolated from cells using TRIzol (Invitrogen) according to the manufacturer’s protocol. First-strand cDNAs were synthesized using iScript cDNA synthesis kit (Bio-Rad, Hercules, CA, USA). Expression of target genes was quantitatively examined using specific primers in combination with SsoFast EvaGreen Supermix (Bio-Rad) in a CFX96 Real-Time PCR Detection System (Bio-Rad). Cyclin D1 and p21 were examined in cells treated with CQ (25 and 50 μM) and NQ (10 and 20 μM). Transcriptional expression of FoxM1, CENP-B, cdc25b, and survivin was determined in cells exposed to CQ (20, 25, and 30 μM), NQ (20, 30, and 40 μM), and thiostrepton (10 μM). MMP-2 and MMP-9 mRNA were detected in cells treated with CQ (25 μM), NQ (40 μM), and thiostrepton (10 μM). GAPDH was used as the reference gene. Primers used for real-time polymerase chain reaction detection are listed in Table 1. Relative expression of genes was normalized against respective controls. The results were calculated by using the 2−ΔΔCt method.
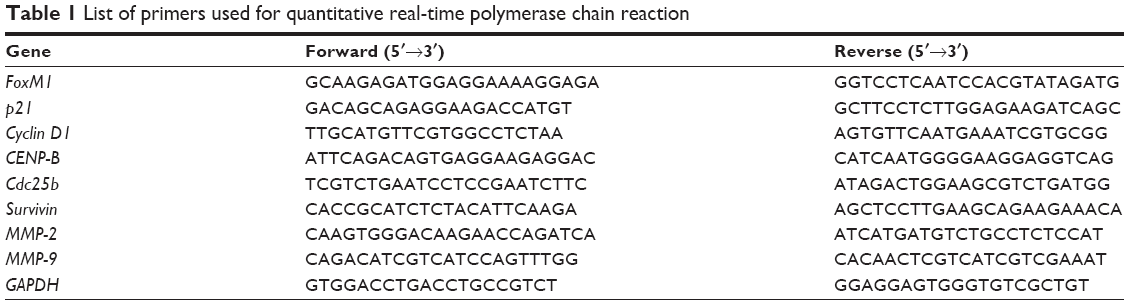 | Table 1 List of primers used for quantitative real-time polymerase chain reaction |
Western blot analysis
After treatment with CQ (20 and 30 μM), NQ (10 and 20 μM), thiostrepton (10 μM), or DMSO, cells were harvested and lysed in RIPA buffer (Cell Signaling Technology, Beverly, MA, USA) in the presence of protease inhibitor cocktail (Cell Signaling Technology). The protein concentration was determined using Bio-Rad protein assay (Bio-Rad). Equal amounts of proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes (Bio-Rad). The membranes were blocked with 5% nonfat milk and incubated with FoxM1 antibody (Cell Signaling Technology) at 4°C overnight. After washing, the membranes were incubated with horseradish peroxidase-conjugated antirabbit antibody (Cell Signaling Technology) for 2hours at room temperature. Signals were detected using the ECL Prime detection system (GE healthcare, Bangkok, Thailand), and β-actin detected with specific antibody (Sigma-Aldrich) was used as the loading control.
Wound healing assay
Cells were seeded to 6-well plates and treated with CQ (20 μM), NQ (10 μM), thiostrepton (2.5 μM), or DMSO. At 24 hours after treatment, cells were scratched using a sterile 200 μL pipette tip. The cells were washed with PBS to remove the detached cells, and incubated in fresh media without compound at 37°C in 5% CO2. Wound closure was monitored at various time points under a microscope. The open area at each time point was quantified by ImageJ, and the ratio of open area at 18hours to that at 0hour was analyzed. Data were collected from at least three independent experiments.
Molecular modeling
Three-dimensional structures of the zinc-coordinated catalytic domain of human MMP-2 and MMP-9 were constructed based on the protein data bank (PDB) codes 3AYU (resolution of 2.00 Å) and 4H3X (resolution of 1.76 Å), respectively. The catalytic domains without the three fibronectin-like modules include the residues Y110–G218 connected with Q393–P449 for MMP-2 and residues F107–G215 connected with Q391–G444 for of MMP-9. Missing side chains were modeled, and hydrogen atoms were added for both protein structures on WHAT–IF web server.38 The atomic coordinates of CQ and NQ were taken from the PDB codes 3KCX and 3AI8, respectively, and then were docked into the active site of MMPs using AutoDock4.2.6,39 in which the rotational bonds of the inhibitors were treated as flexible, whereas those of the protein models were regarded as rigid. Grid boxes were created to cover the substrate-binding site of the protein, where the grid spacing was 0.375 Å, and the coordinating zinc atom in the active site was set as the box center. The Lamarckian Genetic Algorithm (LGA) with 100 runs was used as the search parameter, in which the number of population size was set at 150 and the maximum number of energy evaluations was set to the high level. Three-dimensional models of MMP-inhibitor complexes with the lowest binding energy were visualized on PyMOL.40
Statistical analysis
Data obtained from three independent experiments were expressed as mean ± SD. Statistical analyses were carried out using SigmaPlot 11.0 (Systat Software, Inc., San Jose, CA, USA). Statistical differences were calculated using student’s t-test or ANOVA, and P-value of <0.05 was defined as statistically significant difference.
Results
CQ and NQ reduce viability of human CCA cells
To determine the effects of CQ and NQ (Figure 1) on the viability of the human CCA cells, HuCCT1 and Huh28 cells were exposed to increasing concentrations of CQ (0–30 μM) or NQ (0–40 μM), and cell viability was then measured by MTS assay at 24, 48, and 72 hours after incubation. Significantly, CQ treatment exhibited a dose- and time-dependent decrease in the viability of HuCCT1 and Huh28 cells (Figure 2A). Likewise, NQ significantly reduced the viability of the cell lines tested in a dose- and time-dependent manner (Figure 2B). IC50 values of CQ and NQ determined after treatment for 48 hours are shown in Table 2. Notably, the rapidly dividing HuCCT1 cells (Supplementary Figure S1) were more sensitive to both compounds than the quiescent Huh28 cells (IC50 of 2.84 vs 4.69 μM and IC50 of 3.69 vs 4.49 μM for CQ and NQ, respectively). These results indicate that CQ and NQ exert cytotoxic activity toward CCA cells.
 | Figure 1 Chemical structures of CQ and NQ. |
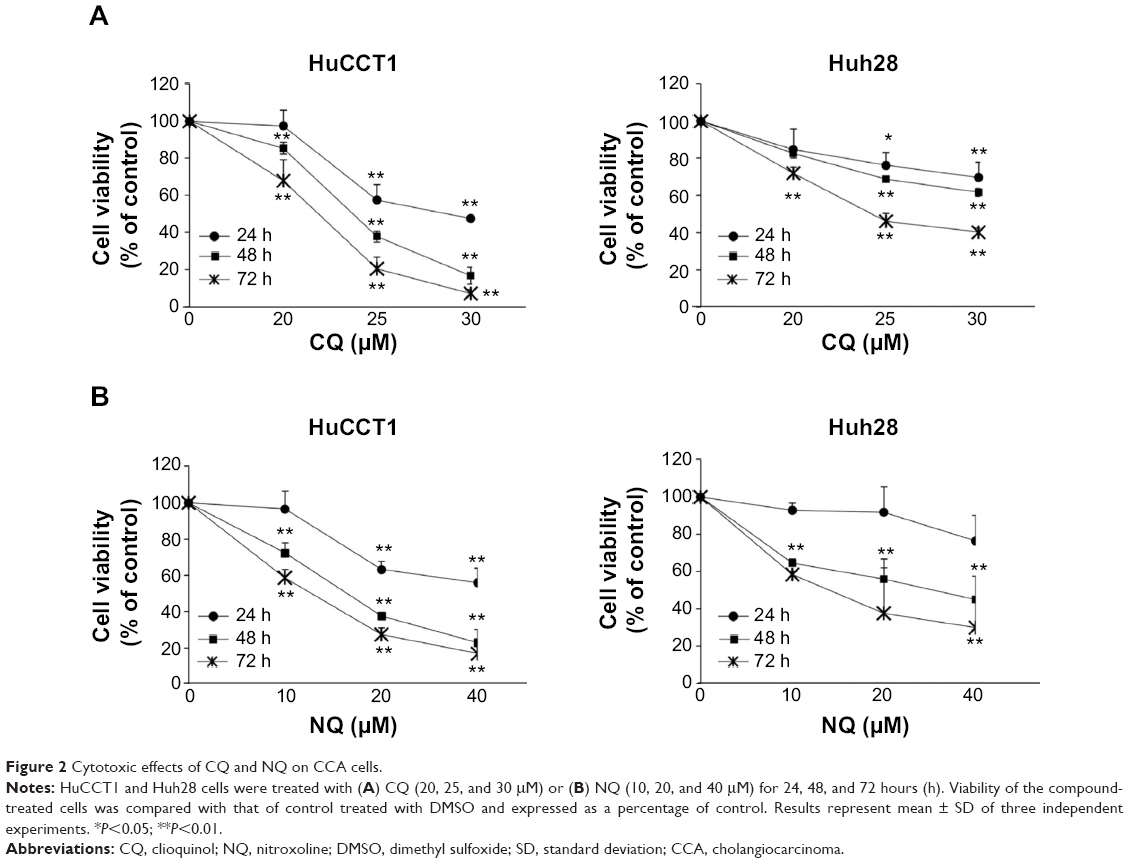 | Figure 2 Cytotoxic effects of CQ and NQ on CCA cells. |
 | Table 2 IC50 values of CQ and NQ for the viability of CCA cells |
CQ and NQ attenuate proliferation and colony formation of CCA cells
To further explore antitumor activity of CQ and NQ, the highly proliferative HuCCT1 cells were utilized for the evaluation of cell proliferation and colony formation after exposure to the agents. HuCCT1 cells were initially treated with low concentrations of CQ (5 and 10 μM) or NQ (2 and 5 μM), and cell proliferation rates were then monitored for 3 consecutive days by MTS assay. It was found that noncytotoxic doses of both CQ and NQ significantly suppressed the proliferation rates of HuCCT1 cells compared with controls treated with DMSO (Figure 3A). Subsequently, clonogenic assay revealed that the presence of CQ (5 and 10 μM) or NQ (2 and 5 μM) effectively reduced colony numbers of HuCCT1 cells in a dose-dependent fashion when compared with DMSO-treated controls (Figure 3B). These results consistently support the inhibition of CCA cell growth by the low doses of CQ and NQ.
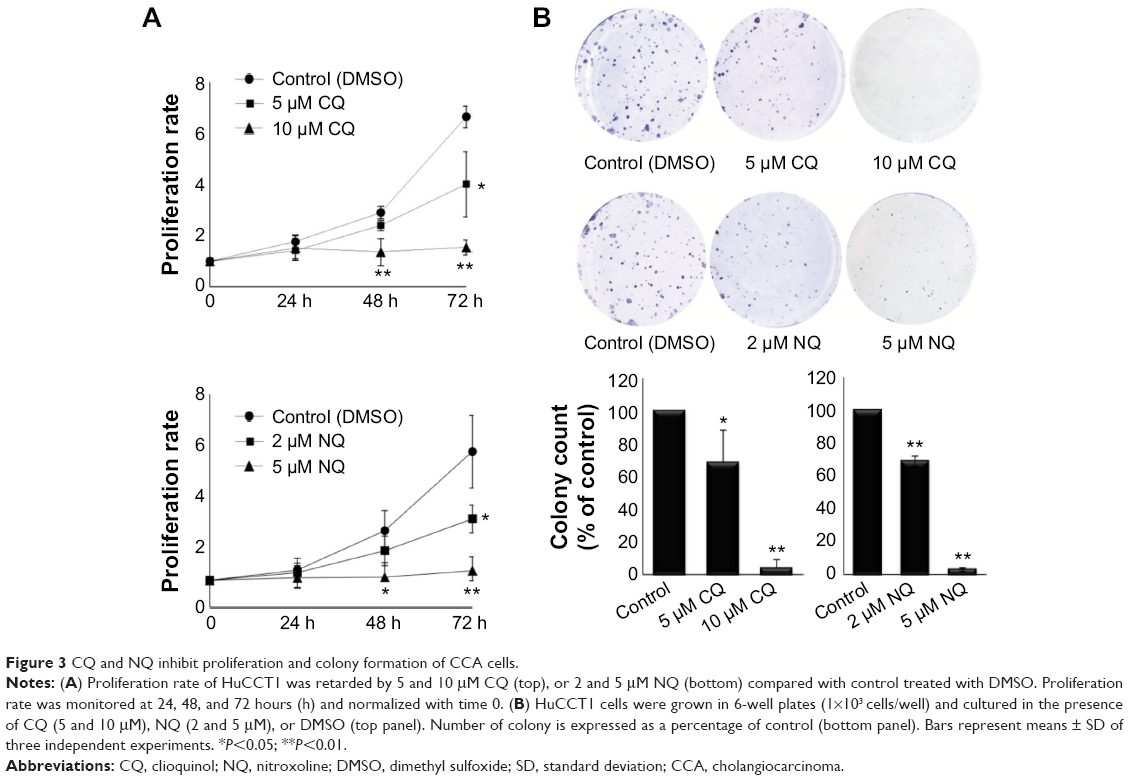 | Figure 3 CQ and NQ inhibit proliferation and colony formation of CCA cells. |
To explain inhibition of CCA cell proliferation after treatment with CQ and NQ, we sought to analyze expression of the cell cycle-associated molecules including cyclin D1 and p21. Quantitative analysis using real-time polymerase chain reaction demonstrated that cyclin D1 mRNA expression was significantly repressed by CQ and NQ in HuCCT1 and Huh28 cells (Figure 4A). In contrast, expression levels of p21 mRNA were enhanced by both compounds in the cell lines tested (Figure 4B). These results suggest that treatment of CQ and NQ can modulate expression of the cell cycle regulatory molecules.
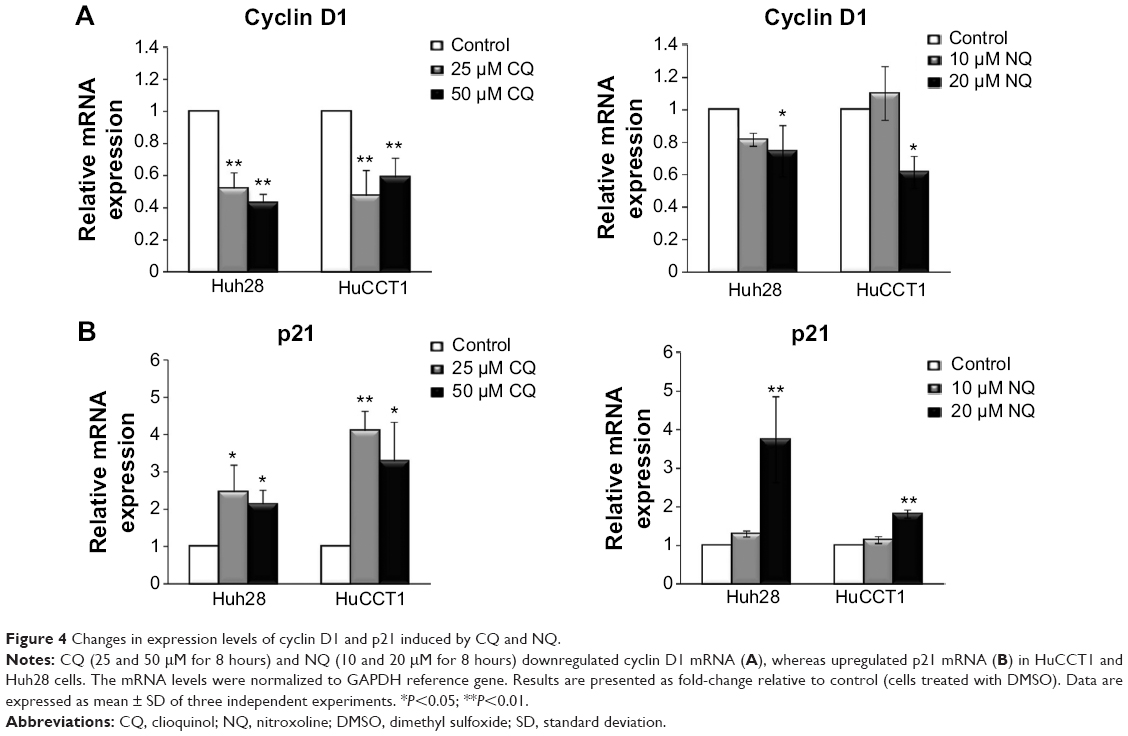 | Figure 4 Changes in expression levels of cyclin D1 and p21 induced by CQ and NQ. |
CQ and NQ downregulate FoxM1 expression
Attenuation of FoxM1 results in inhibition of tumor progression.18,21–23 It has been previously shown that its expression can be targeted by several classes of proteasome inhibitors.24 It was therefore hypothesized whether CQ, which exhibits antitumor actions and acts as proteasome inhibitor, can target FoxM1. Initially, FoxM1 expression in HuCCT1 and Huh28 was determined, and its high expression was clearly observed in HuCCT1 (Supplementary Figure S2A and B). For this reason, HuCCT1 was utilized for subsequent experiments. To explore the suppressive effects of CQ on FoxM1 expression, FoxM1 mRNA in HuCCT1 cells exposed to various concentrations of CQ (20, 25, and 30 μM) for 8 hours was quantitatively assessed using real-time polymerase chain reaction. Interestingly, FoxM1 mRNA was significantly downregulated by CQ in a dose-dependent manner when compared with the control treated with DMSO (Figure 5A). A time-dependent effect of CQ (25 μM) on FoxM1 expression was also observed at varying incubation times (4, 6, and 8 hours) (Figure 5B). These findings prompted us to suspect that NQ, which is CQ analog sharing the common core structure of 8-hydroxyquinoline, may also carry an inhibitory effect on FoxM1. Quantitative real-time polymerase chain reaction revealed that treatment of HuCCT1 with either increasing doses of NQ (20, 30, and 40 μM for 8 hours) or varying incubation times (30 μM for 4, 6, and 8 hours) resulted in a significant decrease in FoxM1 mRNA levels in a dose- and time-dependent fashion (Figure 5C and D). In addition, western blot analysis demonstrated reduction of FoxM1 protein induced by CQ and NQ, further confirming suppressive activity of the agents on FoxM1 expression (Figure 5E). These results provide evidence that CQ and NQ can reduce both transcriptional and translational expressions of FoxM1.
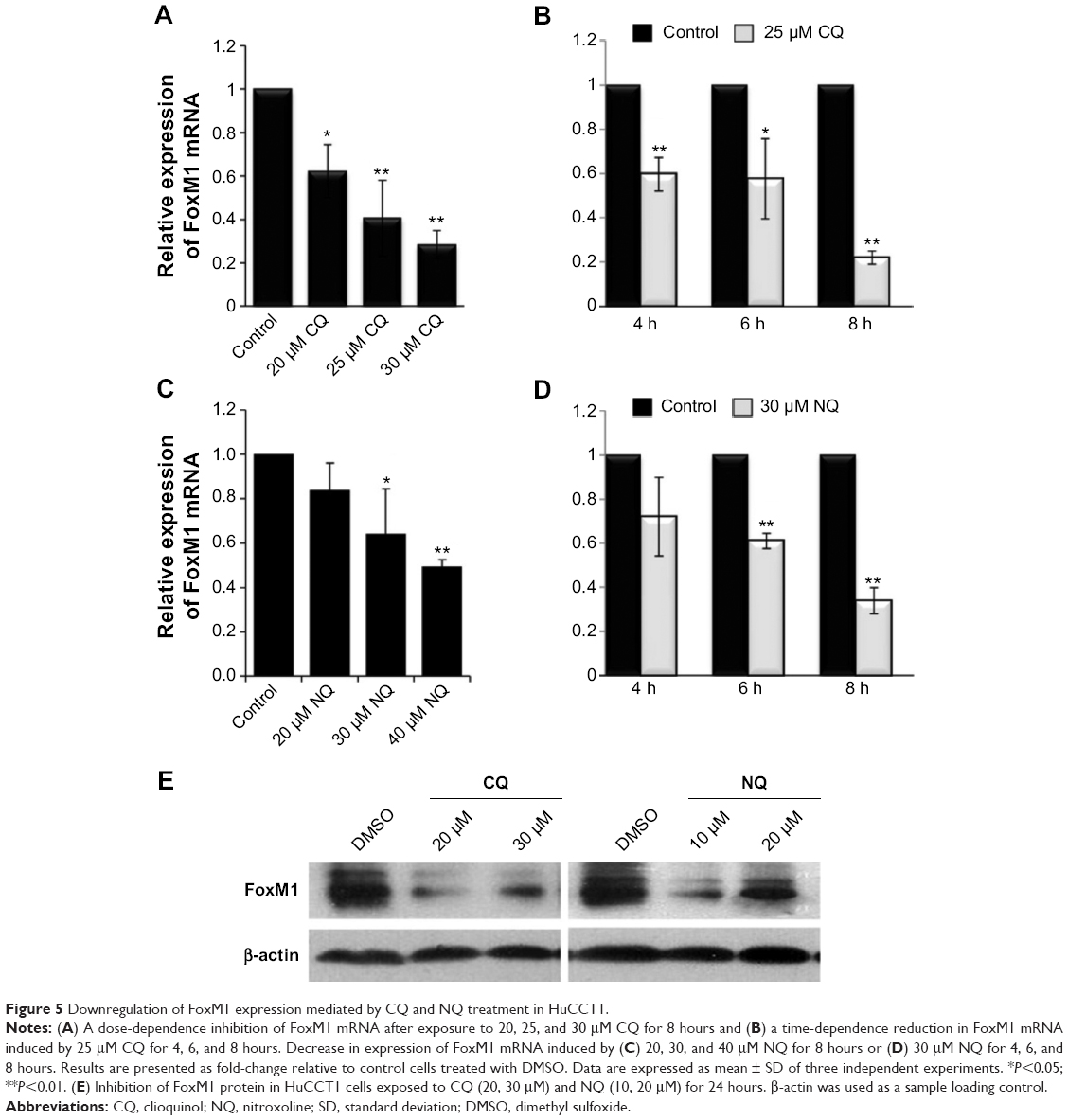 | Figure 5 Downregulation of FoxM1 expression mediated by CQ and NQ treatment in HuCCT1. |
CQ and NQ suppress expression of CENP-B, cdc25B, and survivin
To further verify the inhibition of FoxM1 signaling following the treatment of CQ and NQ, the downstream target genes regulated by FoxM1 including CENP-B, cdc25B, and survivin, were quantitatively examined. Remarkably, mRNA levels of CENP-B, cdc25B, and survivin were significantly decreased in HuCCT1 cells after exposure to either CQ (20, 25, and 30 μM) or NQ (20, 30, and 40 μM) (Figure 6A and B, respectively). These inhibitory effects of CQ and NQ were similar to that of known FoxM1 inhibitor thiostrepton,41 which dramatically reduced the expression levels of FoxM1 (Figure 7A and C) and its downstream targets (Figure 7B). Collectively, these results indicate that CQ, NQ, and thiostrepton can inhibit the expression of FoxM1’s targets concomitant with the downregulation of FoxM1 in CCA cells.
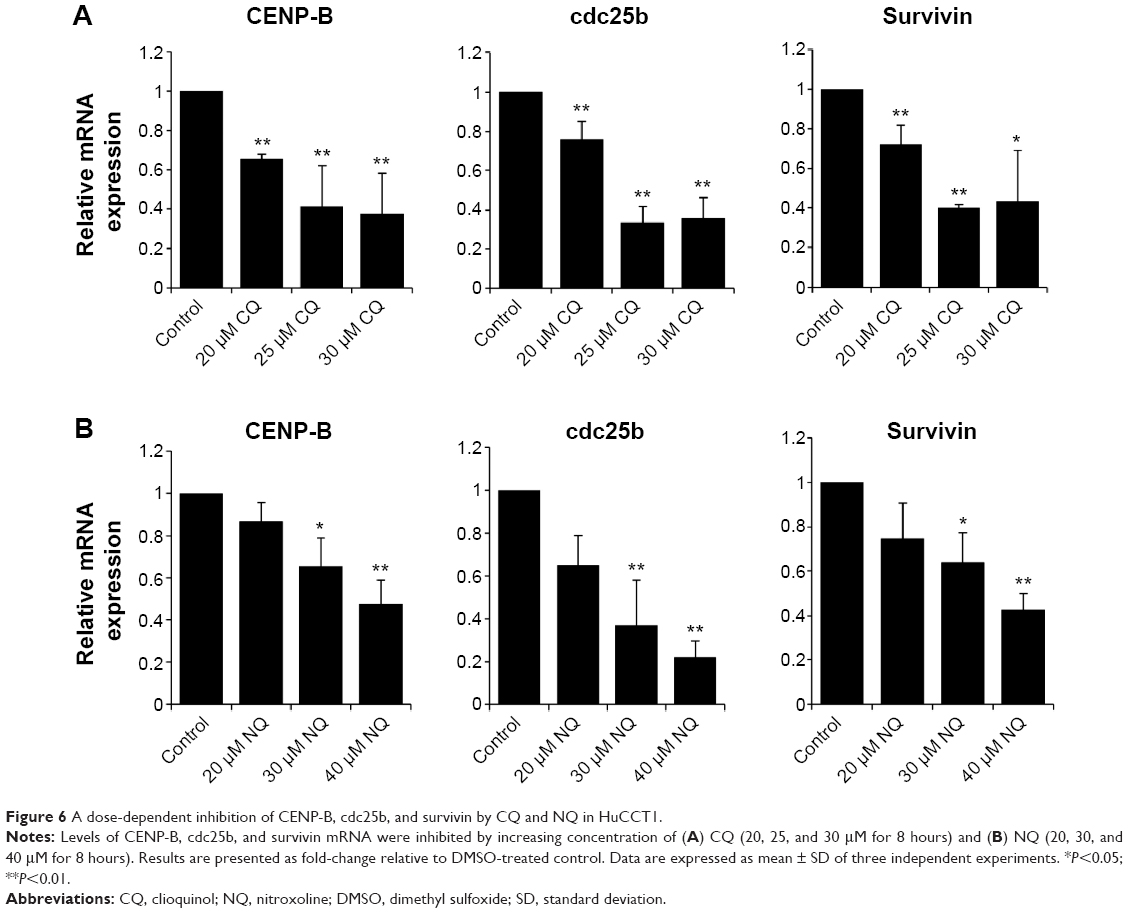 | Figure 6 A dose-dependent inhibition of CENP-B, cdc25b, and survivin by CQ and NQ in HuCCT1. |
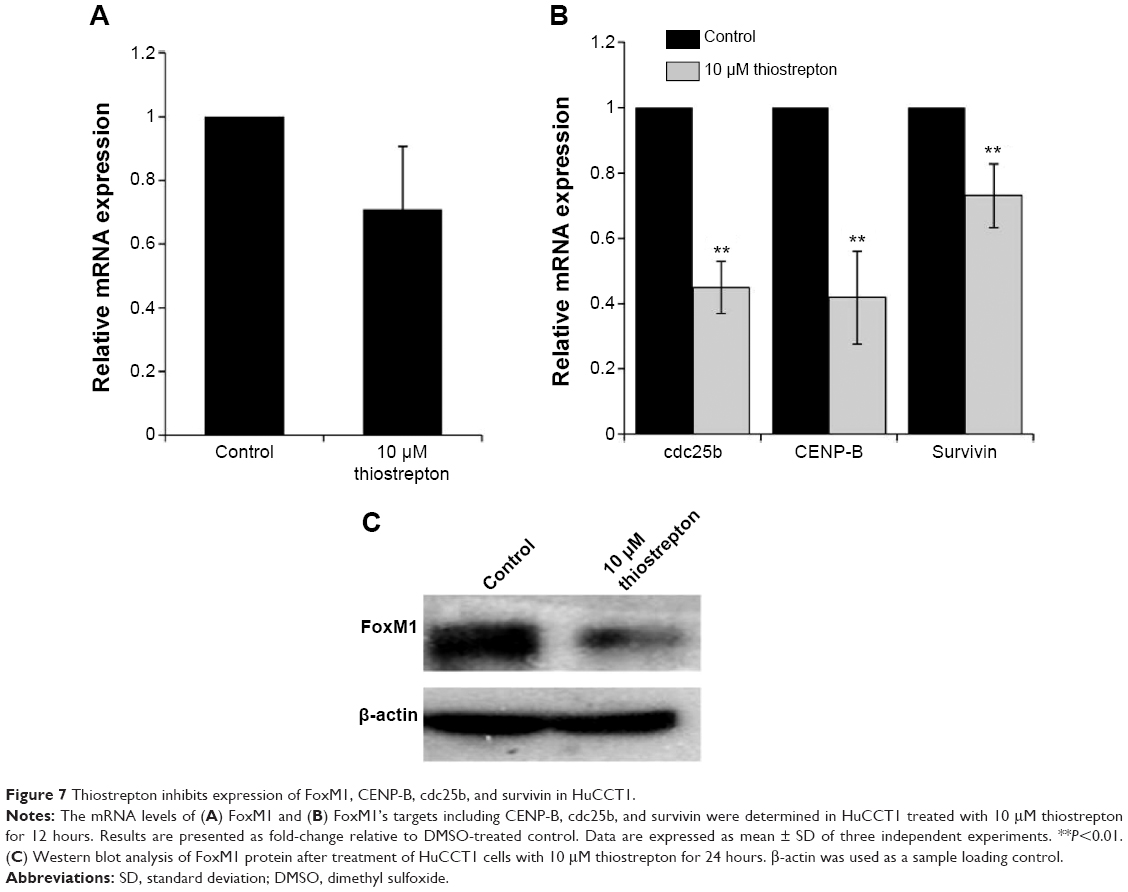 | Figure 7 Thiostrepton inhibits expression of FoxM1, CENP-B, cdc25b, and survivin in HuCCT1. |
CQ and NQ inhibit CCA cell migration
FoxM1 has been implicated in diverse cellular processes, including cell migration. FoxM1 inhibition mediated by CQ and NQ prompted us to further explore the effects of both compounds on migration of CCA cells. The wound healing assay was conducted in HuCCT1 cells that were scratched after treatment with CQ (20 μM), NQ (10 μM), or DMSO (control) for 24 hours. Significant delay of wound closure by CQ and NQ was observed after 18hours (Figure 8A and B). Measurement of the wounded area showed that open area in the controls was dramatically reduced from 100% to 28%, whereas CQ-treated cells only reduced to 78% (Figure 8D). Likewise, open area in NQ-treated cells was reduced from 100% to 44%, whereas that of the control was reduced from 100% to 29% (Figure 8E). Inhibition of wound healing was also observed in HuCCT1 exposed to 2.5 μM thiostrepton, in which its open area was significantly larger than that of control treated with DMSO (84% vs 27%) after 18 hours (Figure 8C and F). These data indicate that CQ, NQ, and thiostrepton attenuate the motility of CCA cells, presumably relating to suppression of FoxM1 expression by the compounds.
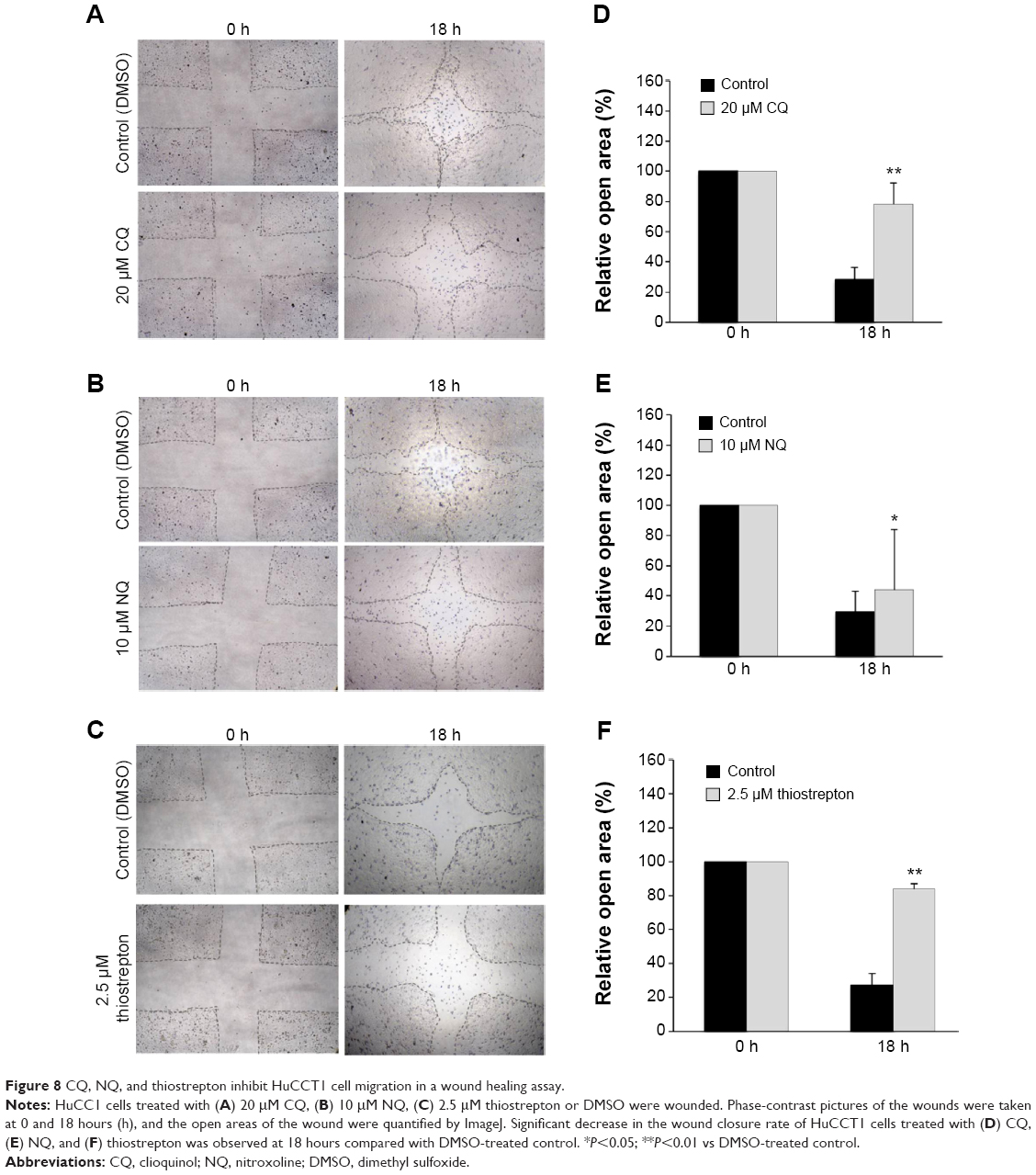 | Figure 8 CQ, NQ, and thiostrepton inhibit HuCCT1 cell migration in a wound healing assay. |
CQ and NQ downregulate MMP-2 and MMP-9 expression
To investigate underlying mechanisms that could be involved in the inhibition of CCA cell motility observed, we next examined expression of MMP-2 and MMP-9, members of the zinc-dependent endopeptidase family, which are critical for cell invasion and migration and regulated by FoxM1.42,43 Compared with the DMSO-treated control, CQ and NQ treatment resulted in a reduction in MMP-2 and MMP-9 expression in HuCCT1 (Figure 9A and B). Similarly, suppression of these two enzymes was also found in thiostrepton-treated HuCCT1 cells (Figure 9C). These suggest that downregulation of MMP-2 and MMP-9 by the compounds may be associated with the inhibition of CCA cell migration, and that could also be partially mediated through FoxM1 inhibition.
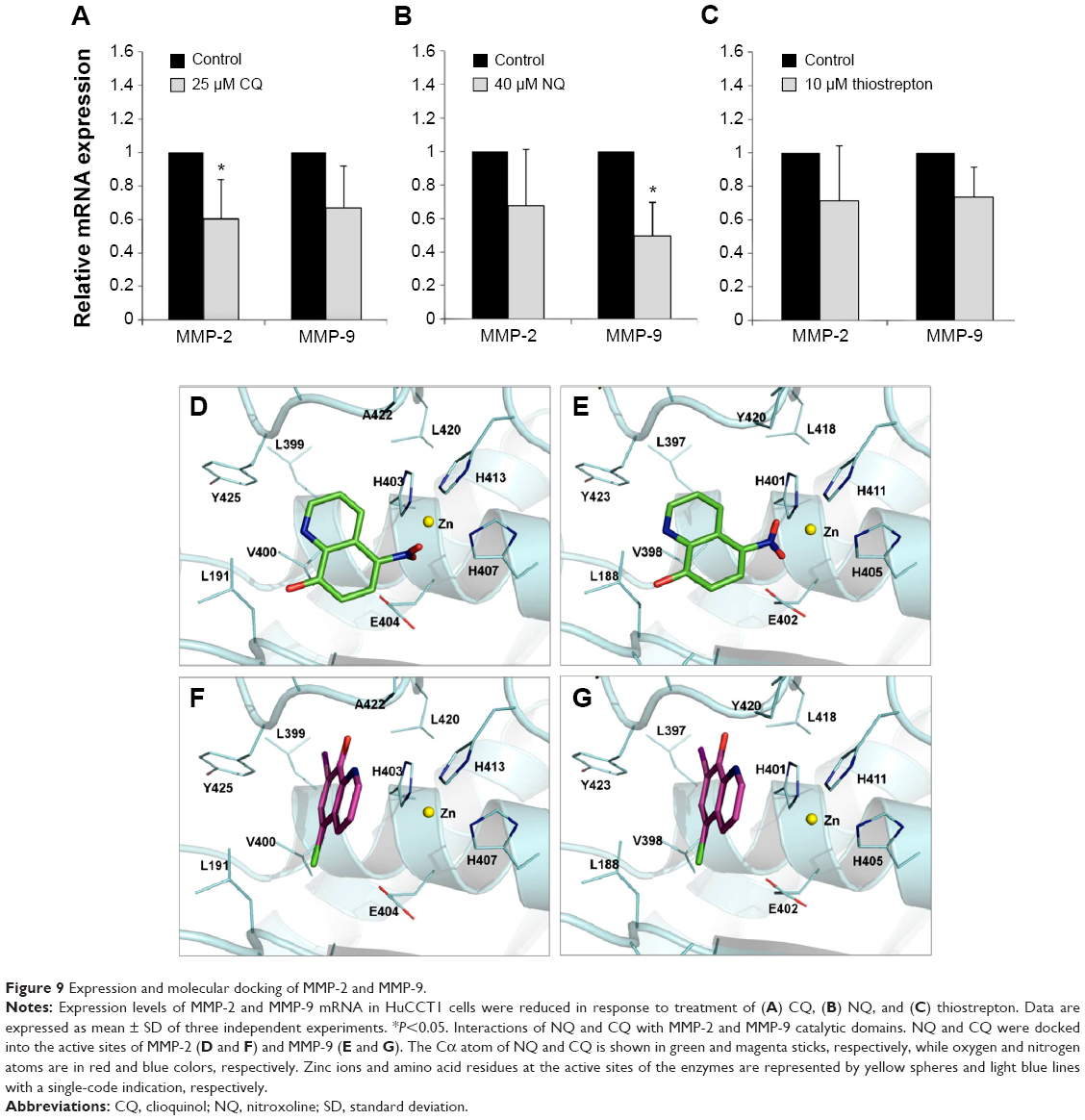 | Figure 9 Expression and molecular docking of MMP-2 and MMP-9. |
CQ and NQ directly bind to the Zn-coordinated active site of MMP-2 and MMP-9
There is evidence that CQ and NQ were found to interact with metal-binding enzymes such as histone deacetylases and methionine aminopeptidases.33,37 Since MMP-2 and MMP-9 contain the catalytic zinc that is coordinated by three histidines at the active site, it would be interesting to investigate whether CQ and NQ can directly interact with the active sites of MMP-2 and MMP-9. We employed a molecular docking method to predict the interaction of the two compounds with the enzymes. Binding mode analyses revealed that the nitro group of NQ obviously interacted with the catalytic zinc as well as three coordinated histidines H403/H407/H413 of MMP-2 and H401/H405/H411 of MMP-9 (Figure 9D and E). In addition, the π–anion interaction between the quinoline ring of NQ and the carboxylic side chain of catalytic aspartates (E404 and E402 of MMP-2 and MMP-9, respectively) was observed. The hydroxyl group of NQ also formed several hydrogen bonds with nearby active site residues. In contrast, CQ bound to the active site of the enzymes with different orientations in which the position of the quinoline ring of CQ was nearly perpendicular to that observed in the NQ docking, and thus the π–cation interaction of the CQ quinoline ring with zinc and π–π interactions of the ring with coordinated histidines seemed to dominate such interaction (Figure 9F and G). Also, many hydrogen bonds via the hydroxyl group and van der Waals contacts via chloride and iodide moieties of CQ were observed. These results suggest that CQ and NQ could interact with MMP-2 and MMP-9, and such interactions may contribute to the inhibition of both enzymes.
Discussion
In recent years, repositioning of the quinoline-based compounds CQ and NQ as an effective agent for treatment of cancer has gained much attention. Although a number of studies have highlighted their promising efficacy in various cancer models, their antitumor activity in CCA has not been explored so far. The present study demonstrates the first evidence suggesting that CQ and NQ could act as anticancer drug against CCA. Herein, CQ and NQ were found to effectively reduce viability, proliferation, and colony formation of CCA cells in a dose-dependent manner. Several factors are involved in controlling cell proliferation. Cyclin D1 has a central role in cell cycle progression, linking to continuing cell proliferation.44 The cyclin-dependent kinase inhibitor p21 (p21WAF1/Cip1) promotes cell cycle arrest, halting cell proliferation.45 Therefore, changes in expression levels of cyclin D1 and p21 in response to treatment of CQ and NQ could be associated with inhibition of CCA cell proliferation observed.
Like most carcinomas, gene expression analyses showed upregulation of FoxM1 in CCA tissues.8,11 FoxM1 has a well-defined function in cell proliferation.12–14 Our finding showed that FoxM1 expression was intensely detected in the rapidly dividing HuCCT1 cells and almost absent in the extremely slowly proliferating Huh28 cells, suggesting that FoxM1 might be one of the factors influencing proliferation of CCA cells. Since FoxM1 not only confers growth advantages on tumor cells but also promotes tumor progression,15–20 compounds that hold suppressive property on FoxM1 signaling would be of great interest as they may offer an alternative choice for treatment of such cancers. In accordance with the previous study that FoxM1 is a common target of proteasome inhibitors,24 our results indicated that CQ, which exhibits the proteasome inhibitory function, potently inhibited the expression of FoxM1 as well as its target genes including cdc25b, CENP-B, and survivin in CCA cells. Moreover, NQ, which is the structurally related analog of CQ, elicited its function on FoxM1 in the same manner. Cdc25b was found to be overexpressed in cancers and associated with a poor prognosis.46 Survivin is an antiapoptotic protein highly detected in numerous cancers, including CCA.47,48 Their suppression induced by CQ and NQ likely supports the inhibition of CCA cell survival and proliferation observed.
Cell migration is one of the initial steps in tumor metastasis and is a critical process that can be inhibited by FoxM1 depletion.21,22 Our results showed that CQ and NQ significantly delayed the migration rates of CCA cells and reduced the expression levels of MMP-2 and MMP-9. It has been known that MMP-2 and MMP-9 can promote the migration of cancer cells through surrounding extracellular matrix via their enzymatic degradation and that their expression can be regulated by FoxM1.42,43,49 The known FoxM1 inhibitor thiostrepton was also tested, and it was observed that thiostrepton not only inhibited FoxM1 expression, but also suppressed MMP-2 and MMP-9 expressions, supporting the link between FoxM1 and the two MMPs. Besides, thiostrepton could significantly attenuate CCA cell migration that might be attributed to deregulation of MMP-2 and MMP-9. Notably, the activity of CQ and NQ resembles that of thiostrepton; we therefore suggest the potentials of CQ and NQ as anticancer drug that can target FoxM1 signaling.
In addition to in vitro experimental investigations, our molecular modeling revealed that CQ and NQ could be well docked to the catalytic domain of MMP-2 and MMP-9, and this binding evidently interfered with the zinc-coordinated active site of the enzymes, suggesting the possibility of the compounds to direct inhibition of MMP catalytic function. Nevertheless, this computational prediction needs to be experimentally validated.
Conclusion
In summary, our findings indicated that the antibiotics CQ and NQ possess anticancer activities that can inhibit CCA cell viability, proliferation, colony formation, and migration. In addition, we reported, for the first time, inhibitory effects of CQ and NQ on FoxM1 signaling. The safe administration of CQ in the clinical trial of Alzheimer’s disease and the safe clinical use of NQ as an antimicrobial drug suggest their promising therapeutic applications for CCA treatment. Further in-depth studies in animal models of CCA are warranted to elucidate the therapeutic efficacies of CQ and NQ. Moreover, further investigations of CQ and NQ effects that directly inhibit FoxM1 transcriptional activity are required to demonstrate their potential function as FoxM1 inhibitor, which would be beneficial for the management of numerous carcinomas highly expressing FoxM1.
Acknowledgment
This research project was supported by Mahidol University, Thailand, and by the Office of the Higher Education Commission, Mahidol University, under the National Research Universities Initiative.
Disclosure
The authors report no conflicts of interest in this work.
References
Matull WR, Khan SA, Pereira SP. Re: impact of classification of hilar cholangiocarcinomas (Klatskin tumors) on incidence of intra- and extrahepatic cholangiocarcinoma in the United States. J Natl Cancer Inst. 2007;99(5):407; author reply 407–408. | ||
Razumilava N, Gores GJ. Cholangiocarcinoma. Lancet. 2014;383(9935): 2168–2179. | ||
Blechacz B, Gores GJ. Cholangiocarcinoma: advances in pathogenesis, diagnosis, and treatment. Hepatology. 2008;48(1):308–321. | ||
Thongprasert S, Napapan S, Charoentum C, Moonprakan S. Phase II study of gemcitabine and cisplatin as first-line chemotherapy in inoperable biliary tract carcinoma. Ann Oncol. 2005;16(2):279–281. | ||
Lee GW, Kang JH, Kim HG, Lee JS, Lee JS, Jang JS. Combination chemotherapy with gemcitabine and cisplatin as first-line treatment for immunohistochemically proven cholangiocarcinoma. Am J Clin Oncol. 2006;29(2):127–131. | ||
Kaestner KH, Knochel W, Martinez DE. Unified nomenclature for the winged helix/forkhead transcription factors. Genes Dev. 2000;14(2):142–146. | ||
Laoukili J, Stahl M, Medema RH. FoxM1: at the crossroads of ageing and cancer. Biochim Biophys Acta. 2007;1775(1):92–102. | ||
Jinawath N, Chamgramol Y, Furukawa Y, et al. Comparison of gene expression profiles between Opisthorchis viverrini and non-Opisthorchis viverrini associated human intrahepatic cholangiocarcinoma. Hepatology. 2006;44(4):1025–1038. | ||
Pilarsky C, Wenzig M, Specht T, Saeger HD, Grutzmann R. Identification and validation of commonly overexpressed genes in solid tumors by comparison of microarray data. Neoplasia. 2004;6(6):744–750. | ||
Halasi M, Gartel AL. FOX(M1) news – it is cancer. Mol Cancer Ther. 2013;12(3):245–254. | ||
Obama K, Ura K, Li M, et al. Genome-wide analysis of gene expression in human intrahepatic cholangiocarcinoma. Hepatology. 2005;41(6):1339–1348. | ||
Wang X, Quail E, Hung NJ, Tan Y, Ye H, Costa RH. Increased levels of forkhead box M1B transcription factor in transgenic mouse hepatocytes prevent age-related proliferation defects in regenerating liver. Proc Natl Acad Sci U S A. 2001;98(20):11468–11473. | ||
Laoukili J, Kooistra MR, Bras A, et al. FoxM1 is required for execution of the mitotic programme and chromosome stability. Nat Cell Biol. 2005;7(2):126–136. | ||
Wang IC, Chen YJ, Hughes D, et al. Forkhead box M1 regulates the transcriptional network of genes essential for mitotic progression and genes encoding the SCF (Skp2-Cks1) ubiquitin ligase. Mol Cell Biol. 2005;25(24):10875–10894. | ||
Dai B, Kang SH, Gong W, et al. Aberrant FoxM1B expression increases matrix metalloproteinase-2 transcription and enhances the invasion of glioma cells. Oncogene. 2007;26(42):6212–6219. | ||
Park HJ, Gusarova G, Wang Z, et al. Deregulation of FoxM1b leads to tumour metastasis. EMBO Mol Med. 2011;3(1):21–34. | ||
Zhang Y, Zhang N, Dai B, et al. FoxM1B transcriptionally regulates vascular endothelial growth factor expression and promotes the angiogenesis and growth of glioma cells. Cancer Res. 2008;68(21):8733–8742. | ||
Li Q, Zhang N, Jia Z, et al. Critical role and regulation of transcription factor FoxM1 in human gastric cancer angiogenesis and progression. Cancer Res. 2009;69(8):3501–3509. | ||
Carr JR, Park HJ, Wang Z, Kiefer MM, Raychaudhuri P. FoxM1 mediates resistance to herceptin and paclitaxel. Cancer Res. 2010;70(12):5054–5063. | ||
Kwok JM, Peck B, Monteiro LJ, et al. FOXM1 confers acquired cisplatin resistance in breast cancer cells. Mol Cancer Res. 2010;8(1):24–34. | ||
Wang Z, Banerjee S, Kong D, Li Y, Sarkar FH. Down-regulation of Forkhead Box M1 transcription factor leads to the inhibition of invasion and angiogenesis of pancreatic cancer cells. Cancer Res. 2007;67(17): 8293–8300. | ||
Miao L, Xiong X, Lin Y, et al. Down-regulation of FoxM1 leads to the inhibition of the epithelial-mesenchymal transition in gastric cancer cells. Cancer Genet. 2014;207(3):75–82. | ||
Ahmad A, Wang Z, Kong D, et al. FoxM1 down-regulation leads to inhibition of proliferation, migration and invasion of breast cancer cells through the modulation of extra-cellular matrix degrading factors. Breast Cancer Res Treat. 2010;122(2):337–346. | ||
Bhat UG, Halasi M, Gartel AL. FoxM1 is a general target for proteasome inhibitors. PLoS One. 2009;4(8):e6593. | ||
Radhakrishnan SK, Bhat UG, Hughes DE, Wang IC, Costa RH, Gartel AL. Identification of a chemical inhibitor of the oncogenic transcription factor forkhead box M1. Cancer Res. 2006;66(19):9731–9735. | ||
Prachayasittikul V, Prachayasittikul S, Ruchirawat S, Prachayasittikul V. 8-Hydroxyquinolines: a review of their metal chelating properties and medicinal applications. Drug Des Devel Ther. 2013;7:1157–1178. | ||
Daniel KG, Chen D, Orlu S, Cui QC, Miller FR, Dou QP. Clioquinol and pyrrolidine dithiocarbamate complex with copper to form proteasome inhibitors and apoptosis inducers in human breast cancer cells. Breast Cancer Res. 2005;7(6):R897–R908. | ||
Mao X, Li X, Sprangers R, et al. Clioquinol inhibits the proteasome and displays preclinical activity in leukemia and myeloma. Leukemia. 2009;23(3):585–590. | ||
Chen D, Cui QC, Yang H, et al. Clioquinol, a therapeutic agent for Alzheimer’s disease, has proteasome-inhibitory, androgen receptor-suppressing, apoptosis-inducing, and antitumor activities in human prostate cancer cells and xenografts. Cancer Res. 2007;67(4):1636–1644. | ||
Ding WQ, Liu B, Vaught JL, Yamauchi H, Lind SE. Anticancer activity of the antibiotic clioquinol. Cancer Res. 2005;65(8):3389–3395. | ||
Yu H, Zhou Y, Lind SE, Ding WQ. Clioquinol targets zinc to lysosomes in human cancer cells. Biochem J. 2009;417(1):133–139. | ||
Yu H, Lou JR, Ding WQ. Clioquinol independently targets NF-kappaB and lysosome pathways in human cancer cells. Anticancer Res. 2010; 30(6):2087–2092. | ||
Cao B, Li J, Zhu J, et al. The antiparasitic clioquinol induces apoptosis in leukemia and myeloma cells by inhibiting histone deacetylase activity. J Biol Chem. 2013;288(47):34181–34189. | ||
Cao B, Li J, Zhou X, et al. Clioquinol induces pro-death autophagy in leukemia and myeloma cells by disrupting the mTOR signaling pathway. Sci Rep. 2014;4:5749. | ||
Ritchie CW, Bush AI, Mackinnon A, et al. Metal-protein attenuation with iodochlorhydroxyquin (clioquinol) targeting Abeta amyloid deposition and toxicity in Alzheimer disease: a pilot phase 2 clinical trial. Arch Neurol. 2003;60(12):1685–1691. | ||
Sobke A, Klinger M, Hermann B, et al. The urinary antibiotic 5-nitro-8-hydroxyquinoline (Nitroxoline) reduces the formation and induces the dispersal of Pseudomonas aeruginosa biofilms by chelation of iron and zinc. Antimicrob Agents Chemother. 2012;56(11):6021–6025. | ||
Shim JS, Matsui Y, Bhat S, et al. Effect of nitroxoline on angiogenesis and growth of human bladder cancer. J Natl Cancer Inst. 2010;102(24): 1855–1873. | ||
Rodriguez R, Chinea G, Lopez N, Pons T, Vriend G. Homology modeling, model and software evaluation: three related resources. Bioinformatics. 1998;14(6):523–528. | ||
Morris GM, Huey R, Lindstrom W, et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem. 2009;30(16):2785–2791. | ||
DeLano WL. The PyMOL Molecular Graphics System, Version 1.3r1. 2010; http://pymol.org/educational/. | ||
Bhat UG, Halasi M, Gartel AL. Thiazole antibiotics target FoxM1 and induce apoptosis in human cancer cells. PLoS One. 2009;4(5):e5592. | ||
Xue YJ, Xiao RH, Long DZ, et al. Overexpression of FoxM1 is associated with tumor progression in patients with clear cell renal cell carcinoma. J Transl Med. 2012;10:200. | ||
Ahmed M, Uddin S, Hussain AR, et al. FoxM1 and its association with matrix metalloproteinases (MMP) signaling pathway in papillary thyroid carcinoma. J Clin Endocrinol Metab. 2012;97(1):E1–E13. | ||
Yang K, Hitomi M, Stacey DW. Variations in cyclin D1 levels through the cell cycle determine the proliferative fate of a cell. Cell Div. 2006;1:32. | ||
Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer. 2009;9(6):400–414. | ||
Lavecchia A, Di Giovanni C, Novellino E. Inhibitors of Cdc25 phosphatases as anticancer agents: a patent review. Expert Opin Ther Pat. 2010;20(3):405–425. | ||
Groner B, Weiss A. Targeting survivin in cancer: novel drug development approaches. BioDrugs. 2014;28(1):27–39. | ||
Javle MM, Tan D, Yu J, et al. Nuclear survivin expression predicts poor outcome in cholangiocarcinoma. Hepatogastroenterology. 2004;51(60):1653–1657. | ||
Sato H, Takino T, Okada Y, et al. A matrix metalloproteinase expressed on the surface of invasive tumour cells. Nature. 1994;370(6484):61–65. |
Supplementary materials
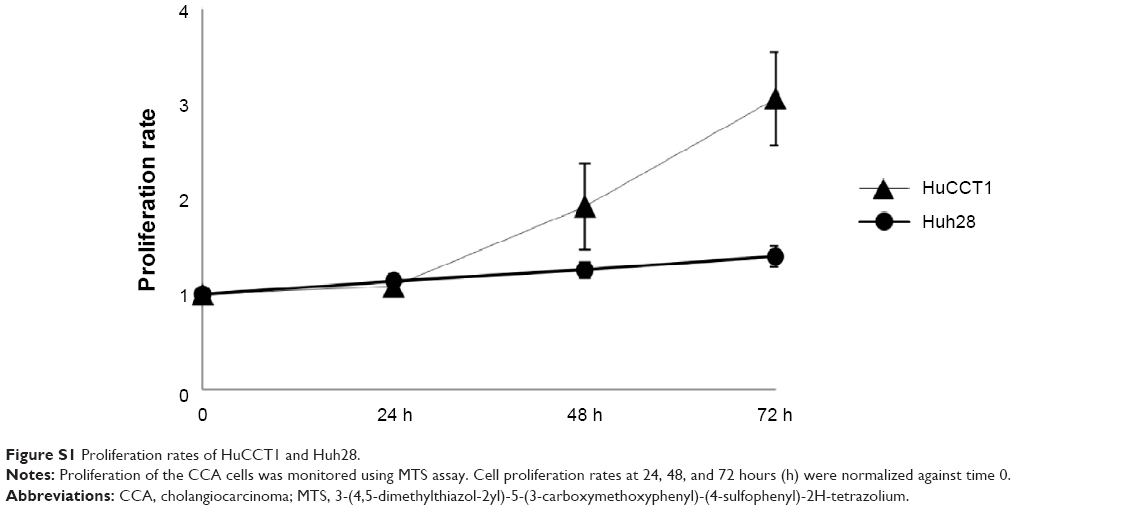 | Figure S1 Proliferation rates of HuCCT1 and Huh28. |
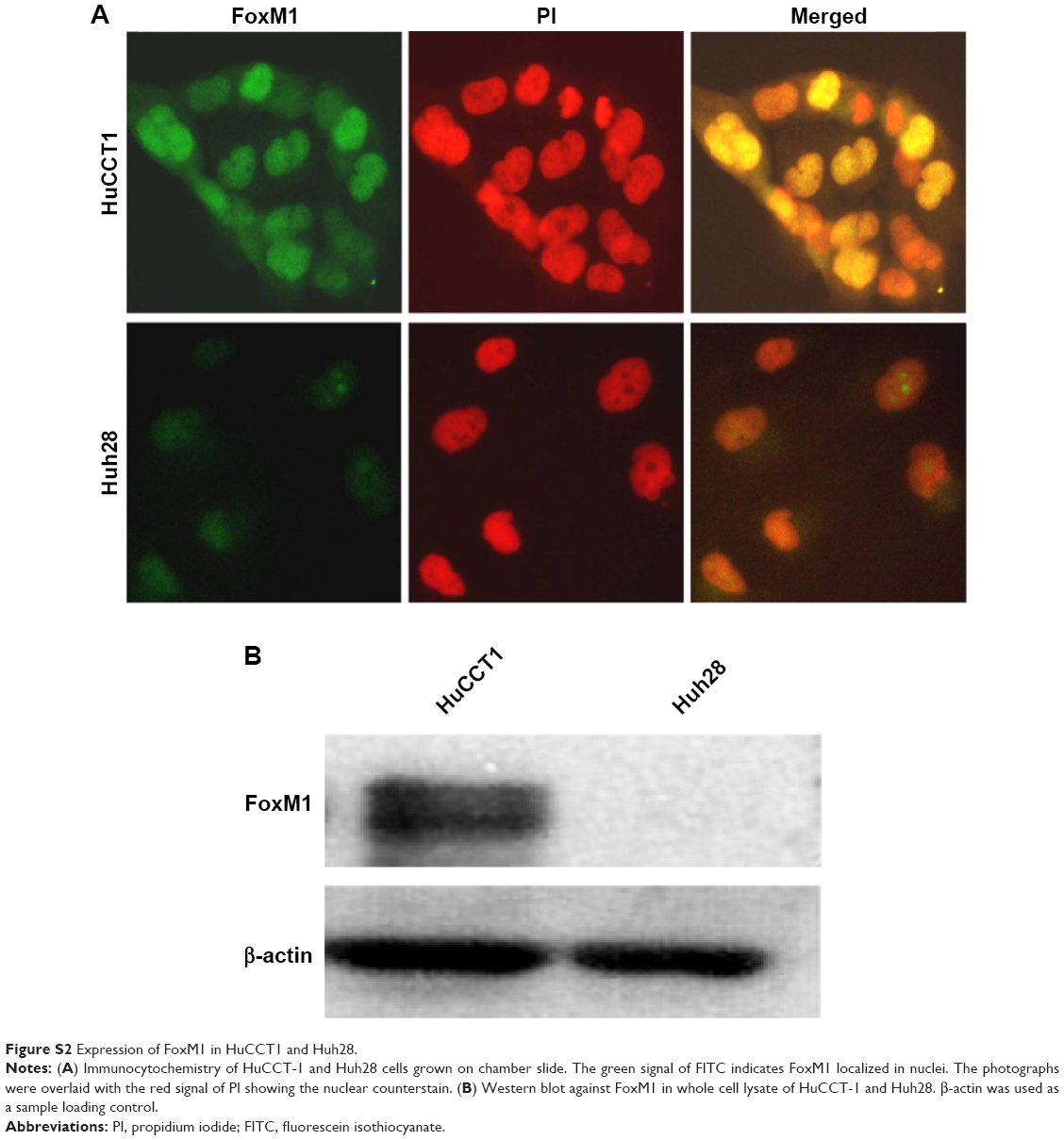 | Figure S2 Expression of FoxM1 in HuCCT1 and Huh28. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.