Back to Journals » OncoTargets and Therapy » Volume 10
Quercetin inhibits epithelial–mesenchymal transition, decreases invasiveness and metastasis, and reverses IL-6 induced epithelial–mesenchymal transition, expression of MMP by inhibiting STAT3 signaling in pancreatic cancer cells
Authors Yu D ![]() , Ye T, Xiang Y, Shi Z, Zhang J, Lou B, Zhang F, Chen B, Zhou M
, Ye T, Xiang Y, Shi Z, Zhang J, Lou B, Zhang F, Chen B, Zhou M
Received 12 March 2017
Accepted for publication 20 May 2017
Published 25 September 2017 Volume 2017:10 Pages 4719—4729
DOI https://doi.org/10.2147/OTT.S136840
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Samir Farghaly
Dinglai Yu,1 Tingting Ye,1 Yukai Xiang,1 Zhehao Shi,1 Jie Zhang,1 Bin Lou,1 Fan Zhang,1 Bicheng Chen,1,2 Mengtao Zhou1
1Department of Surgery, The First Affiliated Hospital, Wenzhou Medical University, Wenzhou, Zhejiang Province, People’s Republic of China; 2Zhejiang Provincial Top Key Discipline in Surgery, Wenzhou Key Laboratory of Surgery, Wenzhou, Zhejiang Province, People’s Republic of China
Abstract: Quercetin, a flavone, is multifaceted, having anti-oxidative, anti-inflammatory, and anticancer properties. In the present study, we explored the effects of quercetin on the epithelial–mesenchymal transition (EMT) and invasion of pancreatic cancer cells and the underlying mechanisms. We noted that quercetin exerted pronounced inhibitory effects in PANC-1 and PATU-8988 cells. Moreover, quercetin inhibited EMT and decreased the secretion of matrix metalloproteinase (MMP). Meanwhile, we determined the activity of STAT3 after quercetin treatment. STAT3 phosphorylation decreased following treatment with quercetin. We also used activating agent of STAT3, IL-6, to induce an increase in cell malignancy and to observe the effects of treatment with quercetin. As expected, the EMT and MMP secretion increased with activation of the STAT3 signaling pathway, and quercetin reversed IL-6-induced EMT, invasion, and migration. Therefore, our results demonstrate that quercetin triggers inhibition of EMT, invasion, and metastasis by blocking the STAT3 signaling pathway, and thus, quercetin merits further investigation.
Keywords: quercetin, EMT, MMPs, STAT3, pancreatic cancer
Introduction
Pancreatic cancer (PC), a highly lethal disease, is one of the most common digestive malignancies. Most PCs are derived from pancreatic intraepithelial neoplasms, which are microscopic, non-invasive epithelial proliferations within the pancreatic ducts.1 PC is the fourth leading cause of cancer-associated death worldwide.2 In addition, the 5-year survival rates of PC patients are 21.3% for local stage cancer, 8.9% for regional stage cancer, and 1.8% for distant stage cancer.3 Therefore, the molecular mechanism of PC must be clarified and new potential chemotherapeutic anticancer drugs should be identified.
Quercetin (3,3′,4′,5,7-pentahydroxyavone), a dietary bioflavonoid widely distributed in fruits, vegetables, and beverages, possesses low intrinsic toxicity and does not have carcinogenic activity in vivo.4,5 Several studies have demonstrated the anti-tumor effects of quercetin in various cancer cells, including PC. For example, cell proliferation was regulated through quercetin-induced miR-200b-3p in PC,6 and quercetin induced cell apoptosis in human gastric cancer stem cells and breast cancer cells.7,8 Moreover, quercetin inhibited cell viability, migration, and metastasis by modulating Wnt signaling components in a prostate cancer cell line.9 Although quercetin synergizes with epigallocatechin gallate to inhibit self-renewal capacity, invasion, and migration in pancreatic cancer stem cells (CSCs),10 the specific effects and cell signaling pathways involved in the effect of quercetin treatment on pancreatic cells are not fully understood. Thus, we aimed to ascertain the roles of quercetin in controlling the epithelial–mesenchymal transition (EMT), invasion and metastasis of PC, and to explain the potential molecular mechanisms in the context of PC.
EMT is a physiological phenomenon in mammalian embryonic development and a basic biological event that maintains cell and tissue balance. However, EMT is also an important phenomenon in tumorigenesis and cancer development.11 During EMT, epithelial cells lose cell polarity, incapacitate cell-cell tight junctions and adhesive connections, and access infiltration and migration ability.12 Epithelial cells possess the cell morphology and characteristics of interstitial cells. We can detect changes in EMT by observing EMT marker proteins, such as N-cadherin, E-cadherin, Vimentin and Snail.13,14 Recent studies have shown that MMPs and EMT are closely related.15,16 MMPs can be used as markers of EMT and can act as a predisposing factor for EMT.17 MMPs are a family of zinc-binding metalloproteases with the ability to degrade the extracellular matrix (ECM), and play a critical role in tumor cell infiltration and metastasis. In addition, MMPs degrade and destroy the ECM and basement membrane (BM) near the tumor surface, allowing tumor cells to infiltrate the surrounding tissue along the damaged region. MMPs can also promote tumor growth and spread through neovascularization and capillary endothelium.18
The STAT family is a family of proteins encoded by several chromosomes. STATs play an important role in the JAK-STAT signaling pathway mediated by ILs, especially IL-6,19 and are involved in the process of cell growth, proliferation, differentiation, and apoptosis.20 The STAT protein family consists of seven members: STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, and STAT6.21 STAT3 has been shown to be closely associated with the development of tumors.22 Studies have shown that ILs, growth factors, and certain oncoproteins can cause phosphorylation of STAT3 in the cytoplasm, resulting in Y705 and S727 site phosphorylation, homodimerization, and subsequent activation. Activated STAT3 translocates into the nucleus and binds to genomic DNA for transcriptional regulation.23 Activated STAT3 plays a vital role in promoting tumor cell proliferation and survival, tumor invasion, angiogenesis, and immunosuppression.20 IL-6 is one of the most important traditional activators of STAT3.24 IL-6 can induce EMT in various cancer cells and enhance invasion and metastasis through STAT3 signaling, including in PC cells.25,26
In this context, our aim is to demonstrate that quercetin can exert a beneficial effect on PC cells and reverse IL-6-induced EMT, invasion, and metastasis. Our results suggest that quercetin may act through the STAT3 signaling pathway.
Materials and methods
Reagents
Quercetin (Sigma-Aldrich Co., St Louis, MO, USA) was stored at −80°C until dilution in DMSO before use. Fetal bovine serum (FBS), trypsin, RPMI-1640, and DMEM were purchased from Gibco (Thermo Fisher Scientific, Waltham, MA, USA). Anti-N-cadherin, anti-Vimentin, anti-Twist, anti-Slug, anti-MMP-2, and anti-MMP-7 antibodies were purchased from Abcam (Cambridge, UK). Anti-E-cadherin, anti-Zeb1, anti-Snail, anti-STAT3, and anti-phospho-STAT3 antibodies were obtained from Cell Signaling Technology (Danvers, MA, USA). Polymerase chain reaction (PCR) primers were purchased from Synbio Tech (Jiangsu, People’s Republic of China). Power SYBR Green PCR Master Mix was purchased from Applied Biosystems (Foster City, CA, USA) and a Revert Aid First Strand cDNA Synthesis Kit was purchased from Thermo Fisher Scientific.
Cell culture and treatment
The human PC cell line PATU-8988 was obtained from the American Type Culture Collection (Manassas, VA, USA). The human PC cell line PANC-1 was purchased from the Institute of Biochemistry and Cell Biology, the Chinese Academy of Science (Shanghai, People’s Republic of China). PATU-8988 cells were cultured in RPMI-1640, PANC-1 cells were cultured in DMEM, and both cell lines were maintained in a 5% CO2 incubator at 37°C. Both media contained 100 U/mL penicillin, 100 μg/mL streptomycin, and 10% FBS, and culture medium was changed every 2 days. When cells were approximately 80%–90% confluent, PBS with 0.25% trypsin and 0.01% EDTA was used to detach the cells for subculture or experimental treatments. The cell morphology was observed with an inverted microscope.
Cell viability determined with Cell Counting Kit 8 (CCK8) assays
Approximately 10,000 PATU-8988 cells were plated in 96-well plates for a 24 h incubation period. Then, the medium was discarded, and FBS-free medium with different concentrations of quercetin was added into each well. After 24 h, the medium was replaced with 100 μL of FBS-free medium containing 10 μL of CCK8 (Dojindo, Kumamoto, Japan) reagent. Then, the cells were incubated for another 2 h, and the absorbance of each well was observed at 490 nm using an ELISA reader (BioTek, Winooski, VT, USA).
Transwell invasion and migration assays
PATU-8988 cells were cultured using a Transwell chamber (BD Biosciences, San Jose, CA, USA). An 8 μm-pore polyethylene terephthalate membrane was inserted into the chamber. BM matrix (Corning Incorporated, Corning, NY, USA) was placed in the upper chambers for the invasion assays but not the migration assays. RPMI-1640 containing 10% FBS was placed in the lower compartment. After treatment with quercetin, 5,000 PC cells (PATU-8988) in serum-free medium were placed in the upper compartment. The cells were incubated in a 5% CO2 incubator for 24 h. Then, the cells on the upper surface of the filter were scraped off with a cotton swab, and those in the lower compartment were stained with a 0.05% crystal violet solution. The cells in three randomly selected fields on each filter were imaged under an inverted microscope. We divided each image equally into nine pictures and selected three of the nine pictures. Finally, nine pictures were used for the count and the statistical analysis.
Wound healing assays
The cells were detached and cultured in 6 cm culture dishes. When the cells were approximately 80%–90% confluent, we used a sterile p200 pipette tip to scrape a wound of ~500 μm as a linear scratch. The cells were washed three times with PBS, and RPMI-1640 medium containing 1% FBS and different concentrations of quercetin were added. After 24 h, pictures of cell migration were acquired with an inverted microscope at 0, 24, and 48 h. The wound area was analyzed using Image-Pro Plus. The average width = wound area/wound height.
RNA extraction and quantitative real-time PCR analysis
When the cells had grown to 80%–90% confiuence, we added TRIzol Reagent (Ambion, Carlsbad, CA, USA) to extract RNA according to the manufacturer’s instructions. Reverse transcription to obtain cDNA was performed with a RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific). Quantitative analyses were performed using SYBR Green Master Mix on a 7500 Real-Time PCR System (Applied Biosystems). The quantitative real-time PCR data were analyzed using ΔCt values, and β-actin was amplified as an internal standard. Primer sequences are listed in Table 1.
 | Table 1 The primers used for real-time polymerase chain reaction |
Western blot analysis
The cells were lysed in RIPA buffer (Beyotime, Shanghai, People’s Republic of China) containing 10% protease inhibitor (Beyotime) and 1% phosphatase inhibitor (Beyotime) for 40 min. The final cell lysate was centrifuged at 12,000× g, and the supernatant was collected. The protein concentration was measured using a BCA assay (Beyotime). After denaturation, the protein mixture was subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis using a 10% polyacrylamide gel. The separated proteins were transferred to polyvinylidene difluoride membranes and incubated at 4°C overnight with primary antibodies. After being washed, the membranes were incubated with HRP-conjugated secondary antibodies (Bioworld Technology, Inc, St Louis Park, MN, USA) for 2 h at room temperature. The density of the protein bands was captured and measured using enhanced chemiluminescence reagent (Thermo Fisher Scientific) and AlphaEaseFC (StandAlone, AlphaInnotech, SanLeandro, CA, USA).
Immunofluorescence microscopy
The cells were seeded onto 6-well plates containing glass coverslips. After cell treatment, the cells were washed three times with PBS and then immersed in the fixation solution containing 4% paraformaldehyde for 20 min. Then, fixed cells were permeabilized in 0.2% Triton X-100, washed with PBS, and blocked with the addition of 5% goat serum albumin for 1 h at room temperature. The cells were incubated with anti-N-cadherin antibody and anti-Vimentin respectively overnight at 4°C. After three rinses with PBS, the cells were incubated with AlexaFluor 488-conjugated secondary antibody (Thermo Fisher Scientific) to detect N-cadherin. Then, the cells were washed three more times with PBS, and DAPI was added for 5 min to stain cell nuclei. Finally, images were captured with an automated upright microscope system (Leica DM4000B; Leica Microsystems, Wetzlar, Germany).
Statistical analysis
Statistical analyses were performed using the SPSS 19.0 statistical software package (IBM Corporation, Armonk, NY, USA). The results were derived from at least three independent experiments and are expressed as the mean ± standard error of the mean (SEM). Student’s t-test was used to assess significance. A value of P<0.05 was considered to indicate a statistically significant difference.
Results
Quercetin decreases cell viability and inhibits EMT in PC cell lines
At the beginning of our experiments, we observed whether quercetin had an effect on cell viability by using CCK8 assays. After the PATU-8988 and PANC-1 cells were treated with quercetin (0, 20, 40, 80, 160 μM) for 24 h, we performed a CCK8 assay to detect cell viability by measuring the absorbance at 490 nm of each well. Cell viability was decreased with increasing concentration (Figure 1A and B). The IC(50) of quercetin was 131.05 μM in the PATU-8988 and 244.909 μM in the PANC-1 cells.
 | Figure 1 Dose-dependent inhibition of cell viability in PANC-1 and PATU-8988 cells, and of epithelial–mesenchymal transition in PATU-8988 cells. |
In the next experiment, we treated PATU-8988 cells with quercetin at concentrations of 0, 20, 40, and 80 μM for 24 h. Then RT-PCR was performed to detect mRNA levels of important EMT biomarkers of N-cadherin, E-cadherin, and Vimentin. The results showed that quercetin increased mRNA expression levels of E-cadherin and decreased mRNA expression levels of N-cadherin and Vimentin with increasing quercetin concentration (Figure 1C). In addition, we then performed a Western blotting assay to detect the protein level of N-cadherin, E-cadherin, and Vimentin. The results were consistent with the trend observed in the RT-PCR results (Figure 1D).
To more thoroughly verify the effect of quercetin on EMT, we focused on the EMT nuclear transcription factors Zeb1, Twist, Slug, and Snail. We treated PATU-8988 cells with quercetin at concentrations of 0, 20, 40, and 80 μM for 24 h. Then, RT-PCR and Western blotting were performed to detect the mRNA and protein levels of Zeb1, Twist, Slug, and Snail. The results showed that quercetin decreased the mRNA and protein levels of Zeb1, Twist, Slug, and Snail in a dose-dependent manner (Figure 1E and F).
We also performed an immunofluorescence assay to determine the protein level of N-cadherin and Vimentin, and found that N-cadherin and Vimentin expression had decreased obviously (Figure 1G). Taken together, our results revealed that quercetin was able to reverse the EMT process in PATU-8988 cells.
Quercetin inhibits PC cell migration and invasion
There is a close relationship between EMT and MMPs in the process of tumorigenesis. Thus, we investigated the effects of quercetin on PC cell invasion and metastasis. The cells were treated as described previously. Then, we performed RT-PCR to detect the mRNA level of MMP2 and MMP7. The results showed that quercetin decreased the mRNA level of MMP2 and MMP7 in a dose-dependent manner (Figure 2A). We also performed a Western blotting assay to detect the protein levels of MMP2 and MMP7. Quercetin also suppressed the protein levels of MMP2 and MMP7 in a dose-dependent manner (Figure 2B).
 | Figure 2 Quercetin inhibits invasion and metastasis of PATU-8988 cells in a dose-dependent manner. |
MMPs are major contributors to cancer cell invasion and metastasis due to their ability to degrade the ECM and BM. We performed Transwell invasion and wound healing assays to investigate whether quercetin inhibited invasion and metastasis of PC cells. The results showed that invasion and metastasis were significantly inhibited in PC cells treated with quercetin (Figure 2C and D).
Quercetin inhibits activation of p-STAT3 and the IL-6-induced increase in p-STAT3
To investigate the effects of quercetin on the STAT3 pathway, we performed a Western blotting assay for STAT3 and p-STAT3 after treatment with different concentrations of quercetin. The results showed that the protein level of p-STAT3 was obviously decreased and the protein level of STAT3 was not affected (Figure 3A).
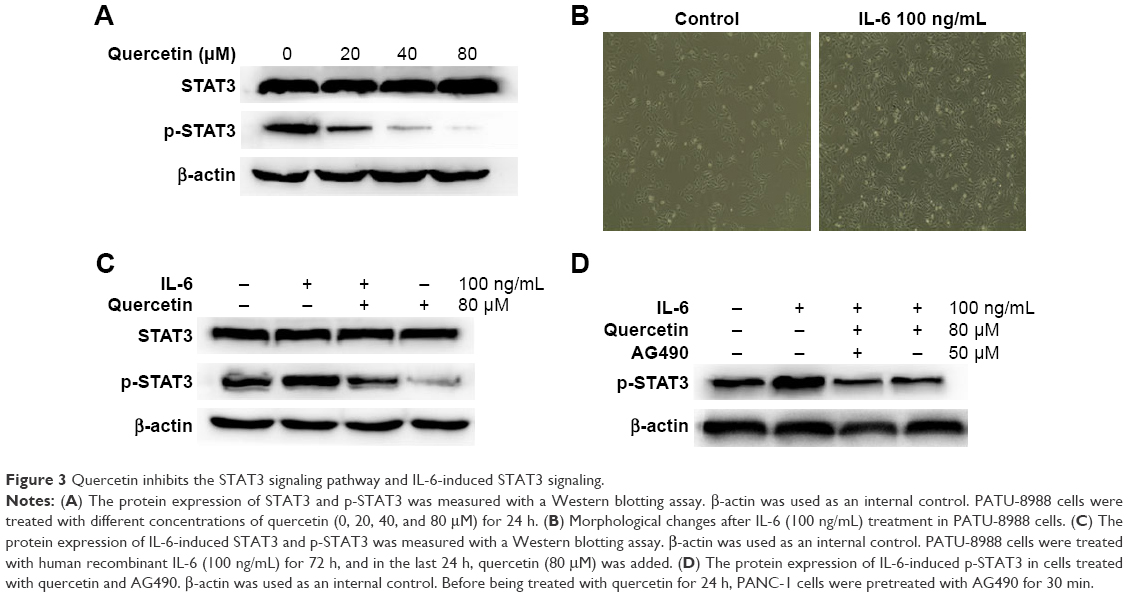 | Figure 3 Quercetin inhibits the STAT3 signaling pathway and IL-6-induced STAT3 signaling. |
IL-6 has been confirmed, in various cells, to activate STAT3 signaling and promote EMT, invasion, and metastasis. In the previously mentioned experiment, we found that quercetin inhibited STAT3 signaling in a dose-dependent manner. Therefore, we investigated whether quercetin could inhibit IL-6-induced activation of STAT3 in PATU-8988 cells. First, we observed changes in cell morphology. There was a transition to a spindle-shaped morphology in the PATU-8988 cells after treatment with IL-6 for 24 h (Figure 3B). We treated the cancer cells with human recombinant IL-6 (100 ng/mL) for 72 h, and in the last 24 h, quercetin (80 μM) was added. The results showed that the level of STAT3 was not changed after treatment with IL-6 or quercetin. However, the increase of p-STAT3 induced by IL-6 was inhibited after treatment with quercetin (Figure 3C). Before being treated with quercetin for 24 h, PATU-8988 cells were pretreated with AG490 (50 μM) for 30 min. There was no obvious difference between the two groups (Figure 3D). The results suggest that quercetin may weaken the IL-6-induced activation of STAT3 and inhibit EMT, invasion, and metastasis by inhibiting STAT3 signaling.
Quercetin reverses the process of IL-6-induced EMT, invasion, and metastasis
IL-6 can induce EMT and promote cell invasion in various cancer cells. We treated PATU-8988 cells with human recombinant IL-6 (100 ng/mL) for 72 h and performed Western blotting and RT-PCR to determine the change in the EMT biomarkers N-cadherin, E-cadherin, and Vimentin; the EMT nuclear transcription factors Zeb1, Twist, Slug and Snail, and MMP2 and MMP7. While E-cadherin was reduced, the remaining indicators were increased (Figure 4A–F), which indicated that IL-6 significantly induced EMT, invasion, and metastasis in PC cells. For the last 24 h, quercetin was added and the effect of IL-6-induced EMT, invasion, and metastasis was reversed after treatment with quercetin (Figure 4A–F).
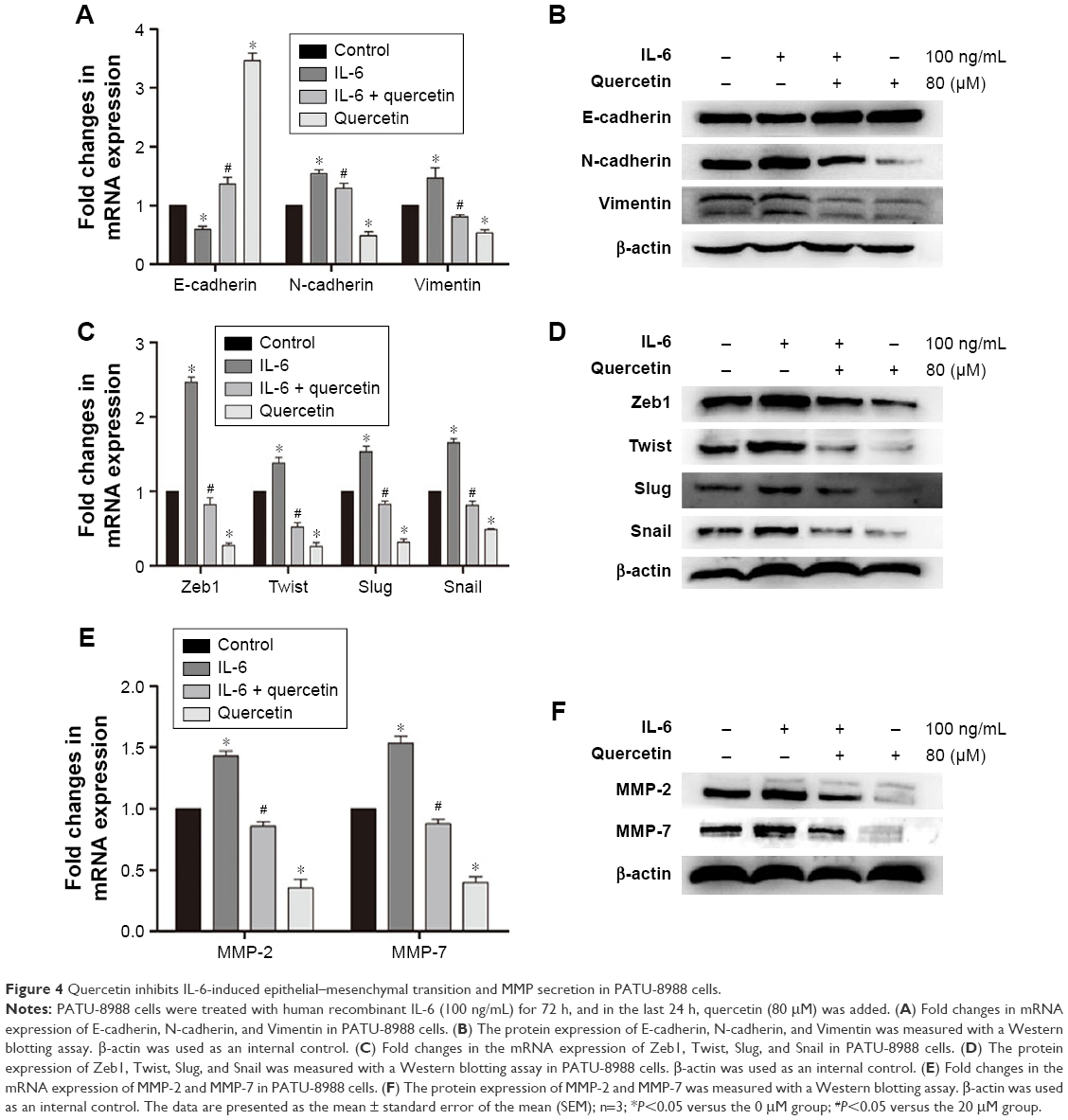 | Figure 4 Quercetin inhibits IL-6-induced epithelial–mesenchymal transition and MMP secretion in PATU-8988 cells. |
Discussion
PC is one of the most invasive tumors of the digestive system and has the worst prognosis. Surgery, the only radical treatment presently offered, is often unavailable when a patient is diagnosed because most patients remain asymptomatic until PC reaches an advanced stage, and there is no effective program for screening patients who may have a high risk of PC.1 Quercetin has emerged as a potential anti-tumor drug. Quercetin induces protective autophagy in gastric cancer cells,27 and quercetin inhibited cell viability, migration, and metastasis in a prostate cancer cell line by modulating Wnt signaling components.28 We hypothesized that quercetin could have a beneficial effect in PC cells via STAT3 signaling. Here, we found that quercetin can reduce the degree of malignancy in PC cells, at least partly, via the STAT3 signaling pathway, and thus, we confirmed that quercetin is a potential therapeutic agent for PC.
EMT is considered an important pathological process that promotes tumor invasion and metastasis.29 EMT was found to occur in PC cell lines and surgically resected PC tissues.30,31 Clinical observations and animal studies have shown that EMT plays an important role in PC progression.32 Therefore, we investigated whether quercetin could reverse the EMT phenotype in PC. The results were consistent with our hypothesis that quercetin has the potential to inhibit EMT in PC. EMT results in degradation of the ECM, and MMPs are the primary mediators of the degradation of the ECM and BM. MMPs play an important role in the process of tumor invasion and metastasis.33 MMPs are highly expressed in PC cells in clinical and experimental models.34 We therefore performed an investigation of the MMP-2, MMP-7 expression levels and an in vitro Transwell invasion assay. The results showed that quercetin effectively inhibited the expression of MMPs and cell invasion.
Although STAT3 is not a housekeeping transcription factor of EMT, it is involved in the expression of EMT-related genes, affecting the phenotype of tumor cells and invasion and metastasis. Activation of STAT3 increased the invasive and metastatic ability of liver cancer cells by mediating the EMT process.35,36 STAT3 signaling enforces MMP expression in various cancer cells and promotes malignant cell invasion and metastasis.37–39 STAT3 is an important oncogene, but unphosphorylated-STAT3 has no effect in the development of cancer. It has been reported widely that p-STAT3, the activation product of STAT3, is likely to contribute importantly to the development of cancer.40 In our experimental results, we observed that the inhibition caused by quercetin of the STAT3 signaling pathway is accompanied by a reversal of the EMT phenotype and MMP secretion. PC cells secrete more IL-6 than cells in tissues adjacent to carcinomas in PC development and progression.41 IL-6, a multi-potent cytokine, binds to sIL-6R to form an IL-6/sIL-6R complex. It then activates gp130 on the cell membrane surface, inducing STAT3 activation.42 IL-6 can activate the STAT3 pathway to promote progression of pancreatic intraepithelial neoplasia and development of PC.43 We used IL-6 to induce EMT in PC cells and investigated whether quercetin reversed the IL-6-induced increase in malignancy via inhibition of the STAT3 signaling pathway. The results are consistent with our speculation that IL-6 can induce EMT and the secretion of MMPs. IL-6-induced EMT and the secretion of MMPs was reversed by quercetin. We also pretreated PATU-8988 cells with a p-STAT3 inhibitor (AG490) for 30 min. The expression of p-STAT3 was inhibited in both groups, and the difference in the expression level was not significant. Taken together, quercetin inhibits EMT, invasion, and metastasis of PC cells by inhibiting the STAT3 signaling pathway. However, we cannot rule out the possibility that quercetin may also act through other mechanisms to inhibit EMT, invasion, and metastasis in PC. This still requires further investigation.
Although the anti-tumor effects of STAT3 were verified in our experiments, the specific mechanisms of how STAT3 regulates EMT, invasion, and metastasis are not fully demonstrated in this article. c-fos and c-jun, which are STAT3 downstream molecules, are early oncogenes. STAT3 activation by phosphorylation causes c-fos and c-jun oncogene activation and expression. Activated STAT3 and c-jun bind to the AP-1 promoter binding site as a heterodimer or with c-fos in a homodimer form, inducing oncogene expression, thus participating in malignant transformation of cells.44 Furthermore, STAT3 plays a vital role in the process of chronic inflammation-mediated tumor progression. The mechanistic link between inflammatory damage and cancer initiation is still unclear. Activation of STAT3 enhances the M2-type polarization of macrophages and helps the formation of tumor-associated phenotypes.45 Tumor-associated macrophages can upregulate the expression of HIF-1 and VEGF with the activation of STAT3, which can significantly promote the formation of tumor blood vessels.46 All in all, what can be affirmed is that quercetin and STAT3 have a beneficial effect in cancer and the relationship between quercetin and STAT3 and other signaling pathways is very complicated and requires further study.
In conclusion, quercetin can inhibit EMT, invasion, and metastasis and reverse the IL-6-induced increase in PC cell malignancy by inhibiting the STAT3 signaling pathway. Our results provide important insight for understanding the mechanisms of the anticancer effect of quercetin. The results of this study suggest that quercetin is a potential anticancer drug. We propose that quercetin treatment may be a new therapeutic strategy that targets EMT, invasion, and metastasis.
Acknowledgments
This research was supported by grants from the Chinese National Natural Science Foundation (no 81370563 and no 81570583) and the Outstanding Youth Fund of Zhejiang Province (no LR14H30001).
Disclosure
The authors report no conflicts of interest in this work.
References
Kamisawa T, Wood LD, Itoi T, Takaori K. Pancreatic cancer. Lancet. 2016;388(10039):73–85. | ||
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5–29. | ||
Simard EP, Ward EM, Siegel R, Jemal A. Cancers with increasing incidence trends in the United States: 1999 through 2008. CA Cancer J Clin. 2012;62(2):118–128. | ||
Havsteen BH. The biochemistry and medical significance of the flavonoids. Pharmacol Ther. 2002;96(2–3):67–202. | ||
Okamoto T. Safety of quercetin for clinical application (Review). Int J Mol Med. 2005;16(2):275–278. | ||
Nwaeburu CC, Abukiwan A, Zhao Z, Herr I. Quercetin-induced miR-200b-3p regulates the mode of self-renewing divisions in pancreatic cancer. Mol Cancer. 2017;16(1):23. | ||
Shen X, Si Y, Wang Z, Guo Y, Zhang X. Quercetin inhibits the growth of human gastric cancer stem cells by inducing mitochondrial-dependent apoptosis through the inhibition of PI3K/Akt signaling. Int J Mol Med. 2016;38(2):619–626. | ||
Ranganathan S, Halagowder D, Sivasithambaram ND. Quercetin suppresses twist to induce apoptosis in MCF-7 breast cancer cells. PLoS One. 2015;10(10):e0141370. | ||
Baruah MM, Khandwekar AP, Sharma N. Quercetin modulates Wnt signaling components in prostate cancer cell line by inhibiting cell viability, migration, and metastases. Tumour Biol. 2016;37(10):14025–14034. | ||
Tang SN, Fu J, Nall D, Rodova M, Shankar S, Srivastava RK. Inhibition of sonic hedgehog pathway and pluripotency maintaining factors regulate human pancreatic cancer stem cell characteristics. Int J Cancer. 2012;131(1):30–40. | ||
Nieto MA, Huang RY, Jackson RA, Thiery JP. EMT: 2016. Cell. 2016;166(1):21–45. | ||
Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2(6):442–454. | ||
Agiostratidou G, Hulit J, Phillips GR, Hazan RB. Differential cadherin expression: potential markers for epithelial to mesenchymal transformation during tumor progression. J Mammary Gland Biol Neoplasia. 2007;12(2–3):127–133. | ||
Wong IY, Javaid S, Wong EA, et al. Collective and individual migration following the epithelial-mesenchymal transition. Nat Mater. 2014;13(11):1063–1071. | ||
Yin Y, Grabowska AM, Clarke PA, et al. Helicobacter pylori potentiates epithelial: mesenchymal transition in gastric cancer: links to soluble HB-EGF, gastrin and matrixmetalloproteinase-7. Gut. 2010;59(8):1037–1045. | ||
Li Y, Yang J, Dai C, Wu C, Liu Y. Role for integrin-linked kinase in mediating tubular epithelial to mesenchymal transition and renal interstitial fibrogenesis. J Clin Invest. 2003;112(4):503–516. | ||
Orlichenko LS, Radisky DC. Matrix metalloproteinases stimulate epithelial-mesenchymal transition during tumor development. Clin Exp Metastasis. 2008;25(6):593–600. | ||
Nagase H, Visse R, Murphy G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc Res. 2006;69(3):562–573. | ||
Sansone P, Bromberg J. Targeting the interleukin-6/Jak/stat pathway in human malignancies. J Clin Oncol. 2012;30(9):1005–1014. | ||
Yu H, Lee H, Herrmann A, Buettner R, Jove R. Revisiting STAT3 signaling in cancer: new and unexpected biological functions. Nat Rev Cancer. 2014;14(11):736–746. | ||
Lim SJ, Li WX. Phospho- and Unphospho-STATs in Signal Transduction and Gene Regulation (STAT). In: Encyclopedia of Signaling Molecules. Springer New York; 2012:1377–1380. | ||
Yu H, Jove R. The STATs of cancer – new molecular targets come of age. Nat Rev Cancer. 2004;4(2):97–105. | ||
Song L, Turkson J, Karras JG, Jove R, Haura EB. Activation of Stat3 by receptor tyrosine kinases and cytokines regulates survival in human non-small cell carcinoma cells. Oncogene. 2003;22(27):4150–4165. | ||
Heinrich PC, Behrmann I, Haan S, Hermanns HM, Müller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signaling and its regulation. Biochem J. 2003;374(Pt 1):1–20. | ||
Rokavec M, Öner MG, Li H, et al. IL-6R/STAT3/miR-34a feedback loop promotes EMT-mediated colorectal cancer invasion and metastasis. J Clin Invest. 2014;124(4):1853–1867. | ||
Wu YS, Chung I, Wong WF, Masamune A, Sim MS, Looi CY. Paracrine IL-6 signaling mediates the effects of pancreatic stellate cells on epithelial-mesenchymal transition via Stat3/Nrf2 pathway in pancreatic cancer cells. Biochim Biophys Acta. 2017;1861(2):296–306. | ||
Wang K, Liu R, Li J, et al. Quercetin induces protective autophagy in gastric cancer cells: involvement of Akt-mTOR- and hypoxia-induced factor 1α-mediated signaling. Autophagy. 2011;7(9):966–978. | ||
Baruah MM, Khandwekar AP, Sharma N. Quercetin modulates Wnt signaling components in prostate cancer cell line by inhibiting cell viability, migration, and metastases. Tumour Biol. 2016;37(10):14025–14034. | ||
Rhim AD, Mirek ET, Aiello NM, et al. EMT and dissemination precede pancreatic tumor formation. Cell. 2012;148(1–2):349–361. | ||
Li Y, VandenBoom TG, Kong D, et al. Up-regulation of miR-200 and let-7 by natural agents leads to the reversal of epithelial-to-mesenchymal transition in gemcitabine-resistant pancreatic cancer cells. Cancer Res. 2009;69(16):6704–6712. | ||
Arumugam T, Ramachandran V, Fournier KF, et al. Epithelial to mesenchymal transition contributes to drug resistance in pancreatic cancer. Cancer Res. 2009;69(14):5820–5828. | ||
Cano CE, Motoo Y, Iovanna JL. Epithelial-to-mesenchymal transition in pancreatic adenocarcinoma. ScientificWorldJournal. 2010;10:1947–1957. | ||
Brooks SA, Lomax-Browne HJ, Carter TM, Kinch CE, Hall DM. Molecular interactions in cancer cell metastasis. Acta Histochem. 2010;112(1):3–25. | ||
Bloomston M, Zervos EE, Rosemurgy AS 2nd. Matrix metalloproteinases and their role in pancreatic cancer: a review of preclinical studies and clinical trials. Ann Surg Oncol. 2002;9(7):668–674. | ||
Zhang CH, Guo FL, Xu GL, Jia WD, Ge YS. STAT3 activation mediates epithelial-to-mesenchymal transition in human hepatocellular carcinoma cells. Hepatogastroenterology. 2014;61(132):1082–1089. | ||
Zhang C, Guo F, Xu G, Ma J, Shao F. STAT3 cooperates with Twist to mediate epithelial-mesenchymal transition in human hepatocellular carcinoma cells. Oncol Rep. 2015;33(4):1872–1882. | ||
Fukuda A, Wang SC, Morris JP 4th, et al. Stat3 and MMP7 contribute to pancreatic ductal adenocarcinoma initiation and progression. Cancer Cell. 2011;19(4):441–455. | ||
Orgaz JL, Pandya P, Dalmeida R, et al. Diverse matrix metalloproteinase functions regulate cancer amoeboid migration. Nat Commun. 2014;5:4255. | ||
Xie TX, Huang FJ, Aldape KD, et al. Activation of stat3 in human melanoma promotes brain metastasis. Cancer Res. 2006;66(6):3188–3196. | ||
Yang J, Chatterjee-Kishore M, Staugaitis SM, et al. Novel roles of unphosphorylated STAT3 in oncogenesis and transcriptional regulation. Cancer Res. 2005;65(3):939–947. | ||
Holmer R, Goumas FA, Waetzig GH, Rose-John S, Kalthoff H. Interleukin-6: a villain in the drama of pancreatic cancer development and progression. Hepatobiliary Pancreat Dis Int. 2014;13(4):371–380. | ||
Jones SA, Horiuchi S, Topley N, Yamamoto N, Fuller GM. The soluble interleukin 6 receptor: mechanisms of production and implications in disease. FASEB J. 2001;15(1):43–58. | ||
Lesina M, Kurkowski MU, Ludes K, et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell. 2011;19(4):456–469. | ||
Ito Y, Sasaki Y, Horimoto M, et al. Activation of mitogen-activated protein kinases/extracellular signal-regulated kinases in human hepatocellular carcinoma. Hepatology. 1998;27(4):951–958. | ||
Gironella M, Calvo C, Fernandez A, et al. Reg3β deficiency impairs pancreatic tumor growth by skewing macrophage polarization. Cancer Res. 2013;73(18):5682–5694. | ||
Kujawski M, Kortylewski M, Lee H, Hermann A, Kay H, Yu H. Stat3 mediates myeloid cell-dependent tumor angiogenesis in mice. J Clin Invest. 2008;118(10):3367–3377. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.