Back to Journals » Journal of Blood Medicine » Volume 13
Pyruvate Kinase Deficiency: Current Challenges and Future Prospects
Authors Fattizzo B ![]() , Cavallaro F, Marcello APML, Vercellati C, Barcellini W
, Cavallaro F, Marcello APML, Vercellati C, Barcellini W
Received 31 May 2022
Accepted for publication 23 August 2022
Published 1 September 2022 Volume 2022:13 Pages 461—471
DOI https://doi.org/10.2147/JBM.S353907
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Bruno Fattizzo,1,2 Francesca Cavallaro,1,2 Anna Paola Maria Luisa Marcello,1 Cristina Vercellati,1 Wilma Barcellini1
1Hematology Unit, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy; 2Department of Oncology and Hemato-Oncology, University of Milan, Milan, Italy
Correspondence: Bruno Fattizzo, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Hematology Unit, Via F. Sforza 35, Milan, 20122, Italy, Tel +39 0255033477, Email [email protected]
Abstract: Pyruvate kinase deficiency (PKD) is a rare autosomal recessive disease marked by chronic hemolytic anemia of various severity and frequent complications including gallstones, splenomegaly, iron overload, and others. Disease phenotype is highly heterogeneous and changes over time with children, adolescents and adult patients displaying different transfusion requirement and rates of complications. The diagnosis relies on the initial clinical suspicion in a patient with chronic hemolysis and exclusion of other more common congenital forms of hemolytic anemias; it is supported by the demonstration of reduced PK enzyme activity, and further confirmed by the detection of (homozygous or compound heterozygous) mutations of PKLR gene. Therapy is mainly supportive, with vitamin supplementation and transfusions (based on symptoms and patient growth rather than on fixed Hb thresholds). Splenectomy is widely performed, although it is less effective than in membrane defects and carries thrombotic and infectious risk. In the last decade, the allosteric PK enzyme activator mitapivat showed dramatic clinical benefit in clinical trials and gene therapy is also being studied to substitute the defective enzyme. In this review, we provide an insight in the current challenges of PKD diagnosis and management and discuss the future application of novel drugs and gene therapy, including a focus on quality of life.
Keywords: pyruvate kinase deficiency, splenectomy, mitapivat, gene therapy
Introduction
Pyruvate kinase deficiency (PKD) is a congenital hemolytic disease characterized by a variable degree of anemia from mild to transfusion dependent and heterogenous clinical features derived from chronic red blood cell (RBC) destruction (gallstones, splenomegaly, iron overload, etc.). Disease phenotype may vary over time with children, adolescents and adult patients displaying different transfusion requirement and rates of complications (Figure 1).1–3 Despite the availability of biochemical assays for enzyme activity and the identification of the several underlying genetic lesions, the primum movens for the diagnosis is still the clinical suspicion and the subsequent reference of patients to tertiary centers. Several confounders further challenge the diagnosis and patients may end up being treated as other congenital hemolytic anemias (CHAs). The latter may have similar phenotypes but may show different outcomes in terms of response to splenectomy and frequency of complications.4 Finally, the advent of novel treatments aimed at restoring PK activity, including the allosteric PK enzyme activator mitapivat and gene therapy, further advocates for a definite diagnosis in these patients.5,6 In this review, we will provide a clinician insight in the current diagnosis and management of PKD, including challenges in disease identification and treatment, and future prospects on novel targeted therapies and gene therapy.
 |
Figure 1 The physiopathology of pyruvate kinase deficiency (PKD), and its clinical features and complications. (A) The Embden-Meyerhof pathway alteration. (B) Different clinical features and complications during infancy, adolescence, and adulthood. Abbreviations: 1,3 DPG, 1,3 biphosphoglyceric acid; 2.3 DPG, 2,3 biphosphoglyceric acid; 2-PG, 2-phosphoglyceric acid; 3-PG, 3-phosphoglyceric acid; ADP, adenosine diphosphate; ATP, adenosine triphosphate; DHAP, dihydroxyacetone phosphate; F1,6P, fructose-1,6-biphosphate; F6P, fructose-6-phosphate; G3P, glyceraldehyde 3-phosphate; G6P, glucose-6-phosphate; NAD, nicotinamide adenine dinucleotide; NADH, nicotinamide adenine dinucleotide+hydrogene; PEP, phosphoenolpyruvate; RBC, red blood cells; yrs, years. |
Epidemiology and Physiopathology
PKD is inherited as an autosomal recessive disease and has an estimated prevalence of 3:1,000,000 to 1:20,000.7,8 Heterozygote frequencies range from 0.15% to 6% in the various populations and have been phylogenetically preserved due to a protective effect against malaria.9–11 As a result, a high PKD frequency is reported in the Middle East and sub-Saharan Africa.12,13 From a physiopathology point of view, RBCs lack nucleus and organelles, and generate energy (adenosine triphosphate, ATP) via the Embden-Meyerhof glycolytic pathway to maintain their shape, flexibility, and rheological properties over their 120-day journey. PK is a key enzyme of the glycolysis, boosting the transformation of pyruvate from phosphoenolpyruvate (PEP) with ATP generation (Figure 1A). PKD results from homozygous or compound heterozygous mutations in the PKLR gene (chromosome 1q22),14–16 including more than 350 lesions, mostly missense (encoding single aminoacid changes), and rarely disruptive mutations (stop codons, frameshifts, and large deletions). The latter are generally associated with a more severe phenotype that affects the enzyme’s structure, stability or catalytic function. Decreased PK activity results in low ATP levels and high levels of the upstream metabolite 2,3‐diphosphoglycerate (2,3‐DPG), that in turn favors O2 release to tissues.1,17,18 As a result, PKD-RBCs are rigid, less flexible, and undergo premature hemolysis in the spleen.
Current Challenges in PKD
Clinical Suspicion and Differential Diagnosis
Diagnosis of PKD is based on the presence of chronic hemolytic anemia, reduced PK enzyme activity, and molecular characterization of the pathogenic mutations. Generally, severe cases are diagnosed at an earlier age, intrauterine complications (growth retardation, hydrops fetalis and prematurity),1 frequent neonatal jaundice (59–90%), and possible transfusion requirement (Figure 1). On the other hand, some subjects may receive PKD diagnosis in late adulthood, possibly due to comorbidities unveiling a mild disease.1,19 Overall, the most frequent symptoms are anemia (90–95% of cases, mild to transfusion-dependent), splenomegaly (80–85%), jaundice (40–70%) and gallstones (30–45%). More rare manifestations include aplastic crises (2–14%), bone deformities (9%), extramedullary erythropoiesis (9%), delayed puberty (8%), hyperpigmentation (6%), leg ulcers, and pulmonary hypertension (2–3%).1,16,19 Hemoglobin values generally range from 6.5 to 11 g/dL and improve after splenectomy (approximately 15 g/l). Concerning hemolytic markers, reticulocytes are elevated (4% −11%) and typically markedly increase after splenectomy (20% −70%);19,20 unconjugated bilirubin is also elevated with a mean value of 60 mol/L (higher if coexistent with Gilbert’s syndrome); and lactate dehydrogenase is usually normal.21 Ferritin is often increased (even >1000 ng/L), regardless transfusion dependency.1,22
The great overlap with the other congenital anemias challenges the differential diagnosis (Figure 2).4 Generally, hemolytic features and increased mean corpuscular volume (MCV) typical of CHAs aid the distinction from hemoglobinopathies that are further investigated by searching for pathologic hemoglobin forms through high performance liquid chromatography (HPLC). The distinction from other CHAs may be more difficult and is supported by evaluation of RBC morphology (peripheral blood smear), osmotic fragility tests, and RBC deformability tests by ektacytometry that are usually normal in PKD and altered in membrane defects. Additionally, PKD is autosomal recessive whilst membrane defects are usually dominant, so that family history is generally not revealing, unless an affected sibling is present. Finally, in patients without family history and onset in adulthood, tests to exclude common causes of acquired hemolytic anemias, particularly the direct anti-globulin test for autoimmune forms, is recommended.
 |
Figure 2 The differential diagnosis of pyruvate kinase deficiency (PKD). Abbreviations: NGS, next generation sequencing; WES, whole exome sequencing; DAT, direct antiglobulin test; HPLC, high performance liquid chromatography. |
After the above-mentioned tests have been performed, RBC enzymatic activities are evaluated leading to PKD diagnosis.2,3 PK enzymatic activity is determined in RBC lysates by spectrophotometry according to Beutler et al, 1984, and is measured as international unit per Hb grams (normal reference range usually defined at each laboratory on healthy blood donors).23 This test is fast but may give false normal results due to incomplete leukocyte/platelets depletion, reticulocytosis, and recent transfusions. Evaluating PK activity in proportion to that of other age-dependent RBC enzymes, such as hexokinase or glucose-6-phosphate dehydrogenase, may disclose relative low enzyme activity and trigger molecular confirmatory test.17,24 Finally, mutations in KLF1, a master regulator of erythropoiesis that modulates the expression of many red cell enzymes including PK, may result in PK deficiency at routine enzyme assays possibly leading to misdiagnosis.25 Genotyping of PKLR is therefore recommended and more than 300 mutations have been described, mostly missense substitutions (70–80%, R510Q in Northern Europe and the United States, R486W in Southern Europe, and R479H mutation among Pennsylvania Amish).26 An effort to perform a genotype–phenotype correlation has been made in the PKD Natural History Study,1 where patients with at least one missense mutation had a milder phenotype (later diagnosis, higher Hb, reduced transfusion need, lower ferritin levels, and less frequent splenectomy) as compared with those with two non-missense mutations. Genetic testing offers several advantages, such as the need for smaller sample volumes, no interference of transfused RBCs, and suitability for prenatal diagnosis that may be performed in the more severe cases. While there are no clear indications for prenatal/pre-gestation screening programs in PKD, genetic counselling may be considered in affected individuals or in case of familiarity.
To Transfuse or Not to Transfuse
Supportive treatment is still the mainstay for PKD management, and routine folic acid supplementation is pivotal to sustain bone marrow reticulocyte compensation. B12 vitamin should also be systematically tested and supplemented if deficient. Concerning transfusions, their use is mainly dependent on the presence of clinical signs and symptoms that vary according to anemia degree and to the compensatory response. In PKD, tolerance of anemia may be higher than in other forms due to both the chronic nature of the disease and to the increased levels of RBC 2.3-DPG content, with consequent increased oxygen release to tissues. Therefore, whilst in thalassemia systematic transfusions are indicated to avoid complications of accelerated erythropoiesis, in PKD transfusion need should be individualized, based on anemia compensation, and its impact on quality of life (QoL). In any case, in a recent study, most PKD patients (84%) had required at least one transfusion in their lifetime.1–3 Transfusion requirement in a single patient may change over time in parallel with anemia compensation. In newborns, transfusion requirement is frequent in the first days of life, possibly suppressing erythropoiesis. This, combined with the physiological hemoglobin nadir, may confound the assessment of anemia severity in neonatal age. It has been therefore recommended to extend the time between transfusions to allow erythropoietin and reticulocyte response in newborns. Additionally, indirect hyperbilirubinemia requires phototherapy in nearly all patients (93%) and exchange transfusions in about half of cases.1 Subsequently, some children require regular transfusions to avoid symptoms whilst others are only intermittently transfused during acute events, particularly infections, and the Hb threshold is that allowing for normal growth/development. Infections, causing both increased hemolysis and suppression of bone marrow compensation, usually decline along with age with a reduction in transfusion need. As a result, about 50% of patients aged ≤5 years are regularly transfused, compared to only 25% of those >5-<12 years and less than 10% of those aged ≥18 years.1,16,20 This is also due to the effect of splenectomy which is usually deferred after the first decade of life as discussed thereafter. Conversely, in the elderly, the accumulation of comorbidities and physiological aging may decrease anemia tolerance triggering transfusion need. However, since intra-patient variation of Hb levels is generally minimal in PKD, clearcut anemia worsening should prompt investigations for additional causes (ie, nutritional deficiencies, evolution into myelodysplasia or marrow infiltration, or presence of accessory spleen in splenectomized patients).
Splenectomy: Friend or Foe?
It has long been known that splenectomy may attenuate anemia and reduce transfusion requirement in about 90% of PKD patients,1–3 with a median rise in Hb of 1.6 g/dL. However, these benefits should be weighed against the well-known infectious and thrombotic risks of splenectomy. It is generally delayed until 5 years of age, and the timing is also decided based on the frequency of RBC transfusions, the presence of allo-immunization, the presence of iron overload (including the efficacy of iron chelation), and the availability and fitness of venous access. Unluckily, assessment of red cell survival, splenic sequestration, and spleen size is not predictive for efficacy of splenectomy in PKD, likely due to liver hemocatheresis. The latter contributes to residual post-splenectomy hemolysis (ie, reticulocytosis and indirect hyperbilirubinemia) in most patients, with consequent persistent jaundice and risk of pigmented gallstones.
The main reasons to perform splenectomy are regular transfusions and severe anemia,1 as also indicated by recent guidelines.27 However, there is a high variability in splenectomy frequencies among the various centers.1 Predictors of Hb response include less severe anemia, with lower total bilirubin, and presence of a more favorable genotype (ie, 2 missense PKLR mutations), so that most severe PKD is less likely to improve. Full splenectomy is recommended since the few reported cases of partial splenectomy had no benefit.28
The risk of post-splenectomy sepsis is mainly related to encapsulated organisms, but also to malaria and babesiosis,29,30 and its frequency was as high as 7% in a recent study.1 Vaccinations (anti-Meningococcal, anti-Pneumococcal, and anti-Haemophilus) may effectively reduce the risk, should be performed at least 2 weeks before splenectomy, and regularly updated based on most recent guidelines. Indications on oral antibiotics for infection prophylaxis are less clearcut and largely variable among Countries. Pediatricians generally recommend that asplenic children receive penicillin prophylaxis until 5 years of age and for at least 1 year following splenectomy. Since vaccinations do not completely abrogate infectious risk, patients’ education to promptly seek for medical attention to receive broad spectrum antibiotics in case of fever (or other suggestive signs/symptoms) is mandatory.2,3,27
Regarding post-splenectomy thrombosis, in PKD the risk is approximately 10%, mainly involving venous district (deep venous thrombosis and pulmonary embolism) and also including unusual sites (portal vein thrombosis and central nervous system thrombosis).20,31,32 Post-splenectomy thrombocytosis can be managed with low dose aspirin until the platelet count is <500 x10^9/L, particularly. Anticoagulant prophylaxis may also be considered in selected cases after careful evaluation of additional risk factors for thrombosis.
Transplant
Hematopoietic stem cell transplant (HSCT) that may virtually cure PKD, has been shown beneficial in animal models,33 and in subsequent reports of PKD patients.34 In a larger series of 16 patients undergoing HSCT in Europe and Asia35 a 74% cumulative survival was reported although with a relevant rate of graft-versus-host disease (GVHD). Additionally, pooling data is particularly difficult given the heterogeneity of donor types, conditioning, GVHD and infection prophylaxis. Altogether, in the absence of clinical trials, recent guidelines favor splenectomy and/or regular red cell transfusions rather than HSCT in PKD.2,3
How to Monitor and Manage Complications
PKD patients may experience several disease-related and treatment-related complications that further challenge clinical course with variable frequency during lifetime. Gallstones may occur at all ages (median 15 years) in up to 30–45% of patients,1,19 are predicted by a more hemolytic pattern with hyperbilirubinemia, and favored by Gilbert syndrome.36,37 In a recent analysis, cholecystectomy was performed simultaneously with splenectomy in 20% of cases, whilst 50% of patients required it after splenectomy due to persistent hemolysis.1 In case of gallstones, simultaneous surgery is therefore recommended. Whilst in their absence the indication to cholecystectomy is less clear, also due to an increase of intrahepatic cholestasis risk post-surgery.
Another frequent and underestimated complication is iron overload, both related to transfusion dependence,19 but also common in patients never/seldomly transfused. Iron overload may also be due to ineffective erythropoiesis and low hepcidin levels, to increased intestinal absorption, or to coinheritance of hemochromatosis genes.22,24 In the PKD Natural History Study, 38% of transfusion independent patients had ferritin >1000 lg/l,22 82% had pathologic liver T2* MRI, and 7% cardiac one, and 10% had received chelation at some point during their life. The recommended monitoring in regularly transfused patients (>6 transfusions/year) includes ferritin levels twice a year, more frequently in those on chelation. The latter is advised after 10–14 RBC transfusions and preceded by a T2* MRI. In non-transfusion dependent patients ferritin levels should be obtained at least every 1–2 years, and MRI at least once, particularly if ferritin >500 lg/l (sensitivity 90%). Such patients may in fact require intermittent chelation.2,3,22 The approach to chelation in PKD is similar to other red cell and iron loading disorders and not specifically standardized for PKD. In non-transfused patients with adequate hemoglobin levels, therapeutic phlebotomy can be considered as an alternative to chelation.
During life, patients may experience sudden Hb drops due to either hemolytic or aplastic crises.1–3 Both are more frequent during infancy and after infections. Hemolytic crises are generally characterized by fatigue, worsening jaundice and hemoglobinuria. Aplastic crises are marked by reticulocytopenia, and possibly pancytopenia, are due to parvovirus B19, and mostly recover in about 10 days, but rarely require longer time and high transfusion need. Positive serology for anti-parvovirus IgM or DNA detection by PCR confirm the diagnosis. In both conditions, anemia may be severe and require transfusion support, and continuous folic acid supplementation.
Other rarer complications include bone deformities, due to hyperplastic bone marrow, and osteopenia, possibly related to iron toxicity, increased bone turnover, and hormone deficiencies. It is therefore advised to monitor vitamin D levels, calcium intake, and to perform bone densitometry at least once.1,20 Extramedullary hematopoiesis is very rare and may be recognized by CT scan showing paravertebral masses;1,38,39 pulmonary hypertension has been reported, should be suspected in case of dyspnea disproportionate to anemia severity, and investigated by echocardiography and cardiologist referral.1,40 Finally, leg ulcers have been seldomly reported,20,41 so that physical examination for cutaneous lesions is recommended in PKD patients.
Although it is not a complication, pregnancy in PKD deserves a mention. It is usually associated with good maternal and fetal outcomes,1 provided close obstetrician/hematologic monitoring. However, up to 10% preterm births and 18% miscarriages have been reported. A preconception evaluation with echocardiogram is advised, as well as continuous folic acid supplementation, whilst iron supplement should be evaluated on a case-by-case basis if deficiency is present. Hemolysis typically worsens and transfusion need increase. The latter should also be tuned on fetal growth.2,3
Future Prospects
Current management of PKD mainly relies on supportive measures, iron chelation, and splenectomy. In the last 10 years, a novel era of therapies directed at boosting enzyme activity, by directly activating PK or by substituting the defective gene, has begun. Published and ongoing clinical trials are summarized in Table 1.
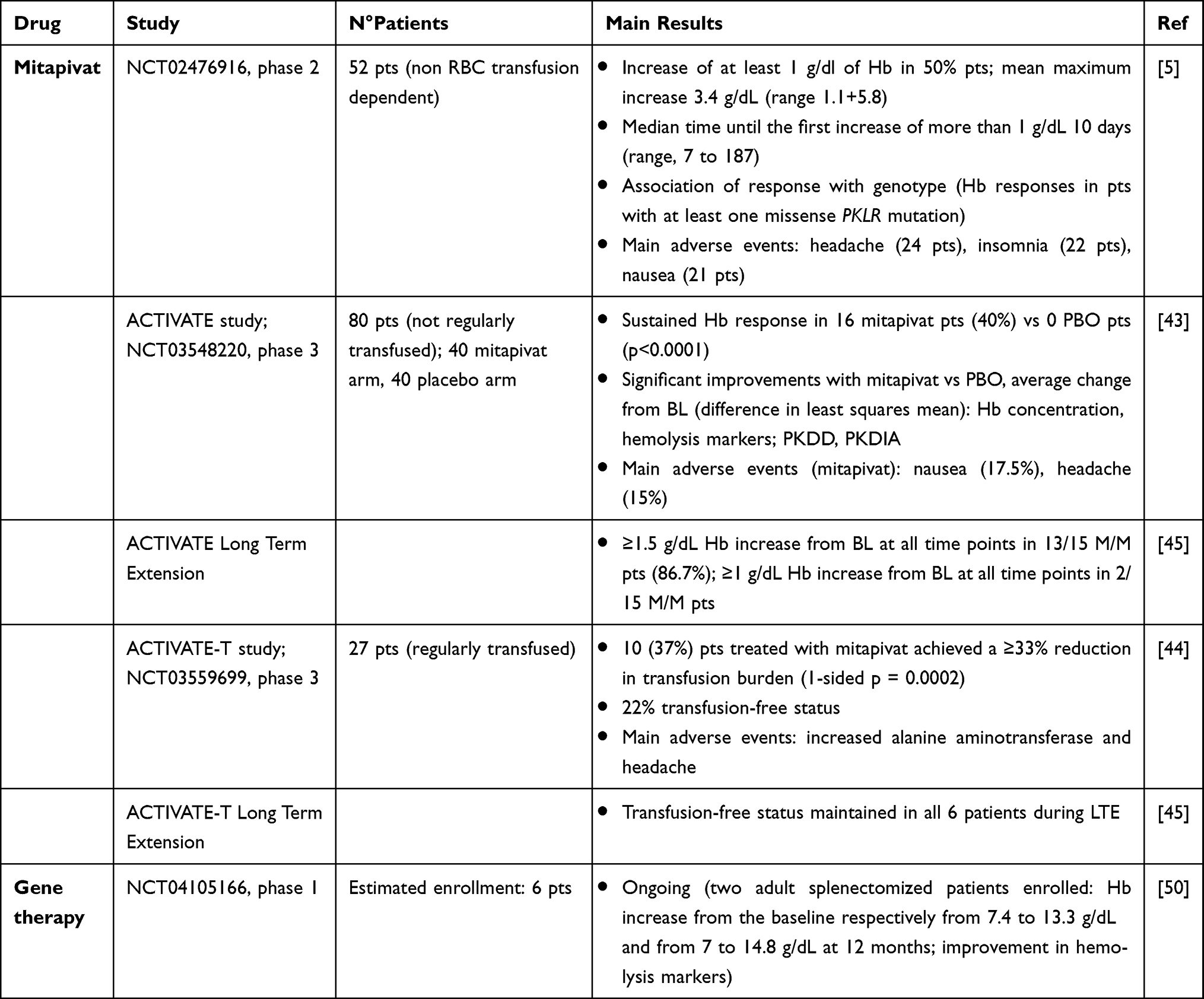 |
Table 1 Future Treatment Prospects in Pyruvate Kinase Deficiency |
Pyruvate Kinase Activators
Mitapivat, previously AG-348, is an oral allosteric activator of RBC PK. In biochemical assays and ex vivo experiments, this small molecule succeeded in increasing wild type and mutant PK enzyme activity.42 Subsequently, a Phase 2 study (NCT02476916)5 enrolled 52 adult patients with PKD not receiving RBC transfusions, who were randomly assigned to receive mitapivat 50 mg or 300 mg twice daily for 24 months. A Hb increase of more than 1 g/dL from the baseline was reported in 26 patients (50%, mean maximum Hb increase of 3.4 g/dL, range 1.1+5.8). Importantly, Hb response was observed only in patients with at least one missense mutation of PKLR (ie, those with residual PK activity). The drug showed a favorable safety profile, with mainly low-grade adverse events (AEs) including headache (24 patients), insomnia (22), and nausea (21), mostly transient and resolving within 7 days of treatment. Further two Phase 3 trials assessed mitapivat in PKD patients. The ACTIVATE study (NCT03548220)43 was a randomized, double-blind, placebo-controlled study evaluating the efficacy of mitapivat in not regularly transfused patients. A 12-week dose escalation period was followed by a 12-week fixed dose one and 16/40 patients in the mitapivat arm (40%) achieved a sustained Hb response versus 0 patients in the placebo one. Patients receiving mitapivat had a mean Hb increase of 1.8 g/dL, an improvement in hemolytic markers, and in patients reported outcomes. Again, AEs were mainly mild and included nausea (17.5%) and headache (15%). Responders patients maintained Hb improvement across the long-term extension (LTE, data cut off at 19.5 months); 17 patients from the placebo arm switched to mitapivat in the LTE, and 6 (35%) achieved a Hb response. The efficacy of mitapivat in regularly transfused patients was evaluated in the open label ACTIVATE-T study (NCT03559699).44 Twenty-seven patients were enrolled and received a dose escalation period of 16 weeks, followed by a fixed dose period of 12 weeks. Ten cases (37%) met the primary endpoint (>33% reduction in transfusion burden), and 6 (22%) became transfusion independent. Mitapivat was again well tolerated, with increased alanine aminotransferase (37%) and headache (37%) as most common AEs. Responders maintained transfusion independence across the LTE study45. Interestingly, mitapivat and etavopivat, a new allosteric PK activator (FT-4202), are currently under study in sickle cell disease (SCD) and thalassemia (NCT04610866, NCT03692052, NCT03815695, NCT04624659, NCT04987489).46–48 These conditions share with PKD features of chronic hemolysis with reduced RBC life span, and ineffective erythropoiesis and further highlight the potential of boosting RBC energy machinery in congenital anemias.
Gene Therapy
Being a recessive inherited disease with a single gene defect involving RBC, PKD is an excellent candidate for gene therapy. In a murine model, PKD hematopoietic stem cells were transfected with a lentiviral vector carrying PK gene and subsequently transplanted in myeloablated PKD mice.49 This led to the restoration of normal glycolytic activity and erythropoiesis, along with reversion of secondary effects of hemolysis. Moreover, no evidence of genotoxicity was noted. Given the promising results in preclinical studies, gene therapy by lentiviral transduction of autologous stem cells and progenitor cells is currently under investigation in an open-label Phase I trial (NCT04105166).50 At the last update, two adult splenectomized patients had been enrolled, with a substantial Hb increase from the baseline in both subjects (from 7.4 to 13.3 g/dL and from 7 to 14.8 g/dL at 12 months, respectively), associated with an improvement in hemolytic markers. Notably, no severe adverse events were reported. Gene therapy is now under evaluation in the pediatric setting.
In the present scenario of PKD therapeutic choices, the indication for gene therapy is certainly a “hot” topic. Hence, an “expanded” definition of severe PKD has recently been proposed by a panel of experts, to facilitate the identification of subjects most suitable for ongoing gene therapy.6
QoL Evaluation in PKD
Along with the emergence of new drugs and therapeutic strategies for PKD, health-related quality of life (HR-QoL) in PKD has been gaining more and more importance in the recent years. HR-QoL is negatively affected not only by signs and symptoms of PKD (such as fatigue, dyspnea, jaundice and splenomegaly), but also by disease complications (eg, iron overload necessitating iron chelation therapy) and transfusion requirement.20 Nevertheless, due to the rarity of the disease and the lack of published HR-QoL data, it’s difficult to define the burden of the disease on patients’ lives. Moreover, understanding HR-QoL of PKD patients and defining specific tools to assess it is crucial to delineate the value and the utility of new treatments. To better explore the burden of disease on patients’ HR-QoL, Grace et al carried out a qualitative interview study of 21 adults with PKD.20 The most common reported signs and symptoms were yellow eyes (19/21), tiredness (18/21), yellow skin (17/21) and low energy (13/21). Appearance, emotional and cognitive states, work, school, sleep and the ability to perform physical, social and leisure activities were all negatively affected by the disease. Moreover, recently published data from the PKD Natural History Study (NCT02053480) described the impact of PKD on HR-QoL and fatigue.51 HR-QoL was measured at 3 different time points (enrollment, 1- and 2-year follow-up) in 254 patients (131 adults and 123 adolescents and children), using tools validated from other anemias (EuroQol 5-Dimension Questionnaire, Pediatric Quality of Life Inventory Generic Core Scale version 4.0, and Functional Assessment of Cancer Therapy-Anemia) and fatigue (Patient Reported Outcomes Measurement Information System Fatigue and Pediatric Functional Assessment of Chronic Illness Therapy-Fatigue). Significantly worse HR-QoL outcomes were reported in regularly transfused adults, children with severe anemia or with non-missense/non-missense genotype, iron overload with chelation requirement and patients with pulmonary hypertension. Jaundice was also reported as a cause of upset both in children and in adults. Salek and colleagues conducted a systematic search of the literature to identify adequate patient reported outcome (PRO)s measures for HR-QoL and fatigue suitable for PKD trials.52 Conditions similar to PDK, such as SCD and thalassemia, were comprised in the research. Based on the results, the European Organisation for Research and Treatment of Cancer Quality-of-life Questionnaire Core 30 (EORTC QLQ-C30) and the Short Form 36-item Health Survey Version 2 (SF-36v2®) were recommended for assessing HR-QoL in PKD. However, being not disease-specific, the EORTC QLQ-C30 and the SF-36v2® may not cover some symptoms of PKD or may include symptoms and impacts not suitable in PKD patients. Therefore, the same Authors proposed two disease-specific PRO measures in 2020.53 Twenty-one PKD patients underwent concept elicitation and then cognitive interviews to develop the PK deficiency daily diary (PKDD) and the PK deficiency-specific impact assessment (PKDIA) survey. The first is a 7-item tool that measure main PK signs and symptoms, whilst the latter is a 14-item measure of PKD impact on patients’ HR-QoL. Subsequently, PKDD and PKDIA were compared with the previously recommended tools EORTC QLQ-C30 and SF-36v2®, reporting only minimal similarities (eg, in the EORTC QLQ-C30, 43% of concepts were similar to the PKDD and 42% to the PKDIA; in the SF-36v2®, 57% of concepts were similar to the PKDD and 17% to the PKDIA). The PKDD and PKDIA appear reliable disease-specific tools to assess PROs and HR-QoL and will likely be implemented in clinical trials of PKD.
Conclusions
The landscape of PKD management is still marked by several challenges, particularly regarding the threshold of clinical suspicion, the sensitivity and availability of diagnostic tests, the management of complications, and the choice of treatment type and timing. Regarding the first, much has been done and much more is still needed to inform clinicians about the differential diagnosis of hemolytic disorders (Figure 2). It is fundamental to differentiate the various CHAs, since treatment may have different efficacy and risks. For instance, splenectomy is generally more effective in membrane defects versus PKD while it is contraindicated in hereditary stomatocytosis for the high thrombotic risk. The clinical suspicion allows to test PK enzymatic activity, particularly in patients without familiarity and unremarkable RBC morphology. Clinicians have to take into account that falsely normal activity may be observed and that the ratio with proximal enzymes activity (ie, hexokinase) may be helpful. The diagnosis is completed by the confirmation of the genetic defect that takes advantages of the use of NGS platforms. The latter may pose several interpretation challenges since genotype/phenotype association is not clearcut and variants of unknown significance continue to emerge. It is also important to reconsider congenital forms (even in adults) after all acquired forms have been excluded and vice versa; and also to take into account the possibility of the coexistence of acquired and congenital forms in the same patient.54 Thereafter, patient management should be individualized considering the different ages, comorbidities, and frequency of complications (gallstone, hemolytic and aplastic crises, and iron overload) (Figure 1B). The need of transfusions, iron chelation, splenectomy, and cholecystectomy will be tuned accordingly. Specifically, transfusions should be triggered by growth retard and symptomatic anemia. Splenectomy is not as effective as in other CHAs, is discouraged during the first 6 years of age, and is usually avoided in elderly comorbid patients for the infectious and thrombotic risks. A further drawback of splenectomy is the poor response observed in subjects with more severe disease and highly disruptive mutations (ie, non missense ones). The allosteric PK stimulator mitapivat is a promising new option with very good responses in the majority of patients; however, responses are generally observed only in patients with at least one missense mutation, whilst those with more severe genotype/phenotype represent an unmet clinical need. Additionally, mitapivat has only been studied in adults and cannot be therefore directly translated into pediatric care. Regarding gene therapy, it might fill the gap in severe cases by substituting the defective enzyme; however, results are very preliminary and there is still uncertainty regarding long-term safety and efficacy. Early evidence seems to favor an early use in pediatric patients, and in young adults not candidates for mitapivat due to unfavorable genotype. On the whole, the timing of patient allocation to splenectomy, mitapivat and gene therapy is another challenge for the treating physician that will require future investigation.
Data Sharing Statement
All data are available within the manuscript and further may be available upon reasonable request to the corresponding author.
Consent for Publication
All Authors approved present submission.
Author Contributions
All authors made a significant contribution to the work reported, in the conception, design, execution, acquisition of data, analysis and interpretation; took part in drafting, revising and critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
No funding sources to declare.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Grace RF, Bianchi P, van Beers EJ., et al. Clinical spectrum of pyruvate kinase deficiency: data from the pyruvate kinase deficiency natural history study. Blood. 2018;131(20):2183–2192.
2. Grace RF, Barcellini W. Management of pyruvate kinase deficiency in children and adults. Blood. 2020;136(11):1241–1249.
3. Grace RF, Mark Layton D, Barcellini W. How we manage patients with pyruvate kinase deficiency. Br J Haematol. 2019;184(5):721–734.
4. Fattizzo B, Giannotta JA, Cecchi N, Barcellini W. Confounding factors in the diagnosis and clinical course of rare congenital hemolytic anemias. Orphanet J Rare Dis. 2021;16(1):415.
5. Grace RF, Rose C, Layton DM, et al. Safety and efficacy of mitapivat in pyruvate kinase deficiency. N Engl J Med. 2019;381(10):933–944.
6. Schwartz JD, Barcellini W, Grace RF, et al. Who should be eligible for gene therapy clinical trials in red blood cell pyruvate kinase deficiency (PKD)?: toward an expanded definition of severe PKD. Am J Hematol. 2022;97(3):E120–E125.
7. Beutler E, Gelbart T. Estimating the prevalence of pyruvate kinase deficiency from the gene frequency in the general white population. Blood. 2000;95(11):3585–3588.
8. Carey PJ, Chandler J, Hendrick A, et al. Northern Region Haematologists Group. Prevalence of pyruvate kinase deficiency in Northern European population in the north of England. Blood. 2000;96(12):4005–4006.
9. Ayi K, Min-Oo G, Serghides L, et al. Pyruvate kinase deficiency and malaria. N Engl J Med. 2008;358(17):1805–1810.
10. Min-Oo G, Fortin A, Tam MF, Nantel A, Stevenson MM, Gros P. Pyruvate kinase deficiency in mice protects against malaria. Nat Genet. 2003;35(4):357–362.
11. Qidwai T, Jamal F, Singh S. Exploring putative molecular mechanisms of human pyruvate kinase enzyme deficiency and its role in resistance against Plasmodium falciparum malaria. Interdiscip Sci. 2014;6(2):158–166.
12. Machado P, Manco L, Gomes C, et al. Pyruvate kinase deficiency in sub-Saharan Africa: identification of a highly frequent missense mutation (G829A;Glu277Lys) and association with malaria. PLoS One. 2012;7(10):e47071.
13. van Bruggen R, Gualtieri C, Iliescu A, et al. Modulation of Malaria Phenotypes by Pyruvate Kinase (PKLR) Variants in a Thai Population. PLoS One. 2015;10(12):e0144555.
14. Zanella A, Fermo E, Bianchi P, Chiarelli LR, Valentini G. Pyruvate kinase deficiency: the genotype-phenotype association. Blood Rev. 2007;21(4):217–231.
15. Zanella A, Bianchi P, Fermo E. Pyruvate kinase deficiency. Haematologica. 2007;92(6):721–723.
16. Grace RF, Zanella A, Neufeld EJ, et al. Erythrocyte pyruvate kinase deficiency: 2015 status report. Am J Hematol. 2015;90(9):825–830.
17. Bianchi P, Fermo E, Lezon-Geyda K, et al. Genotype-phenotype correlation and molecular heterogeneity in pyruvate kinase deficiency. Am J Hematol. 2020;95(5):472–482.
18. Al-Samkari H, van Beers EJ, Morton DH, et al. Characterization of the severe phenotype of pyruvate kinase deficiency. Am J Hematol. 2020. doi:10.1002/ajh.25926
19. Zanella A, Fermo E, Bianchi P, Valentini G. Red cell pyruvate kinase deficiency: molecular and clinical aspects. Br J Haematol. 2005;130(1):11–25.
20. Grace RF, Cohen J, Egan S, et al. The burden of disease in pyruvate kinase deficiency: patients’ perception of the impact on health-related quality of life. Eur J Haematol. 2018;101(6):758–765.
21. Barcellini W, Fattizzo B. Clinical Applications of Hemolytic Markers in the Differential Diagnosis and Management of Hemolytic Anemia. Dis Markers. 2015;2015:635670.
22. van Beers EJ, van Straaten S, Morton DH, et al. Prevalence and management of iron overload in pyruvate kinase deficiency: report from the Pyruvate Kinase Deficiency Natural History Study. Haematologica. 2019;104(2):e51–e53.
23. Beutler E. Red Cell Metabolism. A Manual of Biochemical Methods.
24. Bianchi P, Zanella A. Hematologically important mutations: red cell pyruvate kinase (third update). Blood Cells Mol Dis. 2000;26(1):47–53.
25. Perkins A, Xu X, Higgs DR, et al. KLF1 Consensus Workgroup. Krüppeling erythropoiesis: an unexpected broad spectrum of human red blood cell disorders due to KLF1 variants. Blood. 2016;127(15):1856–1862.
26. Rider NL, Strauss KA, Brown K, et al. Erythrocyte pyruvate kinase deficiency in an old-order Amish cohort: longitudinal risk and disease management. Am J Hematol. 2011;86(10):827–834.
27. Iolascon A, Andolfo I, Barcellini W, et al. Working Study Group on Red Cells and Iron of the EHA. Recommendations regarding splenectomy in hereditary hemolytic anemias. Haematologica. 2017;102(8):1304–1313.
28. Sandoval C, Stringel G, Weisberger J, Jayabose S. Failure of partial splenectomy to ameliorate the anemia of pyruvate kinase deficiency. J Pediatr Surg. 1997;32:641–642.
29. Zahid MF, Bains APS. Rapidly fatal Klebsiella pneumoniae sepsis in a patient with pyruvate kinase deficiency and asplenia. Blood. 2017;130(26):2906.
30. Kristinsson SY, Gridley G, Hoover RN, Check D, Landgren O. Long-term risks after splenectomy among 8149 cancer-free American veterans: a cohort study with up to 27 years follow-up. Haematologica. 2014;99(2):392–398.
31. Lin JN, Chen HJ, Lin MC, et al. Risk of venous thromboembolism in patients with splenic injury and splenectomy. A nationwide cohort study. Thromb Haemost. 2016;115(1):176–183.
32. Chou R, DeLoughery TG. Recurrent thromboembolic disease following splenectomy for pyruvate kinase deficiency. Am J Hematol. 2001;67(3):197–199.
33. Morimoto M, Kanno H, Asai H, et al. Pyruvate kinase deficiency of mice associated with nonspherocytic hemolytic anemia and cure of the anemia by marrow transplantation without host irradiation. Blood. 1995;86(11):4323–4330.
34. Tanphaichitr VS, Suvatte V, Issaragrisil S, et al. Successful bone marrow transplantation in a child with red blood cell pyruvate kinase deficiency. Bone Marrow Transplant. 2000;26(6):689–690.
35. van Straaten S, Bierings M, Bianchi P, et al. Worldwide study of hematopoietic allogeneic stem cell transplantation in pyruvate kinase deficiency. Haematologica. 2018;103(2):e82–e86.
36. Del Giudice EM, Perrotta S, Nobili B, Specchia C. d’Urzo G, Iolascon, A. Coinheritance of Gilbert syndrome increases the risk for developing gallstones in patients with hereditary spherocytosis. Blood. 1999;94:2259–2262.
37. Haverfield EV, McKenzie CA, Forrester T, et al. UGT1A1 variation and gallstone formation in sickle cell disease. Blood. 2005;105:968–972.
38. Plensa E, Tapia G, Juncà J, Pèrez R, Castellà E, Martì S. Paravertebral extramedullary hematopoiesis due to pyruvate kinase deficiency. Haematologica. 2005;90(suppl):ECR32.
39. Rutgers MJ, van der Lugt PJ, van Turnhout JM. Spinal cord compression by extramedullary hemopoietic tissue in pyruvate-kinase-deficiency-caused hemolytic anemia. Neurology. 1979;29(4):510–513.
40. Bachmeyer C, Khalil A, Kerrou K, Girot R, Gounant V. Idiopathic pulmonary arterial hypertension in a patient with pyruvate kinase deficiency and paravertebral extramedullary hematopoiesis. Annals Hematol. 2009;88:603–605.
41. Müller-Soyano A. Tovar de Roura E, Duke PR, et al. Pyruvate kinase deficiency and leg ulcers. Blood. 1976;47(5):807–813.
42. Kung C, Hixon J, Kosinski PA, et al. AG-348 enhances pyruvate kinase activity in red blood cells from patients with pyruvate kinase deficiency. Blood. 2017;130(11):1347–1356.
43. Al-Samkari H, Galactéros F, Glenthøj A, et al. Mitapivat versus Placebo for Pyruvate Kinase Deficiency. N Engl J Med. 2022;386(15):1432–1442.
44. Glenthøj A, van Beers EJ, Al-Samkari H, et al. ACTIVATE-T: a phase 3, open-label, multicenter study of mitapivat in adults with pyruvate kinase deficiency who are regularly transfused. Eur Hematol Assoc. 2021;9:17.
45. Grace RF, Glenthoej A, Barcellini W, et al. Durability of Hemoglobin Response and Reduction in Transfusion Burden Is Maintained over Time in Patients with Pyruvate Kinase Deficiency Treated with Mitapivat in a Long-Term Extension Study. Am Soc Hematol. 2021;138:848.
46. Xu JZ, Conrey A, Frey I, et al. Mitapivat (AG-348) demonstrates safety, tolerability, and improvements in anemia, hemolysis, oxygen affinity, and hemoglobin S polymerization kinetics in adults with sickle cell disease: a Phase 1 dose escalation study. Am Soc Hematol. 2021;138:10.
47. Kuo KH, Layton DM, Lal A, et al. Results from a phase 2, open-label, multicenter study of the oral pyruvate kinase inhibitor mitapivat in adults with non-transfusion-dependent alpha- or beta-thalassemia. Eur Hematol Assoc. 2021;5:92.
48. Brown RCC, Saraf SL, Cruz K, et al. Activation of Pyruvate Kinase-R with Etavopivat (FT-4202) Is Well Tolerated, Improves Anemia, and Decreases Intravascular Hemolysis in Patients with Sickle Cell Disease Treated for up to 12 Weeks. Am Soc Hematol. 2021;138:9.
49. Garcia-Gomez M, Calabria A, Garcia-Bravo M, et al. Safe and efficient gene therapy for pyruvate kinase deficiency. Mol Ther. 2016;24(7):1187–1198.
50. Shah AJ, López Lorenzo JL, Navarro S, et al. Lentiviral mediated gene therapy for pyruvate kinase deficiency: interim results of a global phase 1 study for adult and pediatric patients. Am Soc Hematol. 2021;138:563.
51. Al-Samkari H, van Beers EJ, Morton DH, et al. Health-related quality of life and fatigue in children and adults with pyruvate kinase deficiency. Blood Adv. 2022;6(6):1844–1853.
52. Salek MS, Ionova T, Johns JR, Oliva EN. Appraisal of patient-reported outcome measures in analogous diseases and recommendations for use in Phase II and III clinical trials of pyruvate kinase deficiency. Qual Life Res. 2019;28(2):399–410.
53. Salek S, Boscoe AN, Piantedosi S, et al. Development of the pyruvate kinase deficiency diary and pyruvate kinase deficiency impact assessment: disease-specific assessments. Eur J Haematol. 2020;104(5):427–434.
54. Motta I, Giannotta J, Ferraresi M, et al. Autoimmune hemolytic anemia as a complication of congenital anemias. a case series and review of the literature. J Clin Med. 2021;10(15):3439.
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.