Back to Journals » International Journal of Nephrology and Renovascular Disease » Volume 7
Quantification and localization of M2 macrophages in human kidneys with acute tubular injury
Authors Palmer M, Vichot A, Cantley L, Moeckel G
Received 29 April 2014
Accepted for publication 30 July 2014
Published 7 November 2014 Volume 2014:7 Pages 415—419
DOI https://doi.org/10.2147/IJNRD.S66936
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Professor Pravin Singhal
Matthew B Palmer,1 Alfred A Vichot,2 Lloyd G Cantley,2 Gilbert W Moeckel1
1Department of Pathology, Yale University School of Medicine, New Haven, CT, USA; 2Department of Medicine, Yale University School of Medicine, New Haven, CT, USA
Abstract: This study addresses for the first time the question whether there is significant macrophage population in human kidney sections from patients with acute tubular injury (ATI). We examined therefore the interstitial macrophage population in human kidney tissue with biopsy-proven diagnosis of ATI, minimal change disease (MCD), and MCD with ATI. Kidney biopsies from patients with the above diagnoses were stained with antibodies directed against CD68 (general macrophage marker), CD163 (M2 marker), and HLA-DR (M1 marker) and their respective electron microscopy samples were evaluated for the presence of interstitial macrophages. Our study shows that patients with ATI have significantly increased numbers of interstitial CD68+ macrophages, with an increase in both HLA-DR+ M1 macrophages and CD163+ M2 macrophages as compared to patients with MCD alone. Approximately 75% of macrophages were M2 (CD163+) whereas only 25% were M1 (HLA-DR+). M2 macrophages, which are believed to be critical for wound healing, were found to localize close to the tubular basement membrane of injured proximal tubule cells. Ultra structural examination showed close adherence of macrophages to the basement membrane of injured tubular epithelial cells. We conclude that macrophages accumulate around injured tubules following ATI and exhibit predominantly an M2 phenotype. We further speculate that macrophage-mediated repair may involve physical contact between the M2 macrophage and the injured tubular epithelial cell.
Keywords: macrophages, acute kidney injury, CD163, HLA-DR, CD68, electron microscopy
Introduction
The healing response of mammalian tissues to acute injury is carried out by a complex interplay of diverse cell types including those of the innate immune system. Monocyte-derived tissue macrophages have recently been identified as important regulators of wound healing from early initiation of an inflammatory response to later facilitation of tissue repair and downregulation of inflammation.1 These alternative functions of macrophages are attributed to plasticity of macrophage phenotypes, which have been described as a spectrum from classically activated macrophages (M1 phenotype) to alternatively activated macrophages (M2 phenotype).2 Although the M1–M2 paradigm may be a simplification of the true in vivo picture, it represents a useful model for discerning distinct macrophage functions at different phases of wound healing and tissue repair.
Acute renal tubular injury is a clinicopathologic entity potentially caused by a host of insults to the kidney including ischemia and toxins.3 The resulting injury to renal epithelial cells ranges from mild membrane and cytoplasmic alterations to frank necrosis. The ability of renal tubular epithelium to regenerate after such acute injury can be applied as a model to study the dynamics of wound healing and the roles of various cell types in tissue regeneration.4 Macrophages have been implicated in the process of initiating an inflammatory response as well as providing signals to damaged epithelial cells to proliferate and repair and prevent fibrosis.5,6 The observation that distinct macrophage subsets serve distinct functions in repair after tubular injury suggests that responses more heavily weighted by M2 phenotypes may result in more rapid and complete epithelial regeneration.7 To date, the majority of this data has been obtained using animal models of kidney injury, with almost no quantitative studies of macrophage numbers in human patients with acute tubular injury (ATI). Thus, better understanding the role of macrophage responses to human ATI would support the potential use of macrophage phenotype analysis in biopsies to provide prognostic information, and may ultimately serve to support the idea of manipulating macrophage phenotypes to improve clinical outcomes after renal tubular injury.
To address the role of macrophage responses in the repair of injured human kidney, we have used immunohistochemistry analysis directed against CD68, HLA-DR, and CD-163 to quantify M1 and M2 macrophages respectively in biopsies from patients with ATI alone or minimal change disease (MCD) with or without ATI.
Material and methods
The protocol for case identification, scoring, and history review was approved by the Institutional Review Board at Yale New Haven Hospital. Pathology records were searched to obtain cases with biopsy diagnosis of ATI only or MCD, with or without ATI. Cases that showed any degree of overt interstitial nephritis or interstitial fibrosis were excluded from the study. Data were gathered on renal function and proteinuria levels at the time of biopsy and 1 month postbiopsy.
Immunohistochemistry
Five micron thick sections were cut on charged glass slides. They were then dried, deparaffinized in xylene, rehydrated in graded ethanol to distilled water. The slides were then placed in citrate buffer between 95°C and 101°C for 30 minutes (min). Once the slides had cooled they were placed in Tris-buffered saline (TBS). The slides were rinsed in TBS after each subsequent step. Endogenous peroxidase was quenched with hydrogen peroxide. Slides were then incubated in mouse anti-human CD68 (ThermO Cat# MS397) diluted to 1:600 for 30 min. The antibodies were then detected with Envision + (Mouse) (Dako K4007; Dako Denmark A/S, Glostrup, Denmark). This reaction is visualized with the included DAB in the kit. The slides are then placed in Tris-ethylenediaminetetraacetic acid (EDTA) at 90°C to 101°C for 30 min, then cooled and rinsed in TBS. The second antibody, either HLA-DR (AbCam Cat # ab20181) diluted to 1:500, or CD163 (Cell Marque cat # 163M-16) diluted 1:40, was applied for 30 min. This antibody is then detected with alkaline phosphatase labeled Envision G2 (Dako Cat# K5355). The antibody was detected with Envision secondary antibody-peroxidase reagent (Dako) then visualized with diaminobenzidine and counterstained with hematoxylin. The slides were then dehydrated, cleared, and mounted with resinous mounting medium.
Cases were scored at 200× magnification (one high power field). The numbers of interstitial cells with positive cytoplasmic staining either for CD68, CD163 or HLA-DR, were recorded for ten adjacent high power fields (HPF) and then the average number of positive cells per HPF was calculated.
Electron microscopy
Kidney biopsy tissues were fixed in 2% formaldehyde, 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer, pH 7.4. They were then washed in 0.1 sodium phosphate pH 7.4 and exposed to 1% osmium tetroxide in the same buffer. The samples were then dehydrated through a series of graded ethanols to 100%, and then passed through propylene oxide. Tissue was then infiltrated in 1:1 mixture of propylene oxide, and LX-112 resin, (Ladd Research Industries, Williston, VT, USA). The tissue was finally infiltrated with pure LX-112 resin, transferred to labeled capsules and polymerized overnight at 70°F. Semi-thin, 0.2 μm sections were cut on a Reichert Ultracut E Ultramicrotome, and stained with toluidine blue for evaluation on the light microscope. Suitable areas were then thin sectioned on the same ultramicrotome and sections were collected on copper grids. Sections were then stained with 3% uranyl acetate and lead citrate and viewed utilizing a Carl Zeiss Libra 120 Electron Microscope (Carl Zeiss Meditec AG, Jena, Germany) at 80 KV. Images were taken at an initial magnification of 5,000× with a charge-coupled device (CCD) camera and software (American Medical Technology, Boston, MA, USA).
Statistical analysis
Results are presented as mean ± standard error of the mean of kidney biopsies from 8 patients. Differences were considered statistically significant if P<0.05, using analysis of variance and Bonferroni t-test.
Results
Our human renal biopsy database was searched for recent cases with the histopathological diagnosis of either ATI alone, MCD alone or MCD with ATI. The search yielded five, eight, and seven cases per category, respectively. Serum creatinine and urine protein values at the time of biopsy and at 1 month post-biopsy were retrieved through chart review (Table 1). One month post-biopsy the renal function as assessed by serum creatinine levels was normal or improving towards normalin most patients. At the time of biopsy patients with MCD and ATI had higher urine protein values than patients with MCD alone and the proteinuria was sustained in these patients at the 1 month post-biopsy mark.
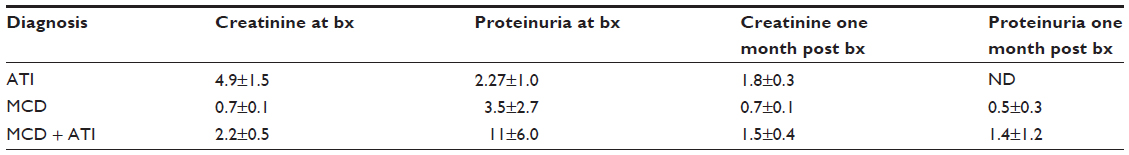 | Table 1 Average values of creatinine (mg/dL) and proteinuria (spot pr:cr ratio) in patients at time of biopsy and 1 month later |
Interstitial macrophage density was determined by counting CD68+ cells/HPF. Alternatively activated (M2) macrophages were quantified by counting the CD163+ sub-population of CD68+ cells and classically activated (M1) macrophages were quantified by counting HLA-DR+ sub-population of CD68+ cells. Patients with ATI showed higher numbers of macrophages, on average 16.3 CD68+ macrophages/HPF, compared to only five CD68+ macrophages/HPF in patients with MCD alone (Figure 1A). The majority of the CD68+ macrophages was also positive for CD163 in both the patients with ATI and patients with MCD (Figure 1A). HLA-DR+ macrophages accounted for 4.5 cells/HPF in ATI patients’ kidneys and for 1.2 cells/HPF in kidneys of patients with MCD (Figure 1A).
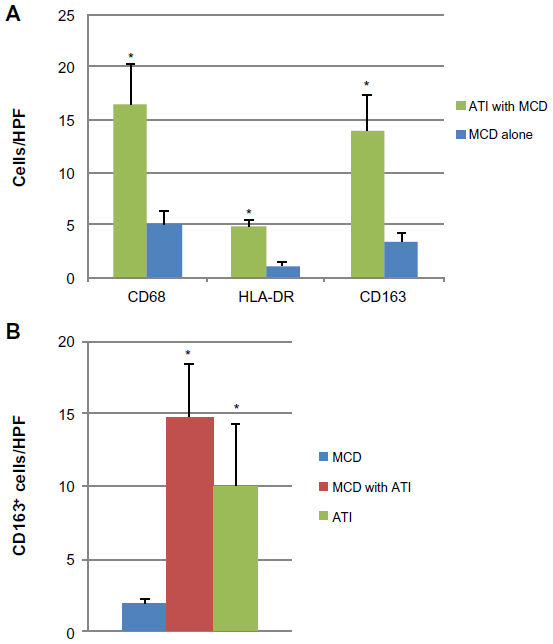 | Figure 1 Macrophages in human kidney tissue. |
Patients with MCD had on average 2.3 CD163+ macrophages per HPF interstitium (Figure 1B). In patients with biopsy diagnosis of concomitant MCD and ATI, the mean number of CD163+ cells per HPF rose to 14.8, a statistically significant increase of approximately 7-fold (P=0.004) (Figure 1B). Cases with ATI alone showed increased numbers of CD163-positive cells compared to MCD. There was no significant change in macrophage density in patients with ATI, regardless of whether they had concomitant MCD (P=0.21) (Figure 1B).
CD163+ M2 macrophages were seen throughout the interstitium and often directly adjacent to injured tubular epithelial cells (Figure 2A and B). HLA-DR positive M1 macrophages were more sparse and often randomly distributed in the interstitium without specific adherence to tubular basement membranes (Figure 2B and C). Ultra structural examination of the peritubular interstitium and peritubular capillaries in patients with ATI showed macrophages migrating from the capillary lumen to the adjacent interstitium (Figure 3A–C) and adhering to the basement membrane of injured tubular epithelial cells (Figure 3D). Thus, the physical adherence of macrophages to the basement membrane of injured tubular epithelial cells may be an important step in the M2-mediated repair process of ATI.
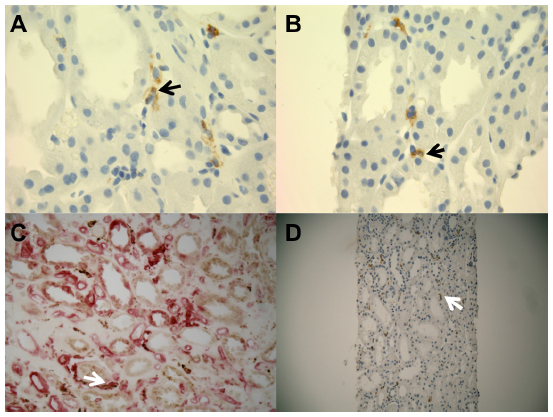 | Figure 2 Representative immunohistochemistry stains for M1 and M2 macrophages in patients with ATI. |
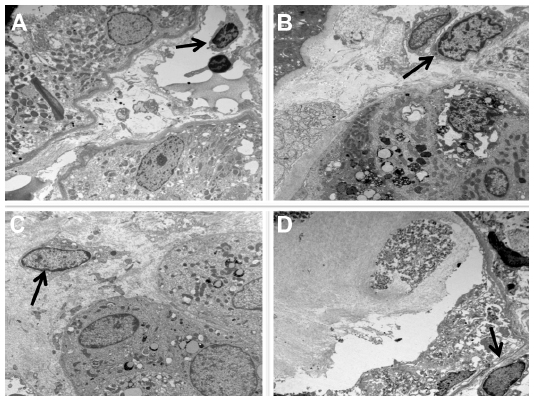 | Figure 3 Macrophage infiltration through the interstitium is directed towards injured tubules. |
Discussion
Our study shows that patients with either ATI alone or MCD with ATI have a significant increase in interstitial macrophage density compared to patients with MCD alone. Based on CD68, HLA-DR and CD163 expression, these are predominately comprised of M2 macrophages, with lesser but significant numbers of M1 macrophages. Although we do not have definitive information on the time interval between the renal insult and the time of biopsy, our chart review demonstrates 5.3±12 days between the clinical diagnosis of AKI (as evidenced by an increased serum creatinine level) and the time of biopsy in these patients. We therefore believe that the biopsy was performed well after the initial insult and most likely during the early repair phase of AKI. These time intervals are similar to animal models of renal ischemia/reperfusion injury, where the M2 macrophage is the predominant macrophage present in the kidney interstitium in the reparative phase of ATI.8 We therefore hypothesize that the M2 macrophage is the predominant macrophage type involved in tubular cell injury repair in humans.
M2 macrophages have been associated with resolving inflammation and inducing repair.9,10 They can express anti-inflammatory cytokine profiles for more than 4 weeks in vitro.11 The mechanisms underlying the protective effects of M2 macrophages include suppression and downregulation of pro-inflammatory cytokines and suppression of M1 macrophages.2 This may explain the relatively low number of M1 macrophages in our patients’ biopsies. Tubular epithelial cells produce cytokines such as CCL2 that induce monocyte mobilization to the kidney following injury.12–14 In experimental models of acute kidney disease, certain cytokines exert M2 macrophage-mediated reparative effects of tubular structure and function.15 Similar mechanisms may play a role in human kidneys following ATI and may lead to extravascular migration of monocytes into the interstitium and subsequent phenotype switch to a M2 predominant macrophage population, which induces epithelial cell repair through stimulation of cytokine excretion.
Previous studies in ischemia/reperfusion models of ATI have documented migration of macrophages to the area of tubular injury.16 CD163 positive M2 macrophages specifically localize close to the tubular basement membrane of injured proximal tubule cells as shown in our immunohistochemistry and ultrastructural studies. We conclude from our results that physical adherence of the M2 macrophage to the injured tubular epithelial cell may play an important role in macrophage-mediated repair mechanisms.
The role of macrophages in tubular injury repair is increasingly recognized.17,18 In situ expansion of specific subtypes of intrinsic macrophages has been recognized as an important mechanism in tubular injury repair.8 Recent advances in molecular immunology and insight into cytokine expression that enhance cell injury repair, have opened possible therapeutic avenues which may lead to novel drug development.18 Further studies in human kidney tissues are needed to assess the role of modified cytokines’ expression patterns that may enhance macrophage-mediated tubular injury repair.
Acknowledgments
We thank Mr Thomas Ardito and Mr Amos Brooks, Department of Pathology, Yale University School of Medicine for their excellent technical skills in preparing the electron microscopy and immunohistochemistry samples respectively. This work was supported in part by an R01 to LGC (DK93771) and RO3 to GWM (DK077700).
Disclosure
The authors have no conflicts of interest to disclose.
References
Alikhan MA, Ricardo SD. Mononuclear phagocyte system in kidney disease and repair. Nephrology (Carlton). 2013;18(2):81–91. | |
Wang Y, Harris DC. Macrophages in renal disease. J Am Soc Nephrol. 2011;22(1):21–27. | |
Bonventre JV. Pathophysiology of ischemic acute renal failure. Inflammation, lung-kidney cross-talk, and biomarkers. Contrib Nephrol. 2004;144:19–30. | |
Bonventre JV, Zuk A. Ischemic acute renal failure: an inflammatory disease? Kidney Int. 2004;66(2):480–485. | |
Li L, Okusa MD. Macrophages, dendritic cells, and kidney ischemia-reperfusion injury. Semin Nephrol. 2010;30(3):268–277. | |
Lee S, Huen S, Nishio H, et al. Distinct macrophage phenotypes contribute to kidney injury and repair. J Am Soc Nephrol. 2011;22(2):317–326. | |
Duffield JS. Macrophages and immunologic inflammation of the kidney. Semin Nephrol. 2010;30(3):234–254. | |
Zhang MZ, Yao B, Yang S, et al. CSF-1 signaling mediates recovery from acute kidney injury. J Clin Invest. 2012;122(12):4519–4532. | |
Herbert DR, Holscher C, Mohrs M, et al. Alternative macrophage activation is essential for survival during schistosomiasis and downmodulates T helper 1 responses and immunopathology. Immunity. 2004;20(5):623–635. | |
Pull SL, Doherty JM, Mills JC, Gordon JI, Stappenbeck TS. Activated macrophages are an adaptive element of the colonic epithelial progenitor niche necessary for regenerative responses to injury. Proc Natl Acad Sci U S A. 2005;102(1):99–104. | |
Wang Y, Wang YP, Zheng G, et al. Ex vivo programmed macrophages ameliorate experimental chronic inflammatory renal disease. Kidney Int. 2007;72(3):290–299. | |
Li L, Huang L, Sung SS, et al. The chemokine receptors CCR2 and CX3CR1 mediate monocyte/macrophage trafficking in kidney ischemia-reperfusion injury. Kidney Int. 2008;74(12):1526–1537. | |
Furuichi K, Wada T, Iwata Y, et al. CCR2 signaling contributes to ischemia-reperfusion injury in kidney. J Am Soc Nephrol. 2003;14(10):2503–2515. | |
Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19(1):71–82. | |
Alikhan MA, Jones CV, Williams TM, et al. Colony-stimulating factor-1 promotes kidney growth and repair via alteration of macrophage responses. Am J Pathol. 2011;179(3):1243–1256. | |
Ysebaert DK, De Greef KE, Vercauteren SR, et al. Identification and kinetics of leukocytes after severe ischaemia/reperfusion renal injury. Nephrol Dial Transplant. 2000;15(10):1562–1574. | |
Huen SC, Cantley LG. Macrophage-mediated injury and repair after ischemic kidney injury. Pediatr Nephrol. Epub January 19, 2014. | |
Williams TM, Little MH, Ricardo SD. Macrophages in renal development, injury, and repair. Semin Nephrol. 2010;30(3):255–267. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.