Back to Journals » Journal of Experimental Pharmacology » Volume 17
Pyrazoline B Induces Oxidative Stress Mediated Toxicity, Cell Cycle Arrest, and Caspase-Independent Apoptosis in BT-474 Human Breast Cancer Cells
Authors Wiraswati HL ![]() , Satriyo PB, Bashari MH
, Satriyo PB, Bashari MH ![]() , Sholikhah EN, Wahyuningsih TD, Ma'ruf IF, Yeh CT, Mustofa M
, Sholikhah EN, Wahyuningsih TD, Ma'ruf IF, Yeh CT, Mustofa M
Received 3 October 2024
Accepted for publication 30 August 2025
Published 13 December 2025 Volume 2025:17 Pages 823—839
DOI https://doi.org/10.2147/JEP.S499127
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Abdelwahab Omri
Hesti Lina Wiraswati,1,2 Pamungkas Bagus Satriyo,3 Muhammad Hasan Bashari,1,2 Eti Nurwening Sholikhah,3 Tutik Dwi Wahyuningsih,4 Ilma Fauziah Ma’ruf,5 Chi-Tai Yeh,6,7 Mustofa Mustofa3
1Department of Biomedical Sciences, Faculty of Medicine, Universitas Padjadjaran, Bandung, 40161, Indonesia; 2Oncology and Stem Cells Working Group, Faculty of Medicine, Universitas Padjadjaran, Bandung, Indonesia; 3Department of Pharmacology and Therapy, Faculty of Medicine Public Health and Nursing, Universitas Gadjah Mada, Yogyakarta, Indonesia; 4Department of Chemistry, Faculty of Mathematics and Natural Sciences, Universitas Gadjah Mada, Yogyakarta, Indonesia; 5Research Center for Pharmaceutical Ingredients and Traditional Medicine, National Research and Innovation Agency, Bogor, Indonesia; 6Department of Medical Research & Education, Taipei Medical University-Shuang Ho Hospital, New Taipei City, Taiwan; 7Department of Medical Laboratory Science and Biotechnology, Yuanpei University of Medical Technology, Hsinchu, Taiwan
Correspondence: Mustofa Mustofa, Department of Pharmacology and Therapy, Faculty of Medicine Public Health and Nursing, Universitas Gadjah Mada, Yogyakarta, 55281, Indonesia, Email [email protected]
Introduction: Luminal subtype B breast cancer represents a clinically challenging subtype, accounting for nearly 40% of all breast cancers. However, clinical outcomes remain suboptimal due to challenges such as poor solubility, resistance, and drug-induced toxicity. In our previous work, a synthesized compound pyrazoline B demonstrated potent toxicity effects towards T47D, 4T1, and Hs578T breast cancer cells, WiDr colorectal cancer cells, and HeLa cervical cancer cells. Building on these findings, we now investigate—for the first time—the therapeutic potential of a lead compound, pyrazoline B, against luminal B breast cancer using the clinically relevant BT-474 model (HER2+/ER+). This study systematically evaluates pyrazoline B’s standalone efficacy and preliminary synergistic interactions with paclitaxel, aiming to address current therapeutic gaps in this high-risk subtype.
Methods: Comprehensive in vitro analysis included proliferation and cell migration (scratch) assays, flow cytometry (apoptosis and cell cycle), ELISA (EGFR/VEGFR-2), and RT-qPCR, complemented by in silico ADME and molecular docking analyses.
Results: Pyrazoline B demonstrated multimodal activity, inducing G0/G1 arrest through Cyclin D1 suppression while reduced EGFR and VEGFR-2 proteins level. The compound triggered caspase-independent cell death via oxidative stress. Additionally, pyrazoline B enhances the inhibitory effect of paclitaxel on the proliferation and migration of cancer cells. ADME predictions revealed that pyrazoline B exhibits more favorable pharmacokinetic properties of than paclitaxel.
Discussion: Our findings established pyrazoline B as a first-in-class multi-target agent against BT-474 luminal B breast cancer, uniquely capable of simultaneously disrupting cell cycle progression, growth factor signaling, and redox homeostasis. Pyrazoline B demonstrates strong potential as a monotherapy, and our initial combination screening showed promising boosting effects when used with existing therapies. Future studies should prioritize: mechanistic synergy studies and in vivo validation to assess translational potential.
Keywords: luminal B breast cancer, BT-474, pyrazoline B, anticancer, cell death, oxidative stress, cell cycle arrest
Introduction
Breast cancer is one of the top five causes of death among women worldwide. In 2020, approximately 2,261,419 new breast cancer cases were reported, with 684,996 deaths worldwide.1 The luminal B subtype accounts for 40% of all breast cancers.2 Luminal B constitutes the most heterogeneous molecular subtype and has aggressive clinical and biological features.3 Luminal B breast cancer is defined by the expression of the estrogen receptor (ER+) or progesterone receptor (PR+) with or without human epidermal growth factor receptor 2 (HER2+/HER2-); it is typically graded higher, grows more quickly, and has poorer outcomes than luminal A.4 Moreover, the subtype of cancer also tends to grow more quickly and have a slightly worse prognosis than luminal A.5 Previous reports reveal a higher proportion of local recurrence and metastases in patients with luminal B breast cancer than in patients in the non-luminal group.6
Treatment options for luminal B breast cancer include surgery, chemotherapy, radiation therapy, hormonal therapy, targeted therapy, and immunotherapy.7 While conventional chemotherapy has shown some success, it is still plagued by issues such as poor water solubility, drug-induced toxicity, therapeutic selectivity, and drug resistance, all of which have proven to be major obstacles.8–18 One chemotherapy drug that remains widely administered despite posing such challenges is paclitaxel. Variations in patient response rates to paclitaxel treatment have been widely documented, highlighting the need for the discovery of novel anticancer agents or therapeutic strategies.19,20
Pyrazoline is a heterocyclic compound with two nitrogen atoms in its ring, produced by the condensation of chalcone with phenylhydrazine. These compounds have a variety of promising pharmacological activities, and present opportunities to carry out various structural modifications on the ring.21–23 For example, in oncology, pyrazoline derivatives have been tested against numerous cancer cell lines and have demonstrated anticancer activity, such as inducing apoptosis and inhibiting cell division and proliferation.24–28 Our previous studies report on N-phenyl pyrazoline derivatives exerting anticancer activity that have been successfully synthesized in our lab.29–31 These compounds show biological activity in cervical cancer cells, including induction of poor survival, reduction of the HeLa’s tumorsphere size, inhibition of cancer cell growth, and reduction of CD133, the marker of cancer stem cells.32,33 In addition, we have shown that an N-phenyl pyrazoline derivates reduces the level of epidermal growth factor receptor (EGFR), a protein that mediates the processes of cell proliferation, differentiation, survival, metastasis, and apoptosis in cervical cancer cells.32–34 We also monitored the inhibitory activity of these compounds in T47D luminal A breast cancer cells, 4T1 and Hs578T triple-negative breast cancer (TNBC) cells, HeLa cervical cancer cells, and in WiDr colorectal cancer cells.34,35 Furthermore, our recent study reported their capacity to improve the sensitivity of paclitaxel in TNBC cells.35 The N-phenyl pyrazoline derivate is therefore a promising anticancer drug and has the potential to be combined with others to increase their efficacy. Since luminal B breast cancers tend to grow faster than luminal A, have a higher proportion of local recurrence and metastases, and have a slightly worse prognosis, we selected BT-474 cell lines representing this cancer subtype as a model to test N-phenyl pyrazoline treatment.
Materials and Methods
Materials
Chemicals were as follows: Paclitaxel (Taxol, PCXL; SRL, India), 2-(4-amidinophenyl)-1H -indole-6-carboxamidine (DAPI; Sigma Aldrich, USA), (methoxyphenyl)-1-phenyl-3-(thiophen-2-yl)-4,5-dihydro-1H-pyrazole (pyrazoline B, pyra B; laboratory of Department of Chemistry-Universitas Gadjah Mada, Indonesia) was synthesized and characterized as previously described and characterized by 1H/13C Nuclear magnetic resonance (NMR), Fourier Transform IR Spectroscopy (FTIR), and Gas chromatography-mass spectrometry (GC-MS) (citation 39), Propidium Iodide (PI; Sigma Aldrich), 2-Methyl-1,4-naphthoquinone (menadione; Sigma Aldrich, USA), N-Acetyl-L-cysteine (NAC; Sigma Aldrich, USA), Pan Caspase Inhibitor (Z-VAD.fmk; Sigma Aldrich, USA), Dimethyl sulfoxide (DMSO; D8418, Sigma Aldrich, USA).
Cell Culture
Human breast cancer BT-474 cell line (ATCC, #HTB-20, USA) represent as luminal B subtype, were cultured in RPMI 1640 medium (SA R8758, Merck, Germany) supplemented with 10% heat-inactivated Fetal Bovine Serum (FBS; F2442, Merck), and 1% Penicillin-Streptomycin (PS; P4458, Merck, Germany) at 37 °C and 5% CO2 atmosphere. For toxicity treatment, cells were incubated with pyrazoline B 140 µM, pyrazoline B (140 µM) combined with paclitaxel (10 µM), pyrazoline B (140 µM) combined with Z.VAD (1 µM) for 24h. DMSO, paclitaxel, and Z-VAD.fmk were used as control. For antioxidant treatment, cells were incubated with prazoline B (140 µM), pyrazoline B (140 µM) combined with antioxidant NAC (5mM) for 24h. Stress agent menadione, the solvent DMSO, and antioxidant NAC were used as control.
Proliferation Assay
Cell proliferation was conducted using 3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide (MTT) assay. First, 1×104 cells were plated in each well of a 96-well plate, then treated with pyrazoline B at different concentrations and incubated for 24h. Next, MTT reagent (M2128; Sigma Aldrich) was added to each well. Incubation was continued for 4h. Once the formazan crystals are formed, they are dissolved in DMSO. The quantity of formazan crystals was determined at 550 nm using a plate reader (Thermo Scientific® Multiscan EX, Singapore).
Clonogenic Assay
For clonogenic assay, BT-474 cells were treated complete medium with pyrazoline B (140 µM), Paclitaxel (50 µM), and co-treatment cell with pyrazoline B (140 µM)-Paclitaxel (50 µM) for 24h incubation. The colony size was observed immediately after 24h incubation, using inverted microscope and was quantified using ImageJ software.36
Cell Migration (Scratch) Assay
Cells were grown in 24-wells plate up to 90% confluency. After the culture medium was discarded, a scratch was made on the monolayer of cells using a p20 pipette tip. The plates were rinsed with Phosphate Buffered Saline (PBS; Gibco) and then incubated with prepared medium containing pyrazoline B (140 µM), Paclitaxel (50 µM), or pyrazoline B-Paclitaxel combination. Medium with DMSO was used as a negative control. Cells were incubated for 24 h and 72 h. The cell migration was observed using an inverted microscope (40x magnification). Cell free area were quantified using ImageJ software.36
Flow Cytometry and Microscopy Assessment
For cell death analysis, PI for the membrane permeabilization and fluorescein-labeled Annexin V (Invitrogen) for the phosphatidylserine exposure were used for cytofluorometric assessment. We also showed a forward angle light scatter (FSC) channel for cell size measurement. In the gating technique, cells were selected using FSC versus side scatter (SSC) to select the cell population. Then, the cells were further gated on live cells versus cell size (PI/FSC) or live cells versus phosphatidylserine exposure (PI/Annexin V). Microscopic analysis is also conducted on cells to assess the presence of morphological changes that serve as indicators of apoptosis, such as cell rounding, membrane blebbing, nuclear condensation, and the formation of apoptotic bodies. The morphology of cells was observed using inverted microscopy (Agilent). Olympus fluorescence microscope BX51 was used to observe nuclear condensation using DAPI staining. The microscope uses a camera connected to a computer and Toupview Software (version x64, 3.7.7892) with 100x magnifications. For cell cycle analysis, the quantitation of DNA content was measured by PI fluorochrome. The cells were fixed with ethanol before staining. In the machine, cell debris was excluded by forward scatter (FSC) and side scatters (SSC). Cytofluorimetric analysis was realized using BD FACSLyric™ Flow Cytometry System (BD Biosciences) equipped with FlowJo™ software (version 10.8; BD Biosciences).
Enzyme-Linked Immunosorbent Assay (ELISA)
Briefly, 40,000 BT-474 cells (in 24 wells plate) were pre-treated with pyrazoline B for 24 h at 37 °C. Cells were then lysed in Radio-Immunoprecipitation Assay Buffer (RIPA; Sigma Aldrich). The expression levels of EGFR or VEGFR-2 proteins of the protein lysates were measured by Human Epidermal Growth Factor Receptor ELISA Kit (EGFR; Cloud-clone Corp, USA) or Human Vascular Endothelial Growth Factor Receptor 2 ELISA Kit (VEGFR-2/KDR; Elabscience, USA) according to the manufacturer’s instructions.
Reverse Transcription Polymerase Chain Reaction (RT-PCR) Analysis
After treatment with pyrazoline B 140 µM, total RNA was isolated from the cells using Qiagen RNeasy Mini Kit (Qiagen, Valencia, CA, USA). Then, mRNA expression was determined by real-time PCR using SensiFAST™ SYBR® No-ROX One-Step Kit (Meridian Biosciences) and specific sequence primer in Agilent AriaMX PCR System (Agilent Technologies, USA). The primers were synthesized by Integrated DNA Technologies (IDT; Singapore). The nucleotide sequences (5′ to 3′) of the forward primers and reverse primers are described in Table 1. The PCR conditions were as follows: 45 °C for 10 min for reverse transcription; 95 °C for 2 min; 40 cycles of 95 °C for 5s, and 60 °C for 20s. Data were analyzed by the 2-ΔΔCT method, using the house-keeping gene β–Actin as the internal control.
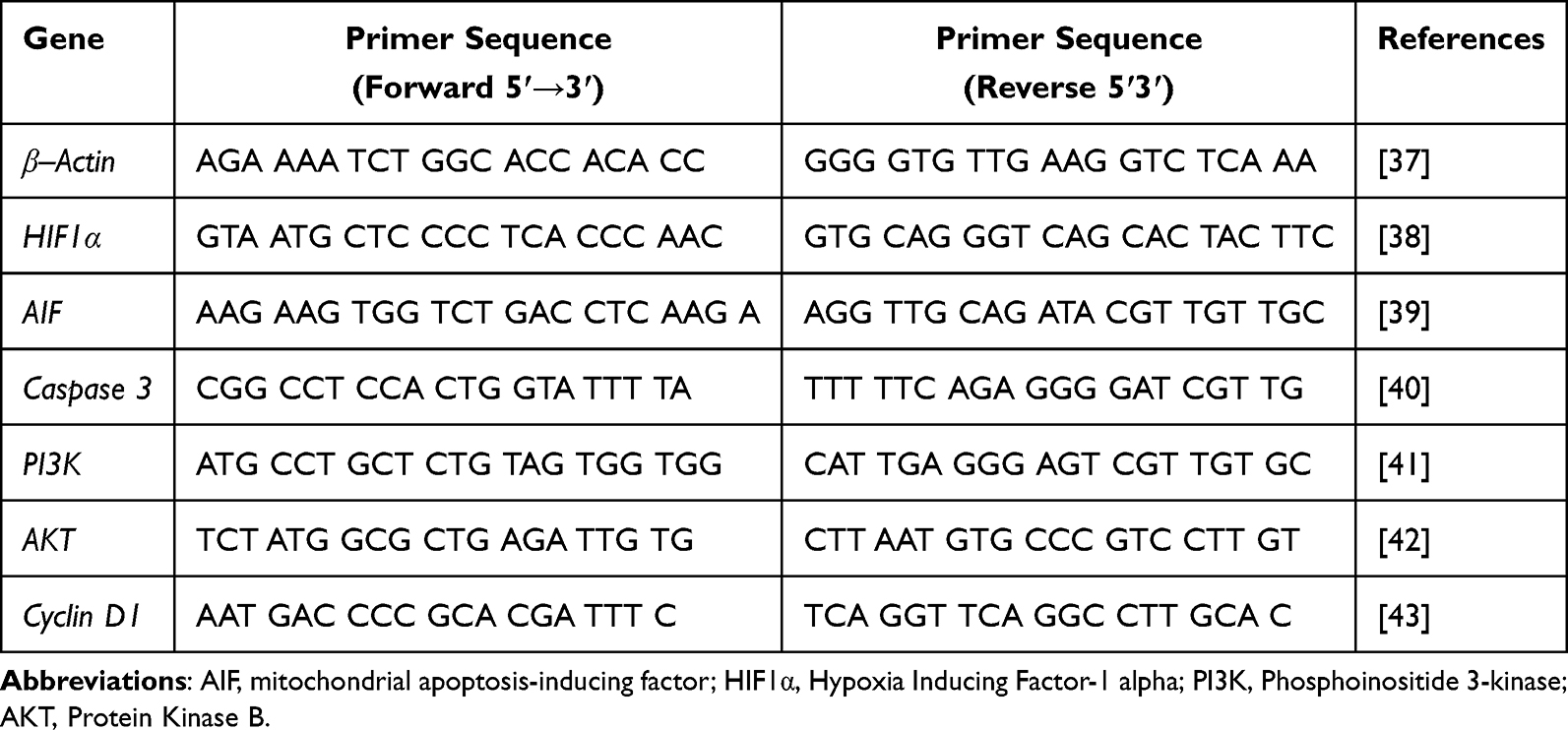 |
Table 1 Oligonucleotide Primers |
Statistical Analysis
All assays were conducted in triplicate on two or three dependent experiments, and the results were presented as the mean ± standard deviation. The ImageJ version 1.51j8 software was utilized for quantifying the data from the migration assay and colony formation assays. T For statistical significance, p-value calculations were carried out using the t-test tool in Microsoft Excel.
Molecular Docking Study and Absorption, Distribution, Metabolism and Excretion (ADME) Analysis
Protein structure was downloaded from Protein Data Bank (PDB).44 Ligands were built using MarvinSketch (http://www.chemaxon.com) or downloaded from PDB.44 Molecular docking was performed using UCSF Chimera45 and Autodock Vina.46,47 Gridbox of Flavin Adenine Dinucleotida (FAD) were set as follows: center_x = −21.00, center_y = 35.00, center_z = −24.00, size_x = 15.00, size_y = 25.00, size_z = 20.00, Gridbox of Nicotinamide Adenine Dinucleotide (NAD) were set as follows: center_x = −33.00, center_y = 31.00, center_z = −20.00, size_x = 18.00, size_y = 20.00, size_z = 20.00. Gridbox of cyp3a4 were set as follows: center_x= −15.00, center_y=−11.00, center_z=27.00, size_x=15.00, size_y=15.00, size_z=15.00. Protein and ligand visualization were performed using UCSF chimera45 and ligplot plus.48 ADME analysis was conducted using SWISSADME webserver, a web-based tool that is designed to evaluate the pharmacokinetics, drug-likeness, and medicinal chemistry properties of small molecules.49
Results
Pyrazoline B Inhibits Cell Viability of BT-474 Luminal B Breast Cancer Cells
To investigate the antitumor activity of pyrazoline B, we induced BT-474 cells with various concentrations of the compound, from 7 µM to 4200 µM. The results showed a tendency to decrease cell viability, equivalent to the increasing dose of pyrazoline B (Figure 1). Cell viability was measured by MTT assay, resulting in an IC50 value of 140 µM for 24h incubation. This suggests that pyrazoline B possesses strong activity against BT-474 cells.
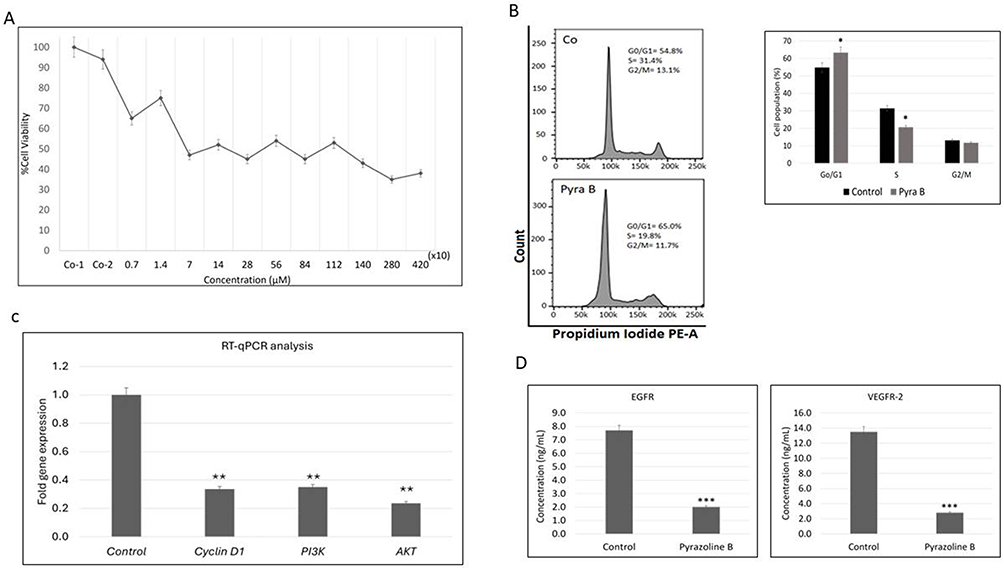 |
Figure 1 Pyrazoline B inhibits BT-474 cell growth. (A) BT-474 cells were cultured for 24h in complete medium with pyrazoline B 7, 14, 70, 140, 280, 560, 840, 1120, 1400, 2800, and 4200 µM, the medium alone (Co-1), or the solvent used to dissolve the compound (or Co-2) for 24h incubation. Cell viability was then assessed by MTT assay and expressed as a percentage of untreated cells. Values represent the mean ±SD of three independent experiments. (B) BT-474 cells were treated with pyrazoline B 140 µM or the solvent (control) for 24h. Flow cytometry was used to assay the cell cycle distribution using PI fluorescence staining. (C) Cells were treated with 140 µM pyrazoline B or solvent (control) for 24h. Total RNA was isolated from the cells using Qiagen RNeasy Mini Kit, then mRNA expression of PI3K, AKT, or Cyclin D1 was determined by real-time PCR in Agilent AriaMX PCR System. (D) Cells were treated with pyrazoline B 140 µM for 24h. The EGFR or VEGFR-2 ELISA kit detected and quantified endogenous EGFR or VEGFR-2 protein levels in the lysate sample. Two independent experiments were carried out in triplicate. Significantly different compared to the control (* p<0.05; **p < 0.01; ***p < 0.001). |
Pyrazoline B Induces G0/G1 Cell Cycle Arrest and Modulates Key Signaling Pathways in BT-474 Cells
After showing the toxicity of pyrazoline B towards cancer cells, we investigated it’s impact on cell cycle progression. First, flow cytometry analysis cell cycle progression of BT-474 cells treated for 24h with 140 µM pyrazoline B showed significant accumulation in the G0/G1 phase from 54.8% in controls to 65% in cells treated by pyrazoline B, suggesting cell cycle arrest (Figure 1B). This observation prompted us to examine Cyclin D1, a critical regulator of G1-S transition. RT-qPCR analysis confirmed downregulation of Cyclin D1 at the transcriptional level (Figure 1C). We next explored upstream regulators of cell cycle control by analyzing the PI3K/AKT pathway, a known mediator of Cyclin D1 expression. Pyrazoline B treatment significantly reduced transcript levels of both PI3K and AKT (Figure 1C), indicating suppression of this crucial survival pathway. Since PI3K/AKT signaling is frequently activated by receptor tyrosine kinases, we measured the protein levels of EGFR and VEGFR-2 using ELISA. ELISA demonstrated decreased expression of both receptors in treated cells (Figure 1D). Overexpression of EGFR or vascular endothelial growth factor receptor 2 (VEGFR-2) has been reported to lead to dysregulation of crucial proteins for cell proliferation, survival, angiogenesis, and metastasis.50,51 This coordinated inhibition of surface receptors (EGFR/VEGFR-2), their downstream effectors (PI3K/AKT), and cell cycle regulators (Cyclin D1) provides a comprehensive mechanistic explanation for pyrazoline B’s anti-proliferative effects.
Pyrazoline B Alone and in Combination with Paclitaxel Inhibits BT-474 Cell Proliferation and Migration
We next evaluated pyrazoline B’s functional effects on cancer cell behaviors. We evaluated the effect of pyrazoline B on BT-474 cell proliferation and migration. The 140 µM doses of pyrazoline B treatment suppressed the size of the colonies relative to the control (Figure 2A and B). Then, to investigate the anti-migratory effect of Pyrazoline B, we applied a wound healing assay on BT-474 cells. The result showed that using 140 µM pyrazoline B suppressed the migration of BT-474 cells (Figure 2C and D). This suppressive effect was seen for up to 72 hours of pyrazoline B incubation. These findings align with and extend our mechanistic data showing pyrazoline B’s ability to induce G0/G1 arrest, downregulate growth factor receptors, and trigger oxidative stress. As a secondary exploration of therapeutic potential, we included preliminary combination studies with paclitaxel. While not the focus of this mechanistic investigation, these assays revealed that pyrazoline B produced comparable anti-proliferative (Figure 2A and B) and anti-migratory (Figure 2C and D) effects to those of paclitaxel monotherapy and demonstrated additive effects when combined with paclitaxel (Figure 2A–D).
 |
Figure 2 Pyrazoline B inhibits proliferation and migration of BT-474 cell line. (A and B) Pyrazoline B inhibits cell proliferation of breast cancer cells. The colony formation assay results revealed the anti-proliferative effect of pyrazoline B in BT-474 cell line. The size of BT-474 cell colonies was decreased induced by pyrazoline B. BT-474 cells were treated for 24h in a complete medium with pyrazoline B 140 µM, paclitaxel 50 µM, 140 µM pyrazoline B-50 µM Paclitaxel combination, or controls, Co-1 (solvent) or Co-2 (medium alone). The solvent control in (B) presented corresponds to Co-1 in (A). The size of colony cells was then observed under an inverted microscope. Results expressed as mean ± SD of assays done at least in triplicate. (C and D) In vitro, scratch wound healing assays showed that the cell migration ability was suppressed by pyrazoline B in a time-dependent manner. BT-474 cells were cultured for 24 and 72h in a complete medium with pyrazoline B 140 µM or solvent (control). Cell migration was then observed under an inverted microscope (40x magnification). (Cancer cell colony area and cell-free area in the scratch assay were quantified using ImageJ software). Three independent experiments were carried out in triplicate. Significantly different to control (*p < 0.05, **p < 0.01). |
Pyrazoline B Induces Apoptosis in a Caspase-Independent Manner
We have previously reported that pyrazoline B induces apoptosis in cervical cancer cells. Here, we wanted to determine whether pyrazoline B causes a similar response in breast cancer cell lines, relating especially to its role in growth inhibition. BT-474 cells were treated with 140 µM pyrazoline B. Apoptotic events were evaluated by microscopy and flow cytometry analysis after 24h incubation with Pyrazoline B. Microscopic observation showed that pyrazoline B causes morphological changes typical of apoptosis, such as cell rounding, apoptotic formation, and nuclear condensation (Figure 3A and B). A decrease in cell size due to shrinkage was also observed by flow cytometry (Figure 3C). Further analysis using PI-annexin V staining showed consistent results. The number of PI-positive cells was increased after pyrazoline B treatment (PI+FSC-, 15.6%) compared to control (PI+FSC-, 2.67%) (Figure 3C). Apoptosis-related biochemical events were determined by evaluating phosphatidylserine exposure using Annexin V staining. The results showed that Annexin V-positive cells increased by 19.9% in pyrazoline B-treated PI-positive cells, compared to 4.5% in controls (Figure 3C). To explore the apoptotic mechanisms of pyrazoline-induced cell death, we next evaluated the response of cells to pyrazoline B induction in the presence of the general caspase inhibitor Z-VAD-FMK. Results showed that Z-VAD-FMK did not reduce the number of PI/Annexin V-positive cells (Figure 3C and D). This indicates that pyrazoline B induces apoptosis in a caspase-independent manner. We then analyzed the expression levels of the caspase 3 and AIF genes. Figure 3E shows that pyrazoline B induces decreased expression of the caspase 3 gene. As depicted in Figure 3E and F, pyrazoline B was found to cause a decline in caspase 3 gene expression, while simultaneously increasing the expression of AIF. These findings offer further evidence for our hypothesis that pyrazoline B triggers BT-474 cell death in caspase-independent manner.
 |
Figure 3 Pyrazoline B induced apoptosis in BT-474 cell lines. (A) Pyrazoline B induced morphological changes typical apoptosis. Cells were treated with 140 µM pyrazoline B or control for 24h. The cell response was observed using an inverted microscope. Yellow arrow indicated round up of the cells, red arrow indicated apoptotic bodies formation, blue arrow indicated membrane blebbing. (B) We were doing fixation on treated cells or control with EtOH. Then, fixed cells were stained with nuclear staining DAPI and were observed using a fluorescence microscope. Red arrow indicated nuclear condensation. (C) Cells were treated with 140 µM pyrazoline B, 1µM Zvad.fmk+ 140 µM pyrazoline B, and 10 μM Paclitaxel+ 140 µM pyrazoline B for 24h. Cells with medium, Zvad.fmk or solvent were used as controls. Drug-induced cell death was quantified by flow cytometry of PI uptake (PI positivity), Phosphatidyl serine exposure (Annexin V positivity), and reduced cell size analysis (low forward light scatter (FSC)). Data are expressed as mean values ± SEM. (D) Further flow cytometry analysis is shown in a graph containing all cells with apoptotic features. The shrinkage cell number represents shrinkage cells with PI-positive and PI-negative (FSC-PI- and FSC-PI+). Phosphatidylserine exposure cells represent all cells with exposed phosphatidylserine in PI-positive and PI-negative cells (AnV+PI− and AnV+PI+). (E and F) Relative mRNA expression of apoptosis-related genes of Caspase-3 or AIF was determined. The expression of apoptosis-related genes in BT-474 was determined using real-time PCR. The graphs indicate the relative mRNA expression in BT-474 treated with pyrazoline B for 24 h. β-actin was used as a control. Two independent experiments were carried out in triplicate. ** Significantly different compared to the control (p < 0.01). |
Pyrazoline B Inhibits Cell Growth and Induces Apoptosis Mediated by Intracellular Oxidative Stress
We have previously indicated that pyrazoline B has a good affinity with enzymes that regulate oxidative stress, such as P450 and mitochondrial apoptosis-inducing factor (AIF). This cellular stress is often associated with inducing apoptosis or inhibiting cell growth. In this study, we evaluated the involvement of pyrazoline B in oxidative stress regulation in BT-474 cells. Incubation of cells with 140 µM pyrazoline B or 10 μM menadione (positive control) for 24h resulted in a significant decrease in cell viability. The addition of extracellular antioxidants (NAC) to pyrazoline B-treated cells abolished cell viability (Figure 4A). In addition, the occurrence of oxidative stress was confirmed by increased expression of the (Hypoxia Inducing Factor-1 alpha) HIF1α gene in cells treated with pyrazoline B (Figure 4B).
 |
Figure 4 Pyrazoline B induced oxidative stress in BT-474 cells. (A) Cells were treated with 140 µM pyrazoline B for 24h in the absence or presence of exogenous antioxidant NAC (5mM). Percentage of cell viability was determined by MTT assay. * Significantly different compared to the control (p < 0.05). #Significantly different compared to 10 ug/mL pyrazoline B (p < 0.05). (B) Cells were treated with 140 µM pyrazoline B or solvent (control) for 24h. Total RNA was isolated from the cells using Qiagen RNeasy Mini Kit, then mRNA expression of HIF1α was determined by real-time PCR in Agilent AriaMX PCR System. Two independent experiments were carried out in triplicate. ** Significantly different compared to the control (p < 0.01). |
We then performed an in silico study to look at the affinity of pyrazoline B for AIF, a mitochondrial protein reported to mediate lethal redox stress and contribute to caspase-independent cell death. The results show that pyrazoline B had greater affinity than the positive control menadione, and slightly lower than another control, thiodione (Table 2). These control compounds were reported by in vitro and in vivo studies to induce caspase-independent cancer cell death.52,53
 |
Table 2 Affinity Energy of Ligands on AIF |
The visual interaction of pyrazoline B and AIF on FAD and NAD binding sites is presented in Figure 5A and B, respectively. More detailed interaction in Figure 5C shows that pyrazoline B forms two hydrogen bonds with Arg284 and Glu452 on the FAD active site of AIF, the same contact residues as AIF-FAD and AIF-thiodione interactions. This is interesting, considering that thiodione is a menadione-conjugate compound reported to be mediated by AIF to play its lethal function in a caspase-independent manner.52,53 In general, all ligands tended towards higher affinity for FAD than NAD binding sites, consistent with previous reports.54 Pyrazoline B interacts using van der Waals bonds on the NAD binding domain of AIF (Figure 5D). The binding of pyrazoline B at the NAD binding site of AIF confirms the role of AIF in facilitating lethal redox cycling, resulting in antioxidant depletion observed in this study.
 |
Figure 5 Molecular docking of pyrazoline B on AIF. (A) Visual interaction of pyrazoline B and AIF on the FAD binding site and (B) on the NAD binding site. FAD (red), pyrazoline B (green), menadione (yellow) and thiodione (blue) visualization on AIF. (C) Contact residues of pyrazoline B on the FAD binding domain of AIF and (D) on the NAD binding domain of AIF. |
Cheminformatic Analysis to Predict Bioavailability of Pyrazoline B
CYP3A4 is a protein present in the small intestine and liver as a barrier against xenobiotics. CYP3A4 substrates show low bioavailability, as such compounds undergo metabolism in the small intestine or liver.55 Paclitaxel has been known to have low oral bioavailability due to its metabolism by Cyp3a4.56 Our in silico analysis also revealed that paclitaxel has a strong affinity against CYP3A4 (−10.1 kcal/mol) (Table 3) involving Arg105 and Arg372 via hydrogen bonding (Figures 6B and 7B). This affinity is higher when compared to erythromycin against CYP3A4 (−8.8 kcal/mol), which involves one hydrogen bond (Ser119) (Figures 6A and 7A). On the other hand, pyrazoline B exerts low affinity toward CYP3A4 (−7.8 kcal/mol) (Table 3) without hydrogen binding (Figures 6C and 7C). Furthermore, absorption, distribution, metabolism, excretion, and toxicity (ADMET) analysis revealed that pyrazoline B (Log S ESOL −5.33) has a higher solubility than paclitaxel (Log S ESOL −6.66) (Figure 8). Therefore, pyrazoline B might show higher oral bioavailability than paclitaxel in future in vivo and clinical trials, since the compound does not appear to be a substrate of CYP3A4 and has a higher solubility.
 |
Table 3 Binding Affinity of Erythromycine (Native Ligand, Pyrazoline B and Paclitaxel Against cyp3a4 |
 |
Figure 6 Molecular docking result of Paclitaxel and pyrazoline B on Cyp3a4 (grey), visualized in the presence of heme fragment (grey stick). (A) Interaction of Cyp3a4 (grey) and erythromycin (yellow), (B) Interaction of Cyp3a4 (grey) and paclitaxel (red), (C) Interaction of Cyp3a4 (grey) and pyrazoline B (green). |
 |
Figure 7 Binding visualization of paclitaxel and pyrazoline B on Cyp3a4. (A) Contact residues of Cyp3a4 on Eryhthromycin’s interaction (native ligand), (B) Contact residues of Cyp3a4 on paclitaxel’s interaction, and (C) Contact residues of Cyp3a4 on pyrazoline B’s interaction. |
 |
Figure 8 Drug-likeness prediction of (A) pyrazoline B and (B) paclitaxel. |
Discussion
Although chemotherapy is widely regarded as a promising strategy for fighting cancer, challenges such as poor bioavailability and low efficacy necessitate an intensive search for substances that can inhibit cell growth and induce cell death. In the present study, we demonstrate the effectiveness of pyrazoline B against BT-474 luminal B breast cancer cells, a finding that has not been reported before. Our results expand the range of known targets for the anticancer activity of pyrazoline B, having previously reported its toxicity towards T47D luminal A breast cancer cells, 4T1 TNBC mouse cells, Hs578T TNBC cells, HeLa cervical cancer cells, and WiDr colorectal cancer cells.34,35 Key findings include induction of G0/G1 cell cycle arrest, inhibition of cell migration and colony formation, and downregulation of critical signaling pathways. Pyrazoline B’s ability to downregulate EGFR is particularly noteworthy, as cancer cells are more sensitive to EGFR downregulation than to direct inhibition of kinase activity.57 This aligns gains additional relevance from recent evidence that EGFR inhibitors are effective in treating various types of cancer, including breast cancer;58 yet, EGFR’s kinase-independent (KID) pro-survival function often drives therapeutic resistance.59–61 While our in silico study suggests pyrazoline B-EGFR affinity,62 future studies should clarify whether pyrazoline B affects the kinase-dependent or -independent activity of EGFR through enzyme inhibition assays.
The observed VEGFR-2 protein regulation adds clinical relevance, given VEGFR-2’s established role in angiogenesis protein63 and its Food and Drug Administration (FDA)-approved inhibitor for multiple types of cancers including, colorectal cancer, renal cell carcinoma, gastrointestinal stromal tumor, differentiated thyroid carcinoma, idiopathic pulmonary fibrosis, hepatocellular carcinoma, soft tissue sarcoma, and progressive neuroendocrine tumors.58,64–66 Although VEGFR-2 inhibitors are still being explored for their potential in treating breast cancer, overexpression of the VEGFR-2 protein has been detected in early and metastatic stages of primary breast cancer.64–66 Moreover, the overexpression of EGFR and VEGFR-2 has been linked to drug resistance and poor prognosis.67,68 The coordinated suppression of EGFR/VEGFR-2 proteins and their downstream PI3K, AKT, and Cyclin D1, suggest that pyrazoline B may disrupt critical crosstalk between these pathways,69,70 potentially improving clinical outcomes for breast cancer patients.69–72 These findings represent an interesting strategy, as many anticancer drugs have been clinically approved for EGFR and VEGFR-2 activity, such as lapatinib, sunitinib, erlotinib, and sorafenib.65,73
Inducing cancer cell death is believed to prevent recurrence and metastasis.74 Currently, chemotherapy drugs including daunorubicin, doxorubicin, and mitomycin C are known to induce cell death.74–76 Many clinical trials today focus on inducing cell death as a means of treating cancer.74 Therefore, drugs that can induce cell death simultaneously with growth/proliferation inhibition are of great interest for fighting cancer cells with a dual action. This study demonstrates that pyrazoline B can induce cell death in BT-474 cells. Microscopic and flow cytometry analysis revealed that pyrazoline B-treated cancer cells undergo apoptosis, characterized by rounding of cells, cell size reduction, apoptotic body formation, and increased number of PI-Annexin V-positive cells compared to controls. We also analyzed caspase dependency in this process. The addition of the general caspase inhibitor Z-VAD-FMK did not restore cell viability, confirming that cells underwent apoptosis in a caspase-independent manner. Caspase independence was also demonstrated through the downregulation of the downstream caspase 3 and the upregulation AIF target genes. While caspase 3 downregulation might superficially suggest reduced apoptotic capacity, the concomitant AIF activation and maintained cell death confirm engagement of alternative termination pathways. This “apoptosis bypass” phenomenon has been documented with other redox-active compounds53,77,78 and may represent an advantage for overcoming caspase-dependent resistance mechanisms. To further our understanding of this pathway, additional studies should be conducted to identify key proteins; this could involve knockdown experiments of AIF or Endonuclease G (EndoG), a protein involved in a caspase-independent mode of apoptosis. These results align with several studies evaluating drug candidates with dual actions, both inducing cell death and inhibiting EGFR/VEGFR-2.72,79 Pyrazoline derivatives have also been previously reported to exhibit these two actions.80
Pyrazoline B-induced intrinsic pathway apoptosis then led us to evaluate the involvement of oxidative stress in this situation. Intrinsic pathways characterized by mitochondrial involvement may be associated with redox cycling or thiol depletion, resulting in the collapse of antioxidant defenses.53,81 It is worth noting that oxidative stress followed by apoptosis is a common feature of chemotherapeutic agents.82 Our study demonstrates that the cytotoxic activity of pyrazoline B was completely abolished by cell supplementation with the antioxidant N-acetyl cysteine. Pyrazoline B also provoked a decrease in cell viability, as shown in comparison with menadione, a stress agent used as a positive control. The increased expression of the HIF-1α gene also confirmed a hypoxic condition. Our findings indicate that the apoptotic pathway and oxidative stress are linked to the inhibition of cell growth and migration. This is consistent with other studies that link the activity of pyrazoline derivatives to the production of reactive oxygen species (ROS), leading to decreased cell proliferation and survival, and inducing cell death.83,84 Together, these findings indicate that pyrazoline B is an excellent anticancer agent against BT-474 cell lines since it inhibits cell growth and proliferation, induces apoptosis, and induces oxidative stress. Since there are two kinds of luminal B cells (Her2+ and Her2-)85 we suggest that future research explore the anticancer activity of pyrazoline B on another subtype of luminal B cancer cell lines. Additionally, future research should include genetic manipulation, such as knockdown studies, to identify the key proteins that interact with pyrazoline B directly.
Considering the toxicity potential of pyrazoline B, we conducted a simulation of its interaction with the CYP3A4 protein, part of the barrier against xenobiotics in the small intestine and liver. Our results indicated a CYP3A4 preference for metabolizing paclitaxel over pyrazoline B, leading to better bioavailability of pyrazoline B. This may be attributable to the structural properties of pyrazoline B, which has a higher solubility than paclitaxel. Structural components of paclitaxel, including its benzoyl group on the amide side chain and its acetyl group, contribute to its low water solubility and have been reported to cause adverse effects in vivo.86 These in silico predictions, while requiring experimental validation, provide mechanistic context for pyrazoline B’s observed effects and prioritize targets for future investigation. This study establishes a foundation for subsequent experimental validation, which we are currently pursuing through AIF knockout/knockdown studies, measurement of redox cycling in purified systems, and in vivo pharmacokinetic analyses.
Anticancer drug exploration against luminal B cancer is a matter of great importance. This subtype of cancer exhibits significant molecular diversity, including variations in clinical behavior and treatment response.87 The presented results demonstrate that pyrazoline B exerts multi-faceted anticancer activity against BT-474 luminal B cells through several distinct mechanisms: inducing of cell death, inhibition of proliferation and migration, and downregulation of EGFR/VEGFR-2 signaling pathways under hypoxic conditions. Interestingly, although not statistically significant, our preliminary data suggest enhanced anti-migratory and anti-proliferative effects when pyrazoline B is combined with paclitaxel than a single regimen.
At the molecular level, pyrazoline B uniquely induces G0/G1 cell cycle arrest, contrasting with paclitaxel’s established G2/M phase blockade.88,89 This differential cell cycle targeting may underlie their observed synergy- a novel finding that, to our knowledge, has not been previously reported. The mechanistic basis for this synergy warrants further investigation, particularly in light of existing combination therapies. For example, while metformin-paclitaxel combination therapy leads to G2 arrest, metformin alone causes cell accumulation at the G1 phase.90 In contrast, co-treatment of ramucirumab and paclitaxel provokes cell cycle arrest at the G0/G1 phase, while ramucirumab alone provokes G0/G1 phase arrest and is ineffective in inhibiting the G2/M phase. Together with paclitaxel, ramucirumab significantly decreased the expression of important complexes for the G2/M phase, such as cdc25A, cdc2, and Cyclin B1.91 These precedents suggest pyrazoline B might similarly influence cyclin D proteins or G2/M regulators when combined with paclitaxel, potentially explaining our observed effects on cell proliferation.
Thus, further research is necessary focusing on monitoring the effects of pyrazoline B-paclitaxel combination therapy on the cell cycle, including investigating the expression of essential proteins in the G0/G1 or G2/M phases and P13K/Akt pathway. Again, we strongly suggest the study of other luminal B cell lines to test pyrazoline B’s wider potency. In addition, further research to monitor the effect of pyrazoline B on potentiating paclitaxel activity in inducing apoptosis of cancer cells would be of interest, given that researchers have reported compounds with dual activity in potentiating paclitaxel.92,93
The differential sensitivity across breast cancer subtypes is particularly interesting. Pyrazoline B works demonstrates about 4 times more potent against luminal A (T47D; IC50 37.78 µM) and 7.5-fold greater potency in TNBC cells (Hs578T; IC50 18.62 µM)34 compared to our luminal B model. This is not surprising when we consider how these cancers behave differently. While luminal cancers typically depend on hormonal signaling pathways, TNBC cells lack these receptors and may be more vulnerable to pyrazoline B’s oxidative stress mechanisms. The intermediate sensitivity in BT-474 cells (HER2+/ER+) suggests that HER2 amplification and growth factor receptor overexpression may activate compensatory survival pathways that require higher drug concentrations.
These findings contribute to growing efforts to enhance the efficacy of paclitaxel through combining it with new drug candidates86,92,94 by targeting multiple cancer hallmarks simultaneously. Such approaches may improve therapeutic outcomes while reducing dose-related toxicity.95,96 Notably, even at higher concentrations, pyrazoline B maintains selectivity over Vero normal kidney cells (IC50 = 489.18 µM)34 reveal a favorable 3.5-fold selectivity window for cancer cells, suggesting potential clinical applicability and its synergy with paclitaxel may offset the limitations of monotherapy potency. Future studies should prioritize comparative analyses across subtypes under identical experimental conditions to optimize subtype-specific dosing strategies, in vivo validation of combination efficacy, and comprehensive toxicity profiling to advance this compound toward clinical application.
Conclusion
This study shows that pyrazoline B exerts multi-modal anticancer activity against BT-474 luminal B breast cancer through inducing G0/G1 phase cell cycle arrest, downregulating EGFR and VEGFR-2 proteins, inhibiting PI3K/AKT/Cyclin D1 signaling pathway, inducing caspase-independent apoptosis via oxidative stress, and inhibiting cell proliferation and migration. Our preliminary combination screening revealed enhanced anti-proliferative and anti-migratory effects when pyrazoline B was paired with paclitaxel, suggesting potential therapeutic synergy. Complementing these findings, in silico analyses indicate pyrazoline B possesses favorable drug-like properties, including superior solubility and bioavailability compared to paclitaxel. Together, our data reveal the potential of pyrazoline B as an anticancer candidate. Further experiments are necessary, for example, to establish the synergy index of these combination regimens in in vitro and in vivo studies.
Ethical Approval
Ethical approval for the study was obtained from Medical Health Research Ethics Committee (MHREC) of Faculty of Medicine, Public Health, and Nursing, Universitas Gadjah Mada – Dr. Sardjito General Hospital (approval number: KE/FK/0919/EC/2022).
Acknowledgments
This study was supported by UGM Research Directorate and UGM Reputation Improvement Team towards World-Class Universities-UGM Quality Assurance Office under Post-Doctoral Batch II program, grant No. 13602/UN1.P.II/Dit-Lit/PT.01.04/2022; and the Directorate of Research and Community Service and Innovation of the Universitas Padjadjaran.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Arnold M, Morgan E, Rumgay H, et al. Current and future burden of breast cancer: global statistics for 2020 and 2040. Breast. 2022;66:15–23. doi:10.1016/j.breast.2022.08.010
2. Metzger-Filho O, Sun Z, Viale G, et al. Patterns of Recurrence and outcome according to breast cancer subtypes in lymph node-negative disease: results from international breast cancer study group trials VIII and IX. J Clin Oncol. 2013;31(25):3083–3090. doi:10.1200/jco.2012.46.1574
3. Bediaga NG, Beristain E, Calvo B, et al. Luminal B breast cancer subtype displays a dicotomic epigenetic pattern. Springerplus. 2016;5:623. doi:10.1186/s40064-016-2235-0
4. Orrantia-Borunda E, Anchondo-Nuñez P, Acuña-Aguilar LE, et al. Subtypes of breast cancer patterns of recurrence and outcome according to breast cancer subtypes in lymph node-negative disease: results from international breast cancer study group trials VIII and IX. 2022.
5. Li ZH, Hu PH, Tu JH, Yu NS. Luminal B breast cancer: patterns of recurrence and clinical outcome. Oncotarget. 2016;7(40):65024–65033. doi:10.18632/oncotarget.11344
6. Assistant S, Biswas R, Shafa S. Molecular subtypes of breast cancer patients according to St Gallen classification. 2020.
7. Loibl S, André F, Bachelot T, et al. Early breast cancer: ESMO clinical practice guideline for diagnosis, treatment and follow-up. Ann Oncol. 2024;35(2):159–182. doi:10.1016/j.annonc.2023.11.016
8. Alves RC, Fernandes RP, Eloy JO, Salgado HRN, Chorilli M. Characteristics, properties and analytical methods of paclitaxel: a review. Crit Rev Anal Chem. 2018;48(2):110–118. doi:10.1080/10408347.2017.1416283
9. Borst P, Schinkel AH. P-glycoprotein ABCB1: a major player in drug handling by mammals. J Clin Invest. 2013;123(10):4131–4133. doi:10.1172/jci70430
10. Fader AN, Rose PG. Abraxane for the treatment of gynecologic cancer patients with severe hypersensitivity reactions to paclitaxel. Int J Gynecol Cancer. 2009;19(7):1281–1283. doi:10.1111/IGC.0b013e3181a38e2f
11. Kenicer J, Spears M, Lyttle N, et al. Molecular characterisation of isogenic taxane resistant cell lines identify novel drivers of drug resistance. BMC Cancer. 2014;14:762. doi:10.1186/1471-2407-14-762
12. Li CM, Lu Y, Chen J, et al. Orally bioavailable tubulin antagonists for paclitaxel-refractory cancer. Pharm Res. 2012;29(11):3053–3063. doi:10.1007/s11095-012-0814-5
13. Oostendorp RL, Buckle T, Lambert G, et al. Paclitaxel in self-micro emulsifying formulations: oral bioavailability study in mice. Invest New Drugs. 2011;29(5):768–776. doi:10.1007/s10637-010-9421-7
14. Sui H, Fan ZZ, Li Q. Signal transduction pathways and transcriptional mechanisms of ABCB1/Pgp-mediated multiple drug resistance in human cancer cells. J Int Med Res. 2012;40(2):426–435. doi:10.1177/147323001204000204
15. Talekar M, Ouyang Q, Goldberg MS, Amiji MM. Cosilencing of PKM-2 and MDR-1 sensitizes multidrug-resistant ovarian cancer cells to paclitaxel in a murine model of ovarian cancer. Mol Cancer Ther. 2015;14(7):1521–1531. doi:10.1158/1535-7163.Mct-15-0100
16. Wei Z, Liang L, Junsong L, et al. The impact of insulin on chemotherapeutic sensitivity to 5-fluorouracil in gastric cancer cell lines SGC7901, MKN45 and MKN28. J Exp Clin Cancer Res. 2015;34(1):64. doi:10.1186/s13046-015-0151-8
17. Wu ZH, Lu MK, Hu LY, Li X. Praziquantel synergistically enhances paclitaxel efficacy to inhibit cancer cell growth. PLoS One. 2012;7(12):e51721. doi:10.1371/journal.pone.0051721
18. Zhou Q, Ye M, Lu Y, et al. Curcumin improves the tumoricidal effect of mitomycin C by suppressing ABCG2 expression in stem cell-like breast cancer cells. PLoS One. 2015;10(8):e0136694. doi:10.1371/journal.pone.0136694
19. Kim SM, Park K, Lim JH, et al. Potential therapeutic agents against paclitaxel-and sorafenib-resistant papillary thyroid carcinoma. Int J Mol Sci. 2022;23(18):10378. doi:10.3390/ijms231810378
20. Sharifi-Rad J, Quispe C, Patra JK, et al. Paclitaxel: application in modern oncology and nanomedicine-based cancer therapy. Oxid Med Cell Longev. 2021;2021:3687700. doi:10.1155/2021/3687700
21. Karrouchi K, Radi S, Ramli Y, et al. Synthesis and pharmacological activities of pyrazole derivatives: a review. Molecules. 2018;23(1):134. doi:10.3390/molecules23010134
22. Kumar S, Bawa S, Drabu S, Kumar R, Gupta H. Biological activities of pyrazoline derivatives--a recent development. Recent Pat Antiinfect Drug Discov. 2009;4(3):154–163. doi:10.2174/157489109789318569
23. Ray P, Salahuddin, Kumar R, Kumar A. Synthesis and pharmacological activity of pyrazoline bearing benzimidazole derivatives an up to date review. Turk J Physiother Rehabil. 2022;32(3):18612–18635. doi:10.3390/molecules23010134
24. Karabacak M, Altıntop MD, Ibrahim Çiftçi H, et al. Synthesis and evaluation of new pyrazoline derivatives as potential anticancer agents. Molecules. 2015;20(10):19066–19084. doi:10.3390/molecules201019066
25. Al-Anazi M, Khairuddean M, Al-Najjar BO, Alidmat MM, Kamal NNSNM, Muhamad M. Synthesis, anticancer activity and docking studies of pyrazoline and pyrimidine derivatives as potential epidermal growth factor receptor (EGFR) inhibitors. Arab J Chem. 2022;15(7):103864. doi:10.1016/j.arabjc.2022.103864
26. Yusuf M, Jain P. Synthetic and biological studies of pyrazolines and related heterocyclic compounds. Arab J Chem. 2014;7(5):553–596. doi:10.1016/j.arabjc.2011.09.013
27. Rana M, Faizan MI, Dar SH, Ahmad T, Rahisuddin. Design and synthesis of carbothioamide/carboxamide-based pyrazoline analogs as potential anticancer agents: apoptosis, molecular docking, ADME assay, and DNA binding studies. ACS Omega. 2022;7(26):22639–22656. doi:10.1021/acsomega.2c02033
28. Wang H, Zheng J, Xu W, et al. A new series of cytotoxic pyrazoline derivatives as potential anticancer agents that induce cell cycle arrest and apoptosis. Molecules. 2017;22(10):1635. doi:10.3390/molecules22101635
29. Suma AAT, Wahyuningsih TD, Mustofa M. Synthesis, cytotoxicity evaluation and molecular docking studyof N-phenylpyrazoline derivatives. Indones J Chem. 2019;19(4):1081–1090.
30. Wahyuningsih TD, Suma A, Astuti E. Synthesis, anticancer activity, and docking study of N-acetyl pyrazolines from veratraldehyde. J Appl Pharm Sci. 2019;9:14–20. doi:10.7324/JAPS.2019.90303
31. Suma A, Wahyuningsih TD, Pranowo D. Synthesis and antibacterial activities of N-phenylpyrazolines from veratraldehyde. Mater Sci Forum. 2017;901:124–132. doi:10.4028/www.scientific.net/MSF.901.124
32. Mustofa, Satriyo PB, Suma AAT, Waskitha SSW, Wahyuningsih TD, Sholikhah EN. A potent EGFR inhibitor, N-phenyl pyrazoline derivative suppresses aggressiveness and cancer stem cell-like phenotype of cervical cancer cells. Drug Des Devel Ther. 2022;16:2325–2339. doi:10.2147/dddt.S350913
33. Matiadis D, Sagnou M. Pyrazoline hybrids as promising anticancer agents: an up-to-date overview. Int J Mol Sci. 2020;21(15):5507. doi:10.3390/ijms21155507
34. Chunaifah I, Venilita RE, Tjitda PJP, Astuti E, Wahyuningsih TD. Thiophene-based N-phenyl pyrazolines: synthesis, anticancer activity, molecular docking and ADME study. J Appl Pharm Sci. 2024;14(4):063–071.
35. Satriyo PB, Mustofa M, Wahyuningsih TD, et al. N-phenyl pyrazoline derivative inhibits cell aggressiveness and enhances paclitaxel sensitivity of triple negative breast cancer cells. Sci Rep. 2024;14(1):13200. doi:10.1038/s41598-024-63778-2
36. Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9(7):671–675. doi:10.1038/nmeth.2089
37. Feng W, Liang C, Wang C, Yu X, Li Q, Yang H. Knockdown of ribosomal protein S15A inhibits proliferation of breast cancer cells through induction of apoptosis in vitro. Cytotechnology. 2018;70(5):1315–1323. doi:10.1007/s10616-018-0221-9
38. Aslan C, Maralbashi S, Kahroba H, et al. Docosahexaenoic acid (DHA) inhibits pro-angiogenic effects of breast cancer cells via down-regulating cellular and exosomal expression of angiogenic genes and microRNAs. Life Sci. 2020;258:118094. doi:10.1016/j.lfs.2020.118094
39. Wang Z, Yuan C, Huang Y, et al. Decreased expression of apoptosis-inducing factor in renal cell carcinoma is associated with poor prognosis and reduced postoperative survival. Oncol Lett. 2019;18(3):2805–2812. doi:10.3892/ol.2019.10630
40. Badran A, Tul-Wahab A, Zafar H, et al. Antipsychotics drug aripiprazole as a lead against breast cancer cell line (MCF-7) in vitro. PLoS One. 2020;15(8):e0235676. doi:10.1371/journal.pone.0235676
41. Zhang CZ, Wang XD, Wang HW, Cai Y, Chao LQ. Sorafenib inhibits liver cancer growth by decreasing mTOR, AKT, and PI3K expression. J Buon. 2015;20(1):218–222.
42. Ku M, Kang M, Suh JS, Yang J. Effects for sequential treatment of siAkt and paclitaxel on gastric cancer cell lines. Int J Med Sci. 2016;13(9):708–716. doi:10.7150/ijms.15501
43. Toden S, Okugawa Y, Buhrmann C, et al. Novel evidence for curcumin and boswellic acid-induced chemoprevention through regulation of miR-34a and miR-27a in colorectal cancer. Cancer Prev Res. 2015;8(5):431–443. doi:10.1158/1940-6207.Capr-14-0354
44. Berman HM, Westbrook J, Feng Z, et al. The protein data bank. Nucleic Acids Res. 2000;28(1):235–242. doi:10.1093/nar/28.1.235
45. Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612. doi:10.1002/jcc.20084
46. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461. doi:10.1002/jcc.21334
47. Eberhardt J, Santos-Martins D, Tillack AF, Forli S. AutoDock Vina 1.2.0: new docking methods, expanded force field, and python bindings. J Chem Inf Model. 2021;61(8):3891–3898. doi:10.1021/acs.jcim.1c00203
48. Laskowski RA, Swindells MB. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model. 2011;51(10):2778–2786. doi:10.1021/ci200227u
49. Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7:42717. doi:10.1038/srep42717
50. Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2(2):127–137. doi:10.1038/35052073
51. Silva SR, Bowen KA, Rychahou PG, et al. VEGFR-2 expression in carcinoid cancer cells and its role in tumor growth and metastasis. Int J Cancer. 2011;128(5):1045–1056. doi:10.1002/ijc.25441
52. Joza N, Susin SA, Daugas E, et al. Essential role of the mitochondrial apoptosis-inducing factor in programmed cell death. Nature. 2001;410(6828):549–554. doi:10.1038/35069004
53. Wiraswati HL, Hangen E, Sanz AB, et al. Apoptosis inducing factor (AIF) mediates lethal redox stress induced by menadione. Oncotarget. 2016;7(47):76496–76507. doi:10.18632/oncotarget.12562
54. Wiraswati H, Martoprawiro M, Akhmaloka A, Warganegara F. Apoptosis Inducing Factor (AIF) stabilizes menadione-conjugate product in programmed cell death. Int J PharmTech Res. 2017;10:237–245. doi:10.20902/IJPTR.2017.10430
55. Kato M. Intestinal first-pass metabolism of CYP3A4 substrates. Drug Metab Pharmacokinet. 2008;23(2):87–94. doi:10.2133/dmpk.23.87
56. Munjal NS, Shukla R, Singh TR. Physicochemical characterization of paclitaxel prodrugs with cytochrome 3A4 to correlate solubility and bioavailability implementing molecular docking and simulation studies. J Biomol Struct Dyn. 2022;40(13):5983–5995. doi:10.1080/07391102.2021.1875881
57. Katreddy RR, Bollu LR, Su F, et al. Targeted reduction of the EGFR protein, but not inhibition of its kinase activity, induces mitophagy and death of cancer cells through activation of mTORC2 and Akt. Oncogenesis. 2018;7(1):5. doi:10.1038/s41389-017-0021-7
58. Modi SJ, Kulkarni VM. Vascular endothelial growth factor receptor (VEGFR-2)/KDR inhibitors: medicinal chemistry perspective. Med Drug Discov. 2019;2:100009. doi:10.1016/j.medidd.2019.100009
59. Thomas R, Weihua Z. Rethink of EGFR in cancer with its kinase independent function on board. Front Oncol. 2019;9:800. doi:10.3389/fonc.2019.00800
60. Weihua Z, Tsan R, Huang W-C, et al. Survival of cancer cells is maintained by EGFR independent of its kinase activity. Cancer Cell. 2008;13(5):385–393. doi:10.1016/j.ccr.2008.03.015
61. Rodriguez SMB, Kamel A, Ciubotaru GV, et al. An overview of EGFR mechanisms and their implications in targeted therapies for glioblastoma. Int J Mol Sci. 2023;24(13):11110. doi:10.3390/ijms241311110
62. Wiraswati HL, Bashari MH, Alfarafisa NM, et al. Virtual screening of anticancer activity of chalcones and pyrazolines as potential EGFR, VEGFR, and cytochrome P450 inhibitors. J Pharm Pharmacogn Res. 2023;11(4):699–713. doi:10.56499/jppres23.1591_11.4.699
63. Abhinand CS, Raju R, Soumya SJ, Arya PS, Sudhakaran PR. VEGF-A/VEGFR2 signaling network in endothelial cells relevant to angiogenesis. J Cell Commun Signal. 2016;10(4):347–354. doi:10.1007/s12079-016-0352-8
64. Guo S, Colbert LS, Fuller M, Zhang Y, Gonzalez-Perez RR. Vascular endothelial growth factor receptor-2 in breast cancer. Biochim Biophys Acta Rev Cancer. 2010;1806(1):108–121. doi:10.1016/j.bbcan.2010.04.004
65. Wu P, Nielsen TE, Clausen MH. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol Sci. 2015;36(7):422–439. doi:10.1016/j.tips.2015.04.005
66. Yan J-D, Liu Y, Zhang Z-Y, et al. Expression and prognostic significance of VEGFR-2 in breast cancer. Pathol Res Prac. 2015;211(7):539–543. doi:10.1016/j.prp.2015.04.003
67. Kalyankrishna S, Grandis JR. Epidermal growth factor receptor biology in head and neck cancer. J Clin Oncol. 2006;24(17):2666–2672. doi:10.1200/jco.2005.04.8306
68. Yang F, Tang X, Riquelme E, et al. Increased VEGFR-2 gene copy is associated with chemoresistance and shorter survival in patients with non-small-cell lung carcinoma who receive adjuvant chemotherapy. Cancer Res. 2011;71(16):5512–5521. doi:10.1158/0008-5472.Can-10-2614
69. Bancroft CC, Chen Z, Yeh J, et al. Effects of pharmacologic antagonists of epidermal growth factor receptor, PI3K and MEK signal kinases on NF-kappaB and AP-1 activation and IL-8 and VEGF expression in human head and neck squamous cell carcinoma lines. Int J Cancer. 2002;99(4):538–548. doi:10.1002/ijc.10398
70. Hung MS, Chen IC, Lin PY, et al. Epidermal growth factor receptor mutation enhances expression of vascular endothelial growth factor in lung cancer. Oncol Lett. 2016;12(6):4598–4604. doi:10.3892/ol.2016.5287
71. Abd El-Meguid EA, Naglah AM, Moustafa GO, Awad HM, El Kerdawy AM. Novel benzothiazole-based dual VEGFR-2/EGFR inhibitors targeting breast and liver cancers: synthesis, cytotoxic activity, QSAR and molecular docking studies. Bioor Med Chem Lett. 2022;58:128529. doi:10.1016/j.bmcl.2022.128529
72. Pal HC, Sharma S, Strickland LR, et al. Delphinidin reduces cell proliferation and induces apoptosis of non-small-cell lung cancer cells by targeting EGFR/VEGFR2 signaling pathways. PLoS One. 2013;8(10):e77270. doi:10.1371/journal.pone.0077270
73. Le Y, Gan Y, Fu Y, et al. Design, synthesis and in vitro biological evaluation of quinazolinone derivatives as EGFR inhibitors for antitumor treatment. J Enzyme Inhib Med Chem. 2020;35(1):555–564. doi:10.1080/14756366.2020.1715389
74. Lim B, Greer Y, Lipkowitz S, Takebe N. Novel apoptosis-inducing agents for the treatment of cancer, a new arsenal in the toolbox. Cancers. 2019;11(8):1087. doi:10.3390/cancers11081087
75. Green DR, Kroemer G. Pharmacological manipulation of cell death: clinical applications in sight? J Clin Invest. 2005;115(10):2610–2617. doi:10.1172/jci26321
76. Ricci MS, Zong WX. Chemotherapeutic approaches for targeting cell death pathways. Oncologist. 2006;11(4):342–357. doi:10.1634/theoncologist.11-4-342
77. Melloul O, Zabit S, Lichtenstein M, Duran D, Grunewald M, Lorberboum-Galski H. Inducing targeted, caspase-independent apoptosis with new chimeric proteins for treatment of solid cancers. Cancers. 2025;17(7):1179.
78. Holze C, Michaudel C, Mackowiak C, et al. Oxeiptosis, a ROS-induced caspase-independent apoptosis-like cell-death pathway. Nat Immunol. 2018;19(2):130–140. doi:10.1038/s41590-017-0013-y
79. Mourad AAE, Farouk NA, El-Sayed E-SH, Mahdy ARE. EGFR/VEGFR-2 dual inhibitor and apoptotic inducer: design, synthesis, anticancer activity and docking study of new 2-thioxoimidazolidin-4one derivatives. Life Sci. 2021;277:119531. doi:10.1016/j.lfs.2021.119531
80. Alkamaly OM, Altwaijry N, Sabour R, Harras MF. Dual EGFR/VEGFR2 inhibitors and apoptosis inducers: synthesis and antitumor activity of novel pyrazoline derivatives. Arch Pharm. 2021;354(4):e2000351. doi:10.1002/ardp.202000351
81. Bolton JL, Trush MA, Penning TM, Dryhurst G, Monks TJ. Role of quinones in toxicology. Chem Res Toxicol. 2000;13(3):135–160. doi:10.1021/tx9902082
82. Saleh AM, Aziz MA, Abdou IM, et al. Cytotoxic activity of the novel heterocyclic compound G-11 is primarily mediated through intrinsic apoptotic pathway. Apoptosis. 2016;21(7):873–886. doi:10.1007/s10495-016-1248-z
83. Halim PA, Hassan RA, Mohamed KO, et al. Synthesis and biological evaluation of halogenated phenoxychalcones and their corresponding pyrazolines as cytotoxic agents in human breast cancer. J Enzyme Inhib Med Chem. 2022;37(1):189–201. doi:10.1080/14756366.2021.1998023
84. Varma RR, Pandya JG, Vaidya FU, Pathak C, Bhatt BS, Patel MN. Biological activities of pyrazoline-indole based Re(I) carbonyls: DNA interaction, antibacterial, anticancer, ROS production, lipid peroxidation, in vivo and in vitro cytotoxicity studies. Chem Biol Interact. 2020;330:109231. doi:10.1016/j.cbi.2020.109231
85. Novita M, Harahap W, Taufik R. Factors affecting the outcome of the operable subtype luminal B breast cancer treatment. Indones J Multidiscip Sci. 2022;2:2093–2107. doi:10.55324/ijoms.v2i3.308
86. Ma Y, Yu S, Ni S, et al. Targeting strategies for enhancing paclitaxel specificity in chemotherapy. Front Cell Dev Biol. 2021;9:626910. doi:10.3389/fcell.2021.626910
87. Choi N, Lee SW, Lim Y, et al. Failure patterns of luminal B breast cancer following postoperative adjuvant radiation therapy. Int J Radiat Oncol Biol Phys. 2017;99(2):E9–E10. doi:10.1016/j.ijrobp.2017.06.613
88. Choi YH, Yoo YH. Taxol-induced growth arrest and apoptosis is associated with the upregulation of the Cdk inhibitor, p21WAF1/CIP1, in human breast cancer cells. Oncol Rep. 2012;28(6):2163–2169. doi:10.3892/or.2012.2060
89. Khing TM, Choi WS, Kim DM, et al. The effect of paclitaxel on apoptosis, autophagy and mitotic catastrophe in AGS cells. Sci Rep. 2021;11(1):23490. doi:10.1038/s41598-021-02503-9
90. Hanna RK, Zhou C, Malloy KM, et al. Metformin potentiates the effects of paclitaxel in endometrial cancer cells through inhibition of cell proliferation and modulation of the mTOR pathway. Gynecol Oncol. 2012;125(2):458–469. doi:10.1016/j.ygyno.2012.01.009
91. Refolo MG, Lotesoriere C, Lolli IR, Messa C, D’Alessandro R. Molecular mechanisms of synergistic action of ramucirumab and paclitaxel in gastric cancers cell lines. Sci Rep. 2020;10(1):7162. doi:10.1038/s41598-020-64195-x
92. França F, Silva PMA, Soares JX, et al. A pyranoxanthone as a potent antimitotic and sensitizer of cancer cells to low doses of paclitaxel. Molecules. 2020;25(24):5845. doi:10.3390/molecules25245845
93. Chen Q, Xu S, Liu S, Wang Y, Liu G. Emerging nanomedicines of paclitaxel for cancer treatment. J Control Release. 2022;342:280–294. doi:10.1016/j.jconrel.2022.01.010
94. Kumar DN, Chaudhuri A, Dehari D, et al. Combination therapy comprising paclitaxel and 5-fluorouracil by using folic acid functionalized bovine milk exosomes improves the therapeutic efficacy against breast cancer. Life. 2022;12(8):1143. doi:10.3390/life12081143
95. Núñez C, Capelo JL, Igrejas G, Alfonso A, Botana LM, Lodeiro C. An overview of the effective combination therapies for the treatment of breast cancer. Biomaterials. 2016;97:34–50. doi:10.1016/j.biomaterials.2016.04.027
96. Lee A, Djamgoz MBA. Triple negative breast cancer: emerging therapeutic modalities and novel combination therapies. Cancer Treat Rev. 2018;62:110–122. doi:10.1016/j.ctrv.2017.11.003
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.