Back to Journals » International Journal of Nanomedicine » Volume 15
PSMA-Targeting Reduction-Cleavable Hyperbranched Polyamide-Amine Gene Delivery System to Treat the Bone Metastases of Prostate Cancer
Authors Ye Y, Zhang L, Dai Y, Wang Z, Li C, Peng Y, Ma D, He P
Received 18 June 2020
Accepted for publication 1 September 2020
Published 28 September 2020 Volume 2020:15 Pages 7173—7184
DOI https://doi.org/10.2147/IJN.S268398
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Yongheng Ye1 ,* Lingli Zhang2 ,* Yuhu Dai,1 Zhi Wang,3 Cuie Li,3 Yue Peng,4 Dong Ma,2 Peiheng He1
1Department of Orthopedic Surgery, Guangdong Provincial Key Laboratory of Orthopaedics and Traumatology, The First Affiliated Hospital of Sun Yat-sen University, Guangzhou, Guangzhou, Guangdong Province 510080, People’s Republic of China; 2Key Laboratory of Biomaterials of Guangdong Higher Education Institutes, Department of Biomedical Engineering, Jinan University, Guangzhou 510632, People’s Republic of China; 3Guangzhou Institute of Biomedicine and Health, Chinese Academy of Sciences, Guangzhou, Guangdong Province 510530, People’s Republic of China; 4Department of Otorhinolaryngology Head and Neck Surgery, Zhuhai People’s Hospital (Zhuhai Hospital Affiliated with Jinan University), Zhuhai, Guangdong Province 519000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Peiheng He
Department of Orthopedic Surgery, Guangdong Prov Key Laboratory Orthopaed & Traumatol, The First Affiliated Hospital of Sun Yat-Sen University, Guangzhou, Guangdong Province 510080, People’s Republic of China
Email [email protected]
Dong Ma
Key Laboratory of Biomaterials of Guangdong Higher Education Institutes, Department of Biomedical Engineering, Jinan University, Guangzhou 510632, People’s Republic of China
Email [email protected]
Objective: This study aimed to develop aptamer-anchored hyperbranched poly(amido amine) (HPAA) for the systemic delivery of miRNA-133a-3p and to evaluate its therapeutic potential against bone metastasis of prostate cancer in vivo and in vitro.
Methods: A glutathione (GSH)-responsive cationic HPAA was prepared by the Michael addition reaction. Furthermore, HPAA-PEG was produced by PEGylation, and then the aptamer targeted to prostate-specific membrane antigen (PSMA) was conjugated to the HPAA-PEG. The obtained HPAA-PEG-APT could form nanocomplexes with miRNA-133a-3p through electrostatic adsorption.
Results: The results of immunocytochemistry indicated that the complexes could target PSMA-expressing LNCaP cells. The ability of HPAA-PEG-APT to facilitate the delivery of miRNA-133a-3p into LNCaP cells was proven, and HPAA-PEG-APT/miRNA-133a-3p demonstrated enhanced antitumor activity, lower cytotoxicity and better biocompatibility in vitro. Moreover, in a mouse tibial injection tumor model, the intravenous injection of the HPAA-PEG-APT/miRNA-133a-3p complex significantly inhibited cancer growth and extended the survival time.
Conclusion: This study provided an aptamer-anchored HPAA-loaded gene system to deliver miRNA-133a-3p for better therapeutic efficacy of bone metastasis of prostate cancer.
Keywords: miRNA, aptamer, hyperbranched polyamide amine, bone metastasis, prostate cancer
Introduction
Prostate cancer (PCa) is a high-incidence tumor of the male reproductive system.1 Early-stage prostate cancer usually has a good prognosis, with a high 5-year survival rate.2 However, advanced prostate cancer frequently metastasizes after chemotherapy, surgery and radiotherapy.3 Bone is the most common distant metastatic site in PCa, mainly due to slow blood flow through the bone marrow and abundant adhesion receptors, growth factors and cytokines.4–6 Serious complications accompany bone metastasis, such as pathological fracture, hypercalcemia, nerve compression syndrome and intractable pain.7 Treatment mainly includes systemic treatment with endocrine therapy and chemotherapy, as well as local treatment for bone metastasis, such as surgery, radiotherapy, and bisphosphonates, which usually lead to drug resistance and harmful side effects.2,3 The emergence of gene therapy has brought a new direction for the treatment of cancer.4,5 The scope of gene therapy has been extended from genes to various nucleic acids such as antisense oligonucleotides (ASOs), siRNA, miRNA and mRNA.6–10 Many lines of evidence have demonstrated that miRNA-based therapies hold great prospects in the treatment of PCa.7,11 In our previous study, miRNA-133a-3p was proven to be a tumor suppressor gene in prostate cancer and downregulates several cytokine receptors associated with PCa growth, including FGFR, EGFR, IGFR and MET, inactivating the PI3K/AKT signaling pathway.12
For gene delivery to the target tissue avoiding nucleic acid enzymatic degradation and immune recognition, the delivery systems were designed with better pharmacokinetics and biodistribution.13,14 Nonviral vectors seem a better choice than viral vectors to avoid the pathogenicity and immunogenicity of viral vectors. Among them, hyperbranched poly(amido amine)s (HPAAs) have attracted attention because they have good endosomal escape ability and can release drugs rapidly.15 In particular, the expression of GSH has been confirmed to be much higher in tumor cells than in normal cells. Therefore, many studies, including our previous work, have developed reduction-cleavable HPAAs containing disulfide bonds, which were anticipated to cause endocellular degradation because of the breakdown of disulfide bonds under high GSH conditions and rapid drug release.16–20
To load genes into specific cells more effectively, targeting gene nanocarriers have been studied. PSMA is expressed at higher levels in advanced PCa, as well as bone metastasis of the disease.21 Many clinical trials have been demonstrated PSMA could be as imaging diagnosis and targeted therapy ability, and thus, PSMA is a potential and promising target for the specific delivery of nanocarriers to PCa.22,23 The A 10–3.2 aptamer (APT) has only 39 nucleotides, its weight is significantly reduced and it has good binding affinity to PSMA. Thus, many research teams have focused on APT-targeting gene delivery and favorable effects were reported.11,24–26
In this study, we synthesized a multifunctional nanocarrier HPAA-PEG-APT/miRNA-133a-3p that can be targeted at the tumor cell (Scheme 1). We examined the capability of HPAA-PEG-APT to deliver miRNA-133a-3p. Additionally, we assess the treatment effect of the HPAA-PEG-APT/miRNA-133a-3p in vitro and in vivo.
 |
Scheme 1 Scheme of the synthesis and the therapeutic effect to tumor of HPAA -PEG -APT/miRNA. |
Experimental Details
Preparation of HPAA Derivative
Materials
CBA was synthesized based on our previous studies.17,18 1-(2-Aminoethyl) piperazine (AEPZ) and dicyclohexylcarbodiimide (DCC) were obtained from Aladdin Industrial Corporation (Shanghai, China). α-Malemidyl-ω-N-hydroxysuccinimidyl polyethyleneglycol (MAL-PEG-NHS, MW:5000) was purchased from Xi’an ruixi Biological Technology Co. Ltd. (Xi’an, China). The branched polyethylenimines (bPEIs) were provided by Sigma-Aldrich. The solvents were provided by Chemical Reagent Factory (Guangzhou, China). A pLenti lentivirus was utilized for transfection and the generation of the LNCaP cell line to construct PSMA-expressing metastatic tumors animal models (OBiO, Shanghai, China). miRNA-133a-3p, negative control-miRNA and the A10-3.2 anti-PSMA aptamer modified with infrared dye Cy5 at the 5ʹ terminus were purchased from RiboBio (Guangzhou, China).
Preparation of HPAA-PEG and HPAA-PEG-APT
HPAA was synthesized as described previously.17,18 First, 200 mg of NHS-PEG-MAL was dissolved in phosphate-buffered solution (PBS, pH 7.4) for HPAA and NHS-PEG-MAL conjugation. Next, a 10:1 dilution of HPAA and NHS-PEG-MAL was conjugated through a specific reaction between the main amino groups of HPAA and NHS groups of the PEG derivative. The synthesis was performed in PBS at ambient temperature in the dark for 2 h. Next, the HPAA-PEG conjugate was purified by ultrafiltration and then was freeze-dried and kept at 4°C until ready for use.
To conjugate the anti-PSMA aptamer with HPAA-PEG, 100 mg of HPAA-PEG was dissolved in PBS (pH 6.5–7.0). Next, 2 mL of DMSO containing 1 mg of APT was added dropwise to the HPAA-PEG and stirred in 30 min, and the compound was reacted at ambient temperature in the dark for 48 h. The HPAA-PEG-APT conjugate was also purified by ultrafiltration, followed by lyophilization and then storage at 4°C.
Characterization
NMR
The HPAA derivatives were characterized by nuclear magnetic resonance (NMR). Two milligrams each of HPAA, HPAA-PEG, NHS-PEG-MAL and HPAA-PEG-APT was dissolved in D2O and NMR. The results were compared using MestReNova software.
Size and Zeta Potential
The changes in the particle size and zeta potential of HPAA derivatives (with or without 10 mM GSH) were characterized by dynamic light scattering (DLS; Zetasizer ZS90, Malvern). One milligram of samples was solubilized in high-purity water and tested at 25°C. The morphology was visualized using a JEM-2010HR transmission electron microscope.
UV Characterization
The amount of APT in HPAA-PEG-APT was measured using UV-vis spectrophotometry, and the absorbance of APT was quantified at 260 nm using free APT as the reference standard.
Gel Electrophoresis
To prepare HPAA-PEG-APT/miRNA complexes, the miRNA-133a-3p gene was mixed with HPAA-PEG-APT buffer at various HPAA-PEG-APT/miRNA mass ratios. For HPAA-PEG-APT, the gene-binding ability was tested by agarose gel electrophoresis. The result was visualized using a UV transilluminator (Bio-Rad, USA).
Cell Lines and Culture
The human PCa cell line LNCaP was provided by The Third Affiliated Hospital of Sun Yat-sen University and was approved to research by ethics committee of The First Affiliated Hospital of Sun Yat-sen University. HUVECs were purchased from Shanghai Chinese Academy of Sciences cell bank (China). LNCaP and HUVECs were cultured as described previously.12
Specificity of the A 10-3.2 Aptamer
The colocalization of Cy5-labeled aptamer and anti-PSMA monoclonal antibodies (Abcam, Cambridge, MA, USA) in LNCaP cells was detected by LSCM.
In vitro Cellular Uptake
LNCaP cells were cultured overnight before transfection in complete DMEM on 24-well plates (5χ104 cells/per well). Next, the cells were supplemented with Opti-MEM (without serum) containing HPAA or HPAA-PEG-APT complexes incubated with FAM-labeled miRNA (miRNA:1 µg/well) at different mass ratios (60, 80, 100, 120:1). Thereafter, all the groups were supplemented with fresh DMEM for 6 h, followed by culture for another 24–48 h. Next, the cells were washed with PBS and digested, following by centrifugation and resuspension using moderate PBS. The cellular uptake was measured by flow cytometry (Accuri C6), and PEI-25k/miRNA was used as the positive control.
Anticancer Effect in vitro
The inhibitory effects of LNCaP cells were observed using the CCK-8 Kit (CCK-8; Beyotime, Shanghai, China). LNCaP cells were incubated in 96-microwell plates at 1 × 104 cells/well for 24 h at 37°C. The cells were mixed with different concentrations (0.02–50 µM) of complexes: HPAA-PEG-APT/miRNA, HPAA/miRNA and HPAA-PEG-APT/NCmiRNA for 72 h. Subsequently, 10 µL of CCK-8 was applied to all wells, followed by incubation for 4 h. The absorbance of different wells was measured using a microplate reader (MULTISKAN MK3), a standard curve was plotted based on the absorbance and cell count, and each sample was measured three times. The IC50 value was counted using GraphPad 8.0 PRISM®.11
Bcl2, EGFR and MET Protein Expression
In total, 5 χ104 LNCaP cells were cultured in 6-well plates and incubated in 5% CO2 at 37°C for 24 h, reaching 8090% confluence. Different samples (HPAA-PEG-APT/NCmiRNA, HPAA/miRNA and HPAA-PEG-APT/miR-133a-3p, PBS) were mixed with the cells and reacted for 72 h (for protein extraction). Western blotting was performed using a previously described standard method.24 Antibodies against Bcl2, EGFR and MET were purchased from Abcam (Cambridge, USA), and EGFR and MET were provided by Cell Signaling Technology.
Cytotoxicity of HPAA Derivatives for LNCaP Cells
The cytotoxicity of HPAA derivatives was observed using the CCK-8 assay in LNCaP and HUVEC lines. In total, 1×104 cells per well of 96-well plates were seeded in DMEM. After 24 h of incubation, fresh media containing HPAA, HPAA-PEG and HPAA-PEG-APT at different concentrations (from 10 to 500 µg/mL) were applied. Next, 100 μL of fresh medium containing 10% CCK-8 was added to all wells, followed by washing with PBS. The absorbance was measured using a microplate reader at a 450-nm wavelength, and the cell viability was defined as the ratio of the treated groups to the control group. Cell viability (%)=Absorbance of the treated group − Absorbance of the control group/Absorbance of the negative control group − Absorbance of the control group χ100%.
Blood Compatibility
One-milliliter PBS solutions containing HPAA, HPAA-PEG, HPAA-PEG-APT, or PEI at different concentrations (0.01 mg/mL, 0.5 mg/mL,1 mg/mL) were prepared, while pure water and the PBS group were used as the positive control and negative control, respectively. Next, 16% hematocrit (v/v) red blood cells (RBCs) suspended in PBS were mixed with 5 mL of PBS containing different concentrations of HPAA derivatives. The positive controls (100% hemolysis caused by PBS containing 5 mL of 0.1% NaCO3 solution) and negative controls (0% hemolysis, only PBS) were prepared. The samples were incubated at 37°C for 1 h, 3 h, 6 h, and 12 h, followed by centrifugation for 5 min at 1000 χ g and measurement of the absorbance of the supernatant at 540 nm using a microplate reader. Finally, the ratio of hemolysis was determined by calculating the optical density (OD): Hemolysis (%)=[(OD of the treated sample – OD of negative control) χ100]/OD of positive control. Each sample was tested in triplicate.
Intraosseous Model of Prostate Cancer and Treatments
All animal procedures were approved by The Institutional Animal Ethics Committee (IAEC) of Sun Yat-sen University and carried out in accordance with the laboratory animal use guidelines of the IAEC. Animal handling followed the national Animal Welfare Law of China. The bone metastasis animal model was induced by injecting LNCaP-luc cells expressing the luciferase gene into the tibia.4 Briefly, 5χ105 LNCaP-luc cells were collected from 25-cm cell culture dishes (Corning, USA) and were resuspended in 20 μL of PBS. The cell suspension was administered into the lumen of the tibia using a 0.1-mL insulin needle. Tumor induction and its progression were investigated by estimating the bioluminescence signal intensity (p/s/cm2/sr). After 5-min intraperitoneal injection of luciferin (200 mg/kg; VivoGloTM Luciferin; Promega, Madison, WI), the mice were imaged using IVIS® Lumina II (PerkinElmer, Waltham, MA). Two weeks following LNCaP cell injection, the mice were separated into 3 groups randomly with 5 mice per group. Next, 0.2-mL mixtures containing PBS (as control), HPAA-PEG-APT/miRNA-133a-3p, and HPAA/miRNA-133a-3p (HPAA derivatives: miRNA= 67 µg: 33 µg) were administered via tail vein twice every 4 weeks. We observed tumor growth by bioluminescence signal intensity. Additionally, hematoxylin and eosin (H&E) staining of the major organs (heart, lung, spleen, liver, kidney and prostate) was performed.
The criteria for the euthanasia of mice were as follows: 20% weight loss; movement difficulty because of tumor burden; tumor ulceration; necrosis; bleeding; dehydration; lethargy; and cachexia. These mice were considered dead in the survival data.
Statistical Analysis
All the data were expressed as means±standard deviation. Analyses of the difference between groups were performed by one-way analysis of variance (ANOVA) using GraphPad Prism 8). P<0.05 was defined as the significance level, * for P <0.05, ** for P <0.01. All statistical tests were two-tailed.
Results and Discussion
Synthesis and Characterization of HPAA Derivatives
In this study, a GSH-responsive HPAA was prepared according to our previous work to form polymeric materials with specific targeting effects and gene codelivery capability,17,18 and then HPAA was bonded with NHS-PEG-MAL through an amino-specific reaction. Furthermore, the anti-PSMA aptamer (APT) was conjugated with HPAA-PEG. Thereafter, the HPAA-PEG-APT vector could deliver miRNA-133a-3p via electrostatic interactions between HPAA and miRNA.
The structure of HPAA derivatives was described by 1H NMR, and the results are shown in Figure 1A. All characterized peaks correspond to its chemical structure, indicating that HPAA derivatives had been synthesized successfully. The peaks of HPAA segments and MAL were from 2.3 to 3.5 and from 5.9 to 6.2, respectively. When the HPAA-PEG was conjugated with APT, the peaks from 5.9 to 6.2 were weakened, proving that some APT replaced the MAL (Figure 1B). These above results showed that HPAA-PEG-APT was synthesized successfully.
 |
Figure 1 Synthesis and characterization of HPAA derivatives (A) 1H NMR spectra of HPAA, HPAA-PEG-MAL and NHS-PEG-MAL. (B) 1H NMR spectrum of HPAA-PEG-MAL and HPAA-PEG-APT. (C) UV-Vis spectrum of APT, HPAA-APT and HPAA. (D) TEM image of HPAA complexes. (E) Particle sizes and (F) potentials of HPAA derivatives (n=3) incubated with or without GSH. *p<0.05, **p<0.05. |
The APT content in HPAA-PEG-APT was tested using UV spectrophotometry. Free APT and HPAA-APT demonstrated a similar characteristic absorption peak at 260 nm, proving that APT was conjugated with HPAA (Figure 1C). A typical transmission electron microscopy (TEM) image of HPAA is shown in Figure 1D, presenting a uniform size distribution of spheres.
The concentration of GSH in tumor cells is significantly higher than that outside the cells.27 The structure of HPAA is rich in disulfide bonds, so HPAA-PEG-APT/miRNA is likely to exhibit excellent degradation performance at high intracellular GSH concentrations. The redox-responsive ability of the HPAA-PEG-APT/miRNA nanoparticles was assessed by measuring the particle size and zeta potentials using Zataszier Nano-ZS with or without 10 mM GSH. In Figure 1E, the particle size of HPAA-PEG-APT incubated with 10 mM GSH for 30 min was smaller than that of HPAA because of the cleavage of the disulfide bond. The GSH-responsive ability of the nanoparticle was helpful to release the gene in the tumor microenvironment. After crossing-linking by AEPZ, the zeta potential of HPAA increased from 0.8 to 25.6 mV because of the aggregation of positive charge. A significant decrease in zeta potential (3.78mV) was observed in HPAA-PEG-APT, which may be caused by the negatively charged groups in NHS-PEG-MAL and APT. With the 10 mM GSH to incubate, there was no difference of the particle zeta potentials between HPAA and HPAA-PEG-APT (Figure 1F). Nanomaterials with neutral potentials have more effective bone marrow localization than those with anionic or cationic potentials because nanomaterials with neutral potentials have reduced interaction with serum proteins, unlike anionic or cationic nanomaterials that are opsonized to the same extent.28–30
The Specificity of Aptamers
Using immunocytochemistry, the chemically synthesized A 10–3.2 aptamer demonstrated its capacity for binding to prostate cancer cells with positive PSMA expression. In LNCaP cells, the Cy5-labeled HPAA-PEG-APT complex colocalized on the cell membrane of LNCaP cells (Figure 2AH).
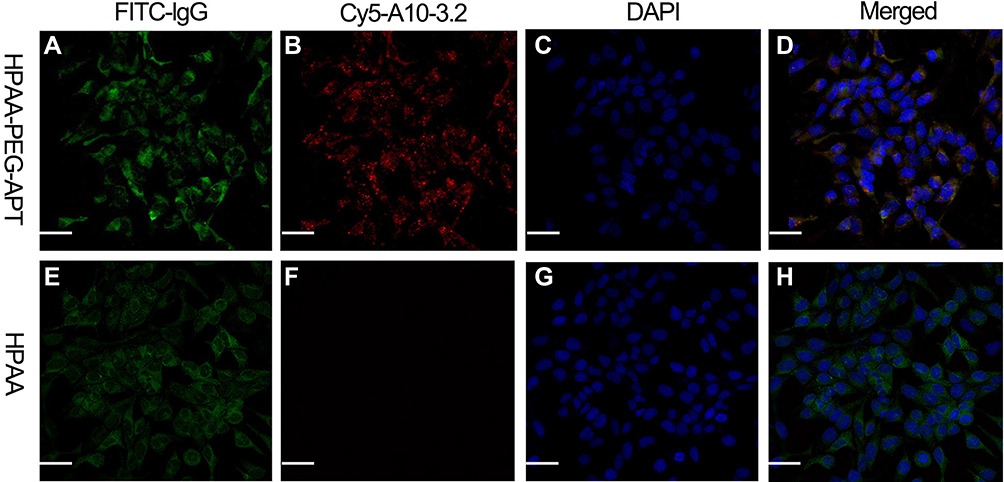 |
Figure 2 The specificity of the aptamer in LNCaP cells. (A and E) Identification of PSMA expression in LNCaP cells via indirect immunofluorescence using anti-PSMA monoclonal antibody and FITC-labeled lgG secondary. (B and F) Identification of Cy5-labeled A 10–3.2 aptamer binding to LNCaP cells using direct immunochemistry. (C and G) cell nuclei with DAPI-stained. (D and H) Merged fluorescence images. Scale bars:50μm. |
In vitro Cellular Uptake
Because of its hyperbranching structure and biodegradation properties, HPAA has also been presented to be an efficient gene delivery carrier in our previous studies.17,18 To evaluate the miRNA delivery capacity of HPAA-PEG-APT, the gel retardation assay was used to detect its gene condensation capacity. When the weight ratio reached 5:1 or more, the miRNA was completely blocked, indicating that it was completely compressed and proving that HPAA-PEG-APT showed good in vitro gene compression ability (Figure 3A). The rate of cellular uptake of HPAA derivatives/miRNA complexes in LNCaP cells was assessed using in vitro cellular uptake assays. The HPAA complexes all showed strong miRNA gene delivery, with cellular uptake rates up to 80% (Figure 3B). For HPAA-PEG-APT, its higher cellular uptake was due to the negative charge of APT reducing the overall positive charge of the HPAA complex and its ability to bind to LNCaP cell surface PSMA antigen targets. However, once the mass ratio exceeded 120:1, its significant cytotoxicity led to a reduced cellular uptake; thus, we chose a mass ratio of 80:1 for subsequent in vitro experiments.
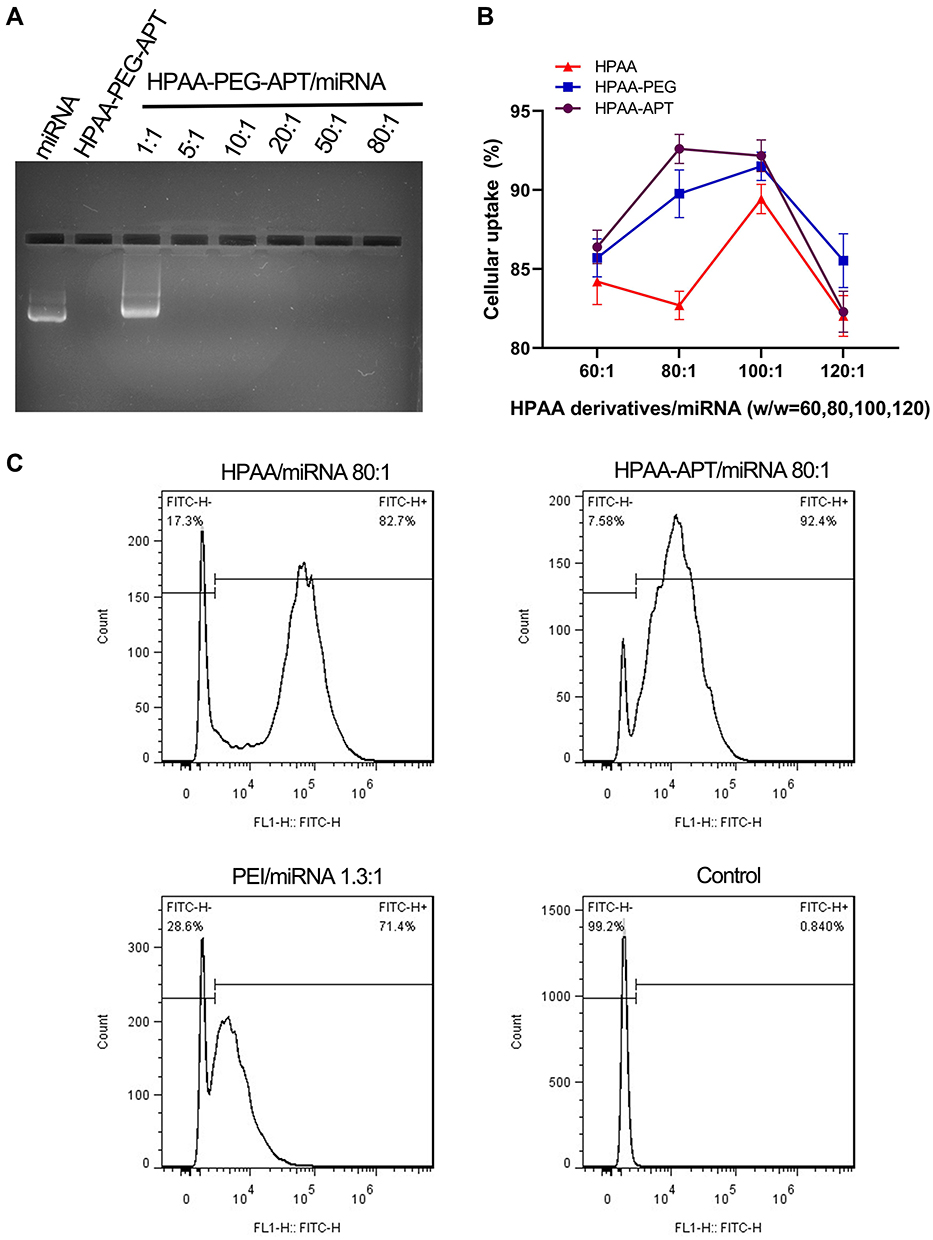 |
Figure 3 The rate of cellular uptake of HPAA derivatives/miRNA complexes in vitro (A) Gel electrophoresis assay of HPAA-PEG-APT/miRNA with different weight ratios. (B) Treated with HPAA derivatives/miRNA (w/w=60,80,100,120) (n=3). (C) Detection of cellular uptake between HPAA and HPAA-APT using FACS. |
Furthermore, flow cytometry revealed the cellular uptake of miRNA-FAM into LNCaP cells was 92.4%±1.2% after coincubation with HPAA-PEG-APT (Figure 3C). By contrast, the cellular uptake of miRNA-FAM into LNCaP cells co-incubated with HPAA was 82.4%±2.3% through flow cytometry (Figure 3C).
Efficacy of Treatments in vitro
Similar to most tumor diseases, the leading cause of mortality of prostate cancer is related to metastasis-related recurrence. Additionally, growing evidence suggests that activation of the PI3K/AKT signaling pathway plays a key factor in tumor metastasis.31–33 In our previous work, miR-133a-3p prevents PCa bone metastasis by specifically targeting diverse cytokine receptors, including EGFR, FGFR1, IGF1R and MET, resulting in inactivation of the PI3K/AKT signaling pathway.12 LNCaP cells were processed with HPAA-PEG-APT/miRNA, HPAA/miRNA and HPAA-PEG-APT/NCmiRNA in various concentrations. The results implied that cell growth was suppressed in a dose-dependent manner (Figure 4A). The IC50 values were 132.8 nM, 621.1 nM, and 2611 nM, respectively. Thus, HPAA-PEG-APT/miRNA has an IC50 value approximately 4.68 times lower than that of HPAA/miRNA, indicating that HPAA-PEG-APT could deliver more miRNAs into LNCaP cells. We further revealed the mechanism by studying the expression level of Bcl-2, MET, and EGFR protein and found that HPAA-PEG-APT efficiently transfects miR-133a-3p into LNCaP cells, specifically inhibiting the expression of the protein according to previous work (Figure 4B).12
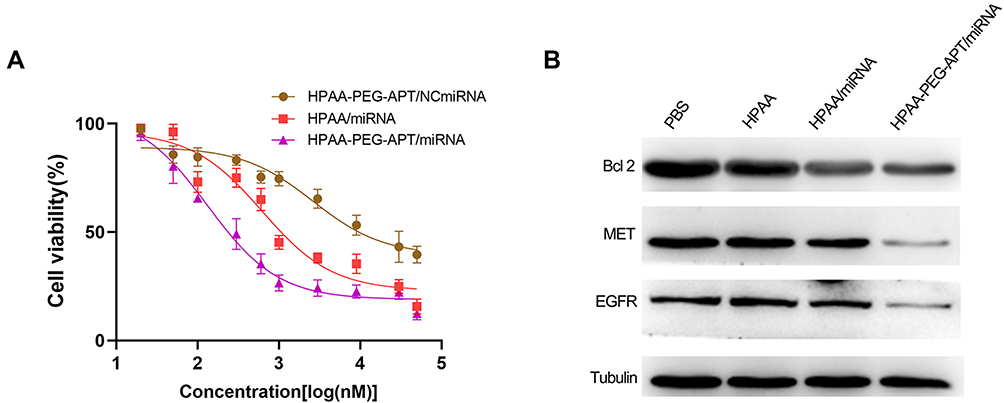 |
Figure 4 In vitro anticancer effects (A) LNCaP Cell viabilities treated with HPAA-PEG-APT/miRNA, HPAA/miRNA and HPAA-PEG-APT/NCmiRNA, respectively (n=3). (B) The quantitative analysis of BCL 2, MET, and EFGR protein expression. |
Biocompatibility
Blood Safety
In vivo, HPAA-PEG-APT should have good blood compatibility to obtain better bioavailability. Spectrophotometric tests of the release of hemoglobin in erythrocytes were performed after treatment with HPAA derivatives to assess the blood compatibility of HPAA derivatives. Figure 5A-D) presents the hemolysis ratios of different HPAA derivative concentrations, with PEI as the control. The results showed that HPAA derivatives had good hemocompatibility. After 12 h incubation and a concentration of HPAA-PEG-APT up to 0.5 mg/mL, the sample still demonstrated nonhemolytic effects related to hemolysis ratios lower than the permissible level of 5%. However, PEI showed severe hemolysis when the concentration of PEI was 0.5 mg/mL, resulting in more than 70% hemolysis.
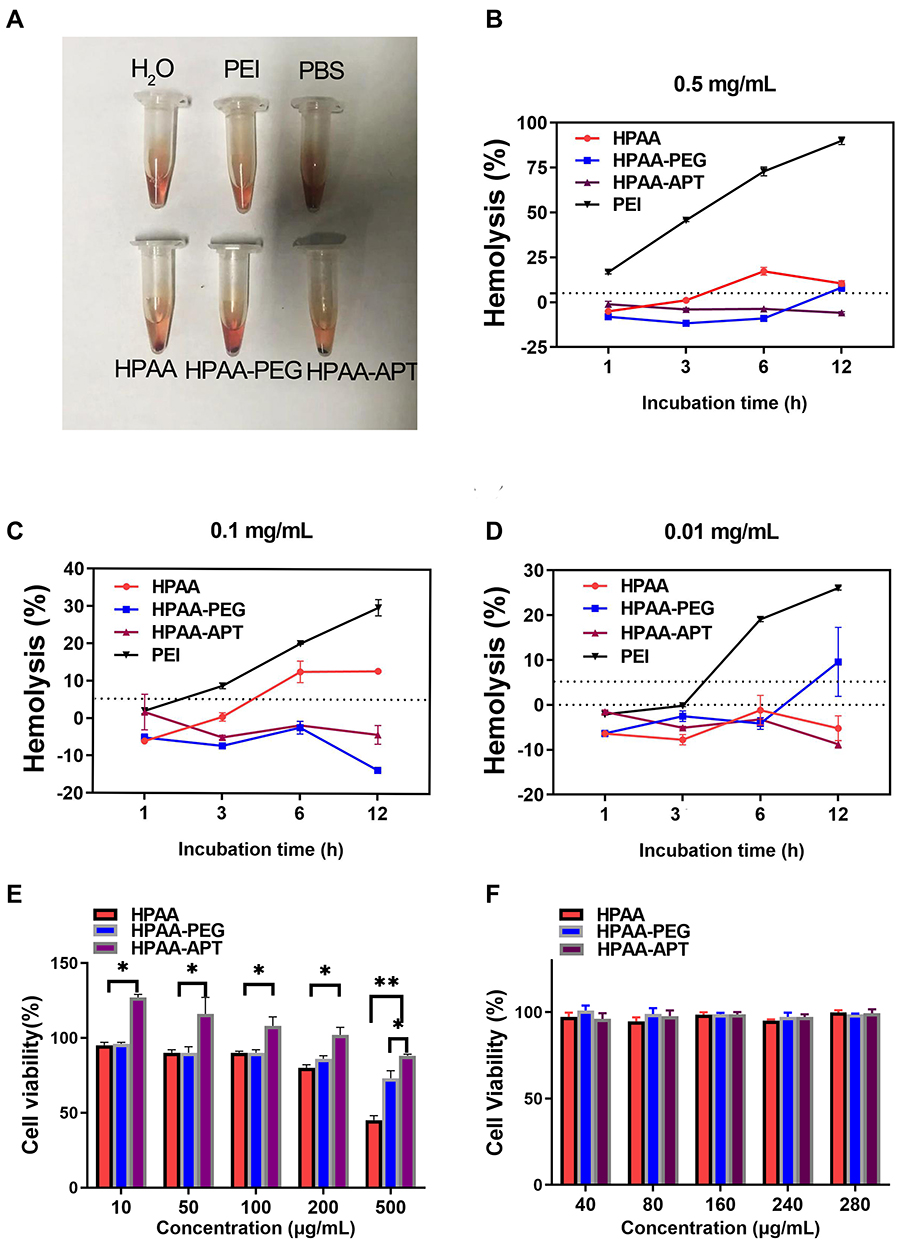 |
Figure 5 The blood safety and toxicity of HPAA derivatives. (A–D) The hemolysis effect of the different concentrations of HPAA derivatives and PEI. (E) CCK-8 results of different concentrations of HPAA derivatives to LNCaP cells. (F) CCK-8 results of different concentrations of HPAA derivatives to HUVEC cells. *p<0.05, **p<0.05. |
Toxicity
When HPAA-PEG-APT was used to treat cancer disease, its cytotoxicity should be evaluated first to confirm safety. The evaluation of cytotoxicity was performed using the CCK-8 assay, and the outcome is displayed in Figure 5. When the concentration of HPAA was more than 0.5 mg/mL, it produced cytotoxicity. By contrast, HPAA-PEG-APT showed little cytotoxicity (Figure 5E). When HPAA-PEG-APT carried the gene into the HUVECs, no cytotoxicity was detected (Figure 5F).
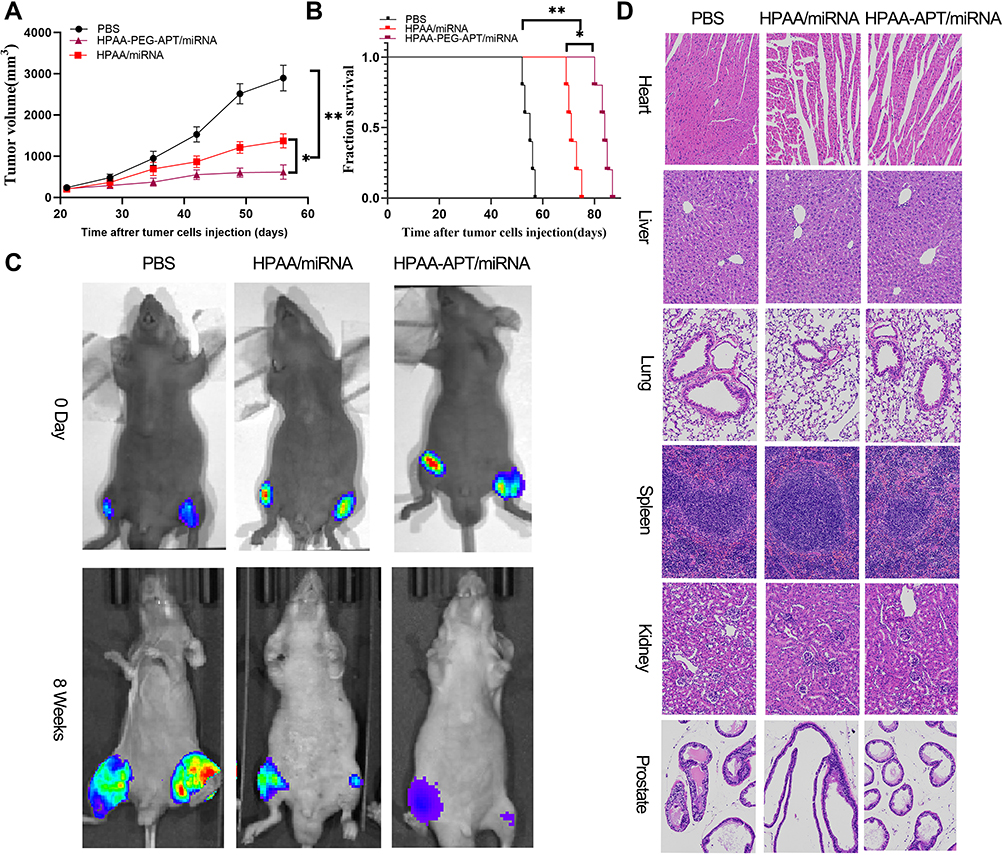 |
Figure 6 In vivo treatment efficacy of HPAA complexes. (A) Tumor growth curve in the different groups. (B) Cumulative survival outcome of in the various treatment groups. (C) Representative bioluminance signal of bone metastasis of a mice at various treatment groups after 8 weeks. (D) representative HE staining of organ histology at various treatment groups. Scale bars:100μm. *p<0.05, **p<0.05. |
Efficacy of Treatments in vivo
The therapeutic effect of HPAA-PEG-APT/miRNA-133a-3p and HPAA/miRNA-133A-3p was explored in a mouse tibial injection tumor model. The representative tumor profile image and tumor growth after different drug treatments are shown in Figure 6. HPAA-PEG-APT/miRNA-133a-3p and HPAA/miRNA-133A-3p demonstrated significant anti-tumor effects. Particularly, the tumors treated with HPAA-PEG-APT/miRNA-133a-3p showed a substantially smaller tumor volume those treated with HPAA/miRNA-133A-3p (Figure 6A). The mice in the HPAA-PEG-APT/miRNA-133a-3p, HPAA/miRNA-133A-3p and control groups died on days 70, 55, and 42, respectively. The anti-tumor effect of the HPAA-PEG-APT/miRNA group was significantly more effective than that of the other groups; 50% of the mice survived more than 83 days, with a maximum survival time of 88 days (Figure 6B). The mice were injected with PBS, HPAA/miRNA-133a-3p and HPAA-PEG-APT/miRNA-133a-3p twice a week for 4 weeks, and the bioluminescent signals of the HPAA/miRNA-133a-3p and HPAA-PEG-APT/miRNA-133a-3p groups were lower than those of the untreated group (Figure 6C). Additionally, the anti-tumor effect of HPAA equipped with APT was more pronounced because of the targeting effect provided by APT, and the nanocomposite can deliver the therapeutic gene to the tumor microenvironment more efficiently in vivo. The in vivo toxicity of HPAA was assessed by H&E staining of vital organs in mice, and no significant difference was found in organ or histology damage between the HPAA and saline groups (Figure 6D), further demonstrating the safety of HPAA-PEG-APT. The above results showed that HPAA-PEG-APT/miRNA-133a-3p is more effective in inhibiting bone resorption.
Conclusion
We successfully constructed HPAA-PEG-APT, which demonstrated good affinity and specificity for PSMA-positive prostate cancer cells and bone metastasis of prostate cancer in vitro and in vivo by loading miRNA-133a-3p. In particular, a reduction-cleavable platform with a gene targeting strategy showed a high-efficiency anti-tumor effect on bone metastasis of prostate cancer tumors and may be a promising gene therapy for cancers.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed on the journal to which the article will be submitted; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest for this work.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7–30. doi:10.3322/caac.21442
2. Adjei IM, Temples MN, Brown SB, Sharma B. Targeted nanomedicine to treat bone metastasis. Pharmaceutics. 2018;10.
3. Saad F, Lipton A, Cook R, Chen YM, Smith M, Coleman R. Pathologic fractures correlate with reduced survival in patients with malignant bone disease. Cancer. 2007;110:1860–1867. doi:10.1002/cncr.22991
4. Altwaijry N, Somani S, Dufès C. Targeted nonviral gene therapy in prostate cancer. Int J Nanomed. 2018;13:5753–5767. doi:10.2147/IJN.S139080
5. Mollaei H, Safaralizadeh R, Rostami Z. MicroRNA replacement therapy in cancer. J Cell Physiol. 2019;234:12369–12384. doi:10.1002/jcp.28058
6. Chen J, Wu Z, Ding W, et al. SREBP1 siRNA enhance the docetaxel effect based on a bone-cancer dual-targeting biomimetic nanosystem against bone metastatic castration-resistant prostate cancer. Theranostics. 2020;10:1619–1632. doi:10.7150/thno.40489
7. Ma D, Liu H, Zhao P, et al. Programing assembling/releasing multifunctional miRNA nanomedicine to treat prostate cancer. ACS Appl Mater Interfaces. 2020;12:9032–9040. doi:10.1021/acsami.9b21707
8. Zhang X, He Z, Xiang L, et al. Codelivery of GRP78 siRNA and docetaxel via RGD-PEG-DSPE/DOPA/CaP nanoparticles for the treatment of castration-resistant prostate cancer. Drug Des Devel Ther. 2019;13:1357–1372. doi:10.2147/DDDT.S198400
9. Wong JKL, Mohseni R, Hamidieh AA, MacLaren RE, Habib N, Seifalian AM. Will nanotechnology bring new hope for gene delivery? Trends Biotechnol. 2017;35:434–451. doi:10.1016/j.tibtech.2016.12.009
10. Gu J, Chen X, Ren X, Zhang X, Fang X, Sha X. CD44-targeted hyaluronic acid-coated redox-responsive hyperbranched poly(amido amine)/plasmid DNA ternary nanoassemblies for efficient gene delivery. Bioconjug Chem. 2016;27:1723–1736. doi:10.1021/acs.bioconjchem.6b00240
11. Wu X, Ding B, Gao J, et al. Second-generation aptamer-conjugated PSMA-targeted delivery system for prostate cancer therapy. Int J Nanomed. 2011;6:1747–1756.
12. Tang Y, Pan J, Huang S, et al. Downregulation of miR-133a-3p promotes prostate cancer bone metastasis via activating PI3K/AKT signaling. J Exp Clin Cancer Res. 2018;37:160.
13. Zhang Y, Wang Z, Gemeinhart RA. Progress in microRNA delivery. J Control Release. 2013;172:962–974. doi:10.1016/j.jconrel.2013.09.015
14. Blanco E, Shen H, Ferrari M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat Biotechnol. 2015;33:941–951. doi:10.1038/nbt.3330
15. Conte R, Valentino A, Di Cristo F, et al. Cationic polymer nanoparticles-mediated delivery of miR-124 impairs tumorigenicity of prostate cancer cells. Int J Mol Sci. 2020;21.
16. Liu T, Li J, Wu X, et al. Transferrin-targeting redox hyperbranched poly(amido amine)-functionalized graphene oxide for sensitized chemotherapy combined with gene therapy to nasopharyngeal carcinoma. Drug Deliv. 2019;26:744–755. doi:10.1080/10717544.2019.1642421
17. Tang Q, Ma X, Zhang Y, Cai X, Xue W, Ma D. Self-sensibilized polymeric prodrug co-delivering MMP-9 shRNA plasmid for combined treatment of tumors. Acta Biomater. 2018;69:277–289. doi:10.1016/j.actbio.2018.01.014
18. Li M, Zhou X, Zeng X, et al. Folate-targeting redox hyperbranched poly(amido amine)s delivering MMP-9 siRNA for cancer therapy. J Materials Chemistry B. 2016;4:547–556. doi:10.1039/C5TB01964H
19. Liu F, Lin L, Sheng S, et al. A glutathione-depleting chemodynamic therapy agent with photothermal and photoacoustic properties for tumor theranostics. Nanoscale. 2020;12:1349–1355. doi:10.1039/C9NR09858E
20. Zhuang Y, Deng H, Su Y, et al. Aptamer-functionalized and backbone redox-responsive hyperbranched polymer for targeted drug delivery in cancer therapy. Biomacromolecules. 2016;17:2050–2062. doi:10.1021/acs.biomac.6b00262
21. Vogel MME, Kroeze SGC, Henkenberens C, et al. Prognostic risk classification for biochemical relapse-free survival in patients with oligorecurrent prostate cancer after [(68)Ga]PSMA-PET-guided metastasis-directed therapy. Eur J Nucl Med Mol Imaging. 2020;47:2328–2338. doi:10.1007/s00259-020-04760-8
22. Hofman MS, Lawrentschuk N, Francis RJ, et al. Prostate-specific membrane antigen PET-CT in patients with high-risk prostate cancer before curative-intent surgery or radiotherapy (proPSMA): a prospective, randomised, multi-centre study. Lancet. 2020;395:1208–1216. doi:10.1016/S0140-6736(20)30314-7
23. Derlin T, Werner RA, Lafos M, et al. Neuroendocrine differentiation and response to PSMA-targeted radioligand therapy in advanced metastatic castration-resistant prostate cancer: a single-center retrospective study. J Nucl Med. 2020;
24. Xu W, Siddiqui IA, Nihal M, et al. Aptamer-conjugated and doxorubicin-loaded unimolecular micelles for targeted therapy of prostate cancer. Biomaterials. 2013;34:5244–5253. doi:10.1016/j.biomaterials.2013.03.006
25. Fan X, Guo Y, Wang L, Xiong X, Zhu L, Fang K. Diagnosis of prostate cancer using anti-PSMA aptamer A10-3.2-oriented lipid nanobubbles. Int J Nanomed. 2016;11:3939–3950. doi:10.2147/IJN.S112951
26. Wu M, Zhao H, Guo L, et al. Ultrasound-mediated nanobubble destruction (UMND) facilitates the delivery of A10-3.2 aptamer targeted and siRNA-loaded cationic nanobubbles for therapy of prostate cancer. Drug Deliv. 2018;25:226–240. doi:10.1080/10717544.2017.1422300
27. Xu X, Wu J, Liu S, et al. Redox-responsive nanoparticle-mediated systemic RNAi for effective cancer therapy. Small. 2018;14:e1802565.
28. Vijayaraghavalu S, Gao Y, Rahman MT, et al. Synergistic combination treatment to break cross talk between cancer cells and bone cells to inhibit progression of bone metastasis. Biomaterials. 2020;227:119558. doi:10.1016/j.biomaterials.2019.119558
29. Adjei IM, Sharma B, Peetla C, Labhasetwar V. Inhibition of bone loss with surface-modulated, drug-loaded nanoparticles in an intraosseous model of prostate cancer. J Control Release. 2016;232:83–92. doi:10.1016/j.jconrel.2016.04.019
30. Arvizo RR, Miranda OR, Moyano DF, et al. Modulating pharmacokinetics, tumor uptake and biodistribution by engineered nanoparticles. PLoS One. 2011;6:e24374. doi:10.1371/journal.pone.0024374
31. Zhang XH, Jin X, Malladi S, et al. Selection of bone metastasis seeds by mesenchymal signals in the primary tumor stroma. Cell. 2013;154:1060–1073. doi:10.1016/j.cell.2013.07.036
32. Jiang WG, Sanders AJ, Katoh M, et al. Tissue invasion and metastasis: molecular, biological and clinical perspectives. Semin Cancer Biol. 2015;35(Suppl):S244–S275. doi:10.1016/j.semcancer.2015.03.008
33. Polivka J
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.