Back to Journals » Drug Design, Development and Therapy » Volume 20
Protective Effect of Salidroside Against Multiple Organs Ischemia-Reperfusion Injury: New Insights From Common and Specific Pharmacological Mechanisms
Authors Chi X ![]() , Zheng S, Guo C, Wang J, Chang Z
, Zheng S, Guo C, Wang J, Chang Z ![]() , Cao X, Jiang L, Zhang Y, Shen W
, Cao X, Jiang L, Zhang Y, Shen W
Received 13 March 2026
Accepted for publication 5 June 2026
Published 12 June 2026 Volume 2026:20 608894
DOI https://doi.org/10.2147/DDDT.S608894
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Tin Wui Wong
Xiansu Chi,1,* Shuang Zheng,1,* Chunyan Guo,2 Jiawei Wang,1 Ze Chang,1 Xiaoxuan Cao,1 Libin Jiang,1 Yunling Zhang,1 Wei Shen1
1Department of Encephalopathy, Xiyuan Hospital of the China Academy of Chinese Medical Sciences, Beijing, People’s Republic of China; 2Graduate School, Beijing University of Chinese Medicine, Beijing, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yunling Zhang, Wei Shen, Department of Encephalopathy, Xiyuan Hospital of the China Academy of Chinese Medical Sciences, No. 1 Xiyuan Caochang Road, Haidian District, Beijing, 100091, People’s Republic of China, Tel +86 010-62835301, Email [email protected]; [email protected]
Abstract: Ischemia-percussion (I/R) injury describes a paradoxical phenomenon in which blood flow restoration following ischemia not only fails to salvage the tissue but also exacerbates tissue injury. Due to the unique physiological functions, cell types, and metabolic requirements of different tissues, their responses and vulnerability to ischemia/reperfusion injuries vary significantly. The limited understanding of complex mechanisms underlying I/R injury in different organs further thwarts the advancement of effective therapeutic interventions, the translational application of promising interventional strategies still faces many obstacles. Salidroside, a natural bioactive constituent of traditional medicinal plants, demonstrates great potential in combating I/R injuries in various organs. Salidroside exerts organ-specific regulatory effects based on the unique characteristics of damage in different organs, simultaneously alleviates I/R injuries by common mechanism include inhibiting oxidative stress, reducing inflammatory responses, regulating cell apoptosis, and inhibiting ferroptosis. To summarize the evidence, we searched the PubMed and Web of Science databases from inception to February 28, 2026, using “salidroside” (and its synonym “rhodioloside”) and “ischemia-reperfusion injury” (and its MeSH term “reperfusion injury”) as core keywords. This narrative review summarizes the common and organ-specific protective mechanisms of salidroside against I/R injury across different organs and points out the limitations of existing studies in terms of evidence quality, dose standardization, thereby providing research directions for further development of related pharmaceutical formulations and their clinical translation.
Keywords: salidroside, ischemia-reperfusion injury, multiple organs, pharmacological mechanism
Introduction
Ischemia-reperfusion (I/R) injury refers to the phenomenon in which, after a period of ischemia, the restoration of blood flow by reperfusion not only fails to salvage the ischemic tissue but instead triggers a series of pathological mechanisms, exacerbates the preexisting reversible injury that occurred during ischemia, activates cell death pathways, and ultimately leads to irreversible tissue destruction.1 Currently, standard clinical treatment strategies for ischemic diseases remain centered on early reperfusion interventions, the fundamental goals of these strategies are to restore blood flow as quickly as possible and to salvage the dying tissue. Paradoxically, reperfusion itself can cause additional damage by eliciting an oxidative stress burst, an inflammatory cascade, calcium overload, mitochondrial dysfunction and so on, thereby inducing or exacerbating tissue injury.2,3 Therefore, although reperfusion therapy greatly reduces mortality from acute ischemic events, the additional injury caused by I/R remains an important factor affecting patient prognosis and, to some extent, weakens the clinical benefit of the treatment. As a “silent killer” underlying major clinical events, I/R injury acts as an important factor contributing to high mortality and disability rates among affected patients, substantial healthcare costs, and immense physical and psychological burden.
The severity and outcome of I/R injury depend on several factors, such as ischemic duration, tissue type, collateral circulation, and reperfusion speed.3–5 It should be clarified that the pathological mechanisms of I/R injury do not occur simultaneously or complete instantaneously. The ischemic phase is primarily characterized by energy metabolism disorders and the reperfusion phase is marked by a burst of reactive oxygen species (ROS) triggering oxidative stress, the infiltration of neutrophils and macrophages, as well as the release of inflammatory cytokines peaking several hours after reperfusion, and multiple cell death, thereby exacerbating tissue damage and organ dysfunction. The temporal dynamics of regulated cell death during I/R injury vary across different organs, for example, in the heart the apoptosis is initiated in the early stage of reperfusion and persists into the later stage, changes in autophagy are also predominantly observed in the early phase; whereas ferroptosis and necrosis dominate the medium-to-long-term stage.6 In contrast, ferroptosis are triggered within the first few hours after cerebral reperfusion, while apoptosis occurs at a later stage.7 Although I/R injury shares some common features across different organs, significant variations still exist in the response and susceptibility to I/R injury due to differences in physiological functions, cell types, metabolic demands, and microvascular structures among organs.8,9 Understanding the mechanisms and characteristics of the I/R process between different organs is crucial for further development of related pharmaceutical formulations and their clinical translation.
Currently, almost no drugs specifically targeting the molecular mechanisms of I/R injury have received regulatory approval.10 Existing therapeutic strategies are primarily in the preclinical research stage, and their clinical translation and application still face numerous shortcomings. A summary of current treatment strategies is provided below. Firstly, a large body of preclinical research has confirmed that inhibitors of the mitochondrial permeability transition pore (including cyclosporine A and TRO40303) can ameliorate I/R injury in multiple organs by preventing or delaying mitochondrial permeability transition pore opening, preserving mitochondrial integrity, and inhibiting cell death. However, the outcomes of clinical translation have been unsatisfactory. In a pivotal Phase III clinical trial for cardiac I/R injury, cyclosporine A failed to improve clinical outcomes in patients with ST-segment elevation myocardial infarction. This failure may be attributed to the high interindividual variability (10%–20%) in peak blood concentration and area under the drug concentration-time curve, as well as the strict dose- and time-dependent nature of its protective effects.11 Similarly, clinical trials of TRO40303 showed no significant differences compared to the placebo group in primary efficacy endpoints such as myocardial infarct size, myocardial salvage index, and left ventricular ejection fraction. Moreover, the TRO40303 group experienced a higher number of adverse events with no definitive causality, raising concerns about its safety.12 Mitochondrial-targeted therapies, such as elamipretide (SS-31, MTP-131, Bendavia), can stabilize mitochondrial structure and restore energy metabolism. However, favorable results were not observed in the Phase II clinical trial and a significant translational gap remains between animal models and clinical trials; simply targeting a single aspect of mitochondrial function may be insufficient to address the complex pathological network of I/R injury in human body.13 Secondly, antioxidants are also important, such as N-acetylcysteine, a classic antioxidant and glutathione precursor, exhibits protective effects against myocardial infarction caused by I/R that vary significantly with the route of administration. To achieve protection, a stable blood concentration of N-acetylcysteine must be maintained early during reperfusion.14 Malonate can inhibit the activity of succinate dehydrogenase, reduce the burst of reactive oxygen species, preserve mitochondrial integrity, and has shown protective effects in I/R injury across multiple organs. However, its neurotoxicity, high dependence on precise administration within the early window of reperfusion (which is difficult to control in clinical practice), and poor cell membrane permeability severely limit its clinical application.15,16
Additionally, natural products are gradually gaining attention from researchers. Various natural products, including phenolic compounds, alkaloids, and saponins, have been shown to exert protective effects by modulating multiple targets, making them promising candidate drugs for future treatment of I/r injury.17 Resveratrol, which possesses anti-inflammatory, oxidative damage-attenuating, and cell integrity-protective effects, exhibiting anti-I/R injury protection in multiple organs including the heart, brain, lung, liver, kidney, and intestine. Pharmacokinetic studies indicate that resveratrol is rapidly metabolized, undergoes extensive conjugation reactions, and is quickly cleared via enterohepatic circulation, leading to low bioavailability and significant interindividual variability, which may be the main factors limiting its clinical application.18 Curcumin can ameliorate multi-organ I/R injury through anti-inflammatory, antioxidant, and anti-apoptotic effects. However, its low solubility, rapid metabolism, along with oral administration potentially causing gastrointestinal discomfort, hepatotoxicity, coagulation inhibition, and hypotensive/hypoglycemic risks, currently restrict its use to adjuvant therapy for ischemic events like myocardial infarction, requiring immediate administration after symptom onset.19–21 Berberine not only ameliorates I/R injury through mechanisms such as antioxidation, anti-apoptosis, anti-inflammation, inhibition of endoplasmic reticulum stress, and promotion of autophagy but also modulates the peripheral immune system. Although clinical evidence remains limited, existing studies suggest berberine has been used in patients with cardiac or cerebral I/R injury, exerting protection by inhibiting excessive inflammatory responses.22 However, the low bioavailability of berberine, which is prone to hydrolytic degradation by gastrointestinal microbiota or enzymes after oral administration, poses a significant challenge for clinical use. Components like quercetin and ginsenosides, among many others, are also being investigated for treating I/R injury. Additionally, the use of traditional Chinese medicine alone or in formulations has shown some therapeutic effects on I/R injury, but its clinical translation has a much longer road ahead.
Moreover, gene therapy and biomimetic nanoparticle delivery systems are emerging as research hotspots and show potential as adjunctive strategies for treating I/R injury. Among these, biomimetic nanoparticles, with their excellent biocompatibility, low immunogenicity, and active targeting capabilities, have become ideal delivery vehicles.23 Gene therapy still faces numerous challenges in terms of delivery efficiency, targeting, long-term safety, and clinical feasibility.3 Both gene therapy and biomimetic nanoparticle delivery systems are still in their early stages and require in-depth mechanistic exploration and rigorous clinical validation; whether they can truly achieve clinical application remains a serious question. Hypothermia therapy is considered capable of acting on multiple targets of I/R injury simultaneously. However, at least six randomized controlled trials have shown negative results for their primary endpoints. The reasons for this failure are likely the clinical impracticality of overcoming the inherent defects of “slow onset, whole-body cooling, and significant side effects”.24 Overall, the translation of existing treatment strategies for I/R injury from laboratory to clinic faces multiple obstacles: significant differences between the idealized conditions of animal and cellular experiments and real-world clinical scenarios; pharmacokinetic deficiencies and safety concerns restricting drug application; and the lack of systematic consideration of interindividual variability and organ-specific characteristics. Consequently, there is an urgent clinical need to explore novel drugs with multi-target regulatory capabilities, favorable safety profiles and achieve stable efficacy with ideal bioavailability.
Salidroside is the primary active compound extracted from the roots of Rhodiola rosea. Rhodiola rosea thrives on alpine rocks and cliffs, grows to heights of 5–40 cm, which is primarily found in the Arctic regions of Europe, Asia, and North America.25,26 The medicinal use of Rhodiola rosea was first documented as early as 77 AD by the Greek physician Dioscorides.27,28 In China, Rhodiola rosea is commonly prescribed to replenish qi, boost physical strength, enhance bodily functions, alleviate fatigue, strengthen immunity, improve anemia, reduce anxiety and depression, treat erectile dysfunction, and promote longevity.27,29–31 The bioactive compounds found in Rhodiola rosea include alkaloids, glycosides, phenols, essential oils, coumarins, sterols, and organic acids.31 Among these compounds, as shown in Figure 1, salidroside has anti-inflammatory, antioxidant, anti-tumor, anti-fatigue, anti-aging, anti-diabetic, anti-apoptotic, and immunomodulatory properties, helps lower blood lipid levels, and improves hemodynamic parameters, to protect multiple systems, including the cardiovascular, nervous, respiratory, digestive, and renal systems and so on.32–34
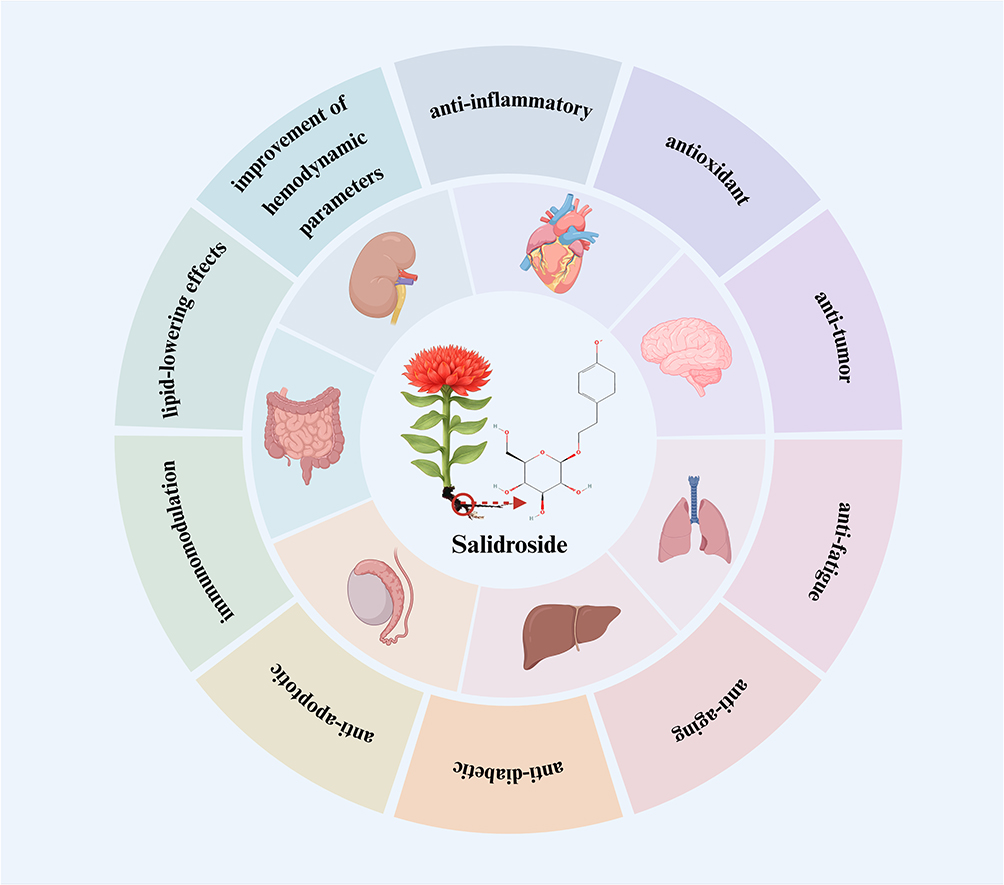 |
Figure 1 Primary pharmacological mechanisms of salidroside on different organs. The main pharmacological mechanisms encompass anti-inflammatory, antioxidant, anti-tumor, anti-fatigue, anti-aging, anti-diabetic, and anti-apoptotic activities, along with immunomodulation, lipid-lowering effects, and the improvement of hemodynamic parameters. |
Nowadays, whether salidroside exerts a protective effect against I/R injury in different organs, as well as the common and specific mechanisms by which it regulates I/R injury across various organs, has not yet been systematically summarized. To summarize the evidence, we conducted a literature search by searching the PubMed and Web of Science databases from inception to February 28, 2026, using “salidroside” (and its common synonym “rhodioloside”) and “ischemia-reperfusion injury” (and its MeSH term “reperfusion injury”) as core keywords. No clinical studies investigating the pure compound salidroside in the context of I/R injury were identified. Therefore, this review focuses on both in vivo and in vitro preclinical studies. Specially, studies using crude Rhodiola rosea extracts or multi-ingredient preparations were excluded from the main analysis, as the presence of other compounds prevents the specific attribution of any observed effects to salidroside alone. The literature review is used only for citation tracing and additional studies are identified by manually screening reference lists. This narrative review aims to integrate recent research findings, summarize the protective roles of salidroside against multi-organ I/R injury, and clarify both its common and specific mechanisms, thereby deepening the understanding of protective effects and its mechanisms of salidroside, and providing a more comprehensive reference for the further pharmacological investigation, formulation development, and clinical translation of salidroside.
Salidroside and Cardiac I/R Injury
The cardiac tissue exhibits the highest oxygen consumption, with energy production almost entirely dependent on aerobic metabolism, rendering it highly sensitive to I/R injury. Following acute myocardial infarction, myocardial cells are more susceptible than other cells to the cessation of oxidative phosphorylation, leading to ATP depletion and triggering intracellular acidosis, calcium overload, and mitochondrial membrane depolarization.35 Coronary angioplasty, thrombolytic therapy, or direct percutaneous coronary intervention enable early revascularization and salvage ischemic myocardium; however, this abrupt reperfusion also triggers multiple mechanisms, including calcium overload, oxidative stress, inflammation, and mitochondrial dysfunction, acts synergistically to worsen myocardial injury, which severely compromises the intended benefits of reperfusion therapy.36 The pathophysiology of cardiac I/R injury is intimately linked to mitochondrial dysfunction, oxidative stress, calcium overload, and inflammatory responses.
The cardioprotective effects exerted by salidroside entails an intricate interplay of mechanisms, primarily achieved through synergistic regulation of multiple biological pathways. Currently, as shown in Figure 2, the protective mechanisms of salidroside against cardiac I/R injury encompass the following aspects: modulating energy metabolism and mitochondrial function, suppressing oxidative stress, alleviating inflammatory responses, inhibiting apoptosis, regulating autophagy, and controlling non-coding RNA networks.
 |
Figure 2 The protective mechanism of salidroside against cardiac ischemia-reperfusion injury. In this figure, different colors represent distinct protective mechanisms of salidroside: The green section shows that salidroside primarily regulates energy metabolism by activating the AMPK pathway and modulates mitochondrial function by preventing Drp1 translocation to mitochondria. The Orange section shows that salidroside alleviates oxidative stress by inhibiting the p38/MAPK pathway. The yellow section shows that salidroside attenuates inflammatory responses by downregulating TLR4 expression and suppressing the downstream NF-κB pathway. The gray section shows that salidroside inhibits apoptosis by suppressing key caspases, upregulating Bcl-2, and downregulating Bax. The blue section shows that salidroside not only inhibits harmful excessive activation of autophagy but also promotes beneficial mitophagy to clear damaged mitochondria and the purple section shows that salidroside also regulates the cardiac non-coding RNA network and promotes cardiac repair. Symbol definitions: A standard arrow (→) indicates activation or positive regulation. A flat-ended arrow (―|) indicates inhibition or negative regulation. An upward arrow (↑) indicates increase, activation, or upregulation. A downward arrow (↓) indicates decrease, inhibition, or downregulation. |
Improvement of Cardiac Energy Metabolism and Mitochondrial Function
Adenosine 5’-monophosphate-activated protein kinase (AMPK) is a central regulator of cellular metabolism and mitochondrial homeostasis,37 and salidroside could enhance cardiac bioenergetics and mitochondrial function by activating the AMPK signaling pathway. Salidroside upregulates peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) and peroxisome proliferator-activated receptor alpha (PPAR-α) through AMPK activation, thereby rebalancing energy metabolism and restoring intracellular ATP and glycogen levels to improve energy supply.38 Furthermore, AMPK stimulation by salidroside suppresses endoplasmic reticulum stress and inhibits mitochondrial fission by preventing the translocation of dynamin-related protein 1 (Drp1) to mitochondria, thereby reducing apoptosis associated with mitochondrial fragmentation.39 Additionally, salidroside mitigates Fe2⁺ accumulation and redox imbalance during cardiac I/R injury via the AMPKα2 pathway, counteracting ferroptosis while preserving mitochondrial energy production and function.40 Since cardiomyocytes have the highest energy consumption, and ATP depletion along with energy metabolism disorder is the main mechanism of cardiac I/R injury, salidroside exerts significant cardioprotective effects by activating the AMPK pathway.
Suppression of Cardiac Oxidative Stress
Oxidative stress is the key pathological mechanism of cardiac I/R injury, and the adverse remodeling it induces can impede the recovery of cardiac function.41 Multiple studies have demonstrated that salidroside serves as an effective inducer of antioxidant enzymes, such as SOD and glutathione peroxidase (GPX), while concurrently reducing malondialdehyde (MDA) levels and inhibiting the ROS generation.41–44 The mechanism by which salidroside repairs I/R injury through its antioxidant effects is mainly associated the inhibition of p38/mitogen-activated protein kinase (MAPK) pathway include downregulating the level of p38, p-p38, JNK, p-JNK, and so on.45 Because a large amount of ROS is explosively generated in the early stage of reperfusion, and the antioxidant capacity of the myocardium during I/R is relatively limited, to address this issue, the effect of salidroside on activating AMPK and ERK, which are sensitive to redox status, can enhance the endogenous antioxidant defense system, while inhibiting p38 MAPK can directly attenuate the source of ROS.
Mitigation of Cardiac Inflammatory Responses
NF-κB signaling dysregulation underlies key processes including inflammation, immunity, proliferation, and differentiation.46 Salidroside inhibits the phosphorylation of key NF-κB pathway components (p65, IκBα, IKKα, and IKKβ), thereby suppressing the subsequent release of pro-inflammatory cytokines like TNF-α, IL-6, and IL-1β.38,43,47 Furthermore, TLR4 serves as an upstream activator of the NF-κB pathway. By down-regulating TLR4 expression and thus inhibiting the downstream NF-κB pathway, salidroside mitigates cardiac I/R injury.47
Inhibition of Cardiac Apoptosis
Apoptosis is a genetically controlled, active, and programmed cell death process, mediated by caspase signaling, inducing a self-contained cell death program characterized by cell shrinkage and nuclear condensation, with plasma membrane integrity maintained until the late stages of the process.48 Salidroside inhibits key caspases in the apoptotic pathway, notably caspase-9 and caspase-3,47,49,50 and modulates the balance between the anti-apoptotic protein Bcl-2 and the pro-apoptotic protein Bax. Moreover, the pro-survival and anti-apoptotic effects of salidroside are mediated by activation of the phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt) signaling pathway.49 Meanwhile, salidroside blocks the phosphorylation of c-Jun N-terminal kinase (JNK) and reduces cytochrome c release, thereby blocking mitochondrial apoptosis.50 Given the extremely weak regenerative capacity of cardiomyocytes, the cell loss caused by apoptosis is difficult to compensate for. Salidroside can maximize the preservation of cardiomyocytes by inhibiting apoptosis.
Modulation of Cardiac Autophagy
Autophagy represents an adaptive alteration to meet the metabolic needs of the cell itself and to renew certain organelles. Salidroside modulates the cardiac autophagy by exhibiting a dual role, not only suppressing the detrimental overactivation of autophagy but also promoting beneficial mitophagy to clear damaged mitochondria.42
Regulation of Cardiac Non-Coding RNA Networks
Salidroside promotes cardiac repair after I/R injury via regulating cardiac noncoding RNA networks. It upregulates microRNA (miR)-21 expression, thereby attenuating oxidative stress and inflammatory responses.43 Furthermore, salidroside suppresses circ0097682, which sequesters miR-671-5p, leading to miR-671-5p release and the subsequent targeting of USP46. This mechanism ultimately reduces cellular damage. Moreover, the effect of salidroside in the myocardium I/R injury is related to circular RNAs circ0000064.42 By regulating the non-coding RNA network, salidroside can simultaneously affect multiple effector processes at the post-transcriptional level.
Salidroside and Cerebral I/R Injury
The brain has a high oxygen demand and is highly sensitive to I/R injury. Post-stroke arterial blood flow obstruction interrupts oxygen supply, causing a sharp decline in adenosine triphosphate (ATP) production. Ionic pump dysfunction readily leads to ion homeostasis disruption, subsequently triggering cytotoxic edema. Although reperfusion therapies like thrombolysis and thrombectomy can salvage brain tissue retaining partial blood flow after stroke, reperfusion may also exacerbate injury.34 Notably, after I/R, restoring blood flow to ischemic tissue triggers a cascade of direct and indirect pathological events that exacerbate oxidative stress,51 activate inflammatory responses, excitatory amino acid toxicity, and calcium overload, compromising the blood-brain barrier. Notably, cerebral I/R injury significantly accelerates neuronal injury and induces neurological dysfunction.52,53
Researches have repeatedly confirmed that salidroside reduces cerebral edema, decreases infarct volume, improves neurological scores,54,55 and exerts neuroprotection within a broad therapeutic time window after cerebral I/R injury.56 As shown in Figure 3, salidroside acts as a neuroprotective agent against cerebral I/R injury by alleviating oxidative stress, inhibiting neuroinflammation, suppressing ferroptosis, inhibiting neuronal apoptosis, modulating autophagy, regulating neural repair and synaptic plasticity, and promoting the dopaminergic system.
 |
Figure 3 The protective mechanism of salidroside against cerebral ischemia-reperfusion injury. Regarding oxidative stress, salidroside increases SOD and GPX levels, decreases MDA and GST levels, and significantly enhances Nrf2 expression, thereby activating the endogenous antioxidant system, which in turn upregulates HO-1 and Trx1. Regarding inflammatory response, salidroside inhibits NLRP3 assembly by suppressing the NF-κB pathway via TLR4, or alternatively modulates the NF-κB pathway through RIP140. It also regulates Th17/Treg cell balance via the STAT3 signaling pathway. Regarding ferroptosis, salidroside inhibits ferroptosis by activating the PINK1/Parkin pathway or by regulating FUNDC1. Regarding neuronal apoptosis, salidroside inhibits apoptosis by activating the PI3K/Akt and SIRT1/FoxO3a pathways, or via the Nrf2/Trx1 axis to suppress ASK1 and MAPK family proteins. Regarding neuronal autophagy, salidroside activates the AMPK/TSC2/mTOR pathway, or activates ERβ/BNIP3-mediated mitophagy. Regarding regulation of neural repair and synaptic plasticity, salidroside promotes neural functional reconstruction by regulating the Notch1/Hes1 pathway, or the FGF2-mediated cAMP/PKA/CREB signaling pathway, and by modulating neurotrophic factors, nerve growth factor, and growth response proteins. Regarding the dopamine system, salidroside can increase dopamine secretion by enhancing tyrosine hydroxylase and reducing MAO-B. Symbol definitions: A standard arrow (→) indicates activation or positive regulation. A flat-ended arrow (―|) indicates inhibition or negative regulation. An upward arrow (↑) indicates increase, activation, or upregulation. A downward arrow (↓) indicates decrease, inhibition, or downregulation. |
Alleviation of Oxidative Stress
In several studies, salidroside can enhance SOD and glutathione-S-transferase activity57–60 while reducing MDA levels and lipid peroxidation, thereby protecting against cerebral I/R injury.58,61 As a key link in the endogenous antioxidant defense of brain tissue, Nrf2 expression is enhanced by salidroside, thereby upregulating heme oxygenase-1 (HO-1) and thioredoxin 1 (Trx1) expression.58,62
Inhibition of Neuroinflammation
Salidroside shows great promise in repairing damaged brain tissue by the inhibition of neuroinflammation, with literature extensively confirming the significant reduction in TNF-α, IL-6, IL-1β, and monocyte chemotactic protein-1 (MCP-1) expression.62–65 One mechanism by which salidroside may function is through the TLR4-NF-κB pathway, thereby inhibiting NLR family pyrin domain-containing 3 (NLRP3) inflammasome assembly,64 or the suppression of NF-κB signaling mediated by receptor-interacting protein 140 (RIP140).63 Moreover, salidroside inhibits complement system activation by reducing complement activation via the lectin (eg., mannose-binding lectin 2) and classical pathways (eg., C1q), thereby decreasing C3 and C3a deposition to inhibit inflammatory responses. At the same time, by acting via the signal transducer and activator of transcription 3 (STAT3) signaling salidroside normalizes the Th17/Treg balance, leading to reduced inflammatory responses.65 The brain is highly sensitive to inflammatory responses. Microglia, as the intrinsic immune cells of the brain, release large amounts of pro-inflammatory cytokines when overactivated, forming a neurotoxic microenvironment. Furthermore, the blood-brain barrier restricts the entry of peripheral anti-inflammatory drugs. By targeting key pathways such as TLR4/NF-κB, NLRP3, and STAT3, salidroside achieves multi-level inhibition of neuroinflammation.
Suppression of Ferroptosis
Because the burst of ROS generated after cerebral I/R readily attacks membrane lipids and triggers a chain reaction of lipid peroxidation, while the endogenous capacity of neurons to resist ferroptosis is extremely limited, ferroptosis will be triggered within the first few hours after reperfusion. On the one hand, salidroside inhibits ferroptosis by activating the PINK1/Parkin signaling pathway;66 on the other hand, salidroside also regulates FUNDC1, a critical bridge between mitophagy and ferroptosis, and facilitates FUNDC1-mediated non-ubiquitin-dependent mitophagy, which suppresses ferroptosis and consequently attenuates cerebral I/R injury.67
Attenuation of Neuronal Apoptosis
It is extensively documented that salidroside modulates the balance of apoptosis-related proteins, notably by increasing the Bcl-2/Bax ratio and suppressing caspase-3 activity, or reducing the levels of the pro-apoptotic protein P53 to inhibit the neuronal apoptosis in the brain.68–71
Salidroside can not only activate the PI3K/Akt and SIRT1/FoxO3a pathways to inhibit apoptosis and promote neuronal survival,68,69 thereby exerting neuroprotective effects, but also suppress ASK1 and MAPK family proteins through the Nrf2/Trx1 axis, thereby interrupting the apoptotic signaling cascade.58
Modulation of Neuronal Autophagy
Salidroside could activate the AMPK/TSC2/mTOR signaling or activate ERβ/BNIP3-mediated mitochondrial autophagy, thereby mitigating cellular damage in brain tissue.72,73 AMPK is activated by salidroside upon ATP depletion in the brain to initiate autophagy via TSC2-mediated inhibition of mTORC1, whereas salidroside through ERβ/BNIP3 pathway to selectively mediate mitophagy, thereby meeting the urgent need for efficient clearance of damaged organelles.
Regulation of Neural Repair and Synaptic Plasticity
Through the Notch1/Hes1 pathway, salidroside promotes neural repair by stimulating the expression of brain-derived neurotrophic factor (BDNF) and nerve growth factor (NGF).74 Furthermore, salidroside improves synaptic plasticity by upregulating the expression levels of early growth response protein (EGR)1, EGR2, EGR4, and activity-regulated cytoskeleton-associated protein (ARC), as well as via the fibroblast growth factor-2 (FGF2)-mediated cAMP/PKA/CREB signaling pathway.56,70 Cerebral I/R injury leads to extensive synaptic loss and disruption of neural network connectivity, salidroside could promote the reconstruction of neural network function by activating these above nerve growth factors.
Promotion of the Dopaminergic System
Salidroside modulates the dopaminergic system, increasing levels of dopamine and its metabolites (3,4-dihydroxyphenylacetic acid and homovanillic acid), enhances tyrosine hydroxylase expression, and improves neurological function.54 At the same time, salidroside and its derivatives (eg., p-benzoyl salidroside) are found to competitively inhibit the activity of monoamine oxidase B, reduce neurotransmitter degradation, and protect dopaminergic neurons.56,75
Salidroside and Pulmonary I/R Injury
I/R injury of the lung is a serious complication after trauma, embolism, and cardiothoracic surgery, such as cardiopulmonary bypass and pulmonary transplantation. Pulmonary I/R injury is characterized by endothelial dysfunction, capillary leakage, and inflammatory response; among them, damage to the endothelial cells of pulmonary capillaries is characteristic of pulmonary I/R injury.75 After pulmonary I/R, burst generation of ROS triggers lipid peroxidation and oxidative stress, subsequently, complement activation, chemokine release, and upregulation of adhesion molecules promote massive sequestration and activation of neutrophils in pulmonary capillaries, thereby exacerbating inflammation.76,77 Alveolar epithelial cells are directly exposed to hyperoxia and ROS attack after I/R, making them susceptible to ferroptosis and leading to protein-rich fluid leakage into the alveoli and subsequent pulmonary edema.78 The above mechanisms collectively cause decreased lung compliance, pulmonary hypertension, and ventilation-perfusion mismatch, and may progress to acute lung injury or even ARDS.79
Currently, research on pulmonary I/R treatment with salidroside remains relatively scarce. The reason may be that the clinical scenarios of pulmonary I/R are relatively confined, with a relatively small patient population, resulting in a weaker driving force for drug development. Based on current research findings, salidroside primarily mitigates pulmonary I/R injury by inhibiting ferroptosis. Salidroside could enhance cellular resistance to lipid peroxidation by activating the core antioxidant pathway Nrf2/SLC7A11/GPX4, thereby directly suppressing ferroptosis.80 Furthermore, salidroside alleviates inflammatory responses and suppresses ferroptosis, reduce pulmonary cell apoptosis by inhibiting the pro-inflammatory JAK2/STAT3 signaling pathway.81 In addition, another in vivo and in vitro study confirmed that salidroside enhances the anti-ferroptosis effect by mediating the MAFK/PTOV1-AS2/miR-525-5p/ACE2 molecular pathway.82 Through the synergistic effects of these pathways, salidroside collectively reduces oxidative damage, diminishes inflammatory cell infiltration, and suppresses ferroptosis, and thereby significantly ameliorates the pulmonary histopathological changes and pulmonary dysfunction.
Salidroside and Hepatic I/R Injury
I/R injury of the liver is the leading cause of death after liver surgery and transplantation.83 The liver’s functionality is intricately linked to a consistent supply of oxygen, making it particularly susceptible to hepatic hypoxia in the event of blood flow obstruction. During reperfusion, there is a notable increase in oxygen concentration, which can lead to an overproduction of ROS. Concurrently, the intracellular antioxidant defense mechanisms, including glutathione and superoxide dismutase (SOD), become compromised and are unable to effectively neutralize the excess ROS. This situation further exacerbates oxidative stress within the liver.84 Then, liver sinusoidal endothelial cells are damaged by ROS and inflammatory mediators, leading to the diminished production of nitric oxide, vasoconstriction, and microvascular obstruction. This results in the “no-reflow” phenomenon, exacerbating hypoxia within the ischemic zone. Particularly, Oxidative stress acts as a trigger, activating hepatic Kupffer cells and monocyte-derived macrophages, and culminating in the release of diverse pro-inflammatory cytokines, chemokines, and other signaling molecules, triggering an intense local and systemic inflammatory response, thereby resulting in hepatic failure, systemic inflammatory response syndrome (SIRS), and multiple organ dysfunction syndrome (MODS;85,86).
Current research suggests that salidroside mainly alleviates hepatic inflammatory storms and suppresses oxidative stress in the liver to repair the hepatic I/R injury.
Alleviation of Hepatic Inflammatory Storms
By antagonizing TLR4, salidroside blocks the downstream NF-κB/NLRP3 pathway. This interference curtails the assembly of the NLRP3 inflammasome and the subsequent maturation and release of inflammatory factors, thereby markedly alleviating inflammatory damage.87 Notably, the liver is rich in Kupffer cells, which, once activated after reperfusion, trigger an inflammatory storm via the TLR4/NF-κB/NLRP3 pathway, while the JAK2/STAT3 pathway amplifies this effect. Salidroside inhibits the above two pathways, thereby blocking the initiation and amplification of inflammation in hepatic I/R injury.88
Suppression of Hepatic Oxidative Stress
Due to the highly active metabolism of hepatocytes, the endogenous antioxidant defense system is crucial for maintaining the cellular redox balance. Salidroside can activate Nrf2, the master node of the antioxidant system, via upstream activation of Akt signaling and inhibition of GSK-3β, leading to Nrf2 nuclear translocation and subsequent upregulation of HO-1 and NQO-1.88,89 Consequently, salidroside elevates the activity of antioxidant enzymes (eg., SOD, catalase, GPX) and lowers MDA content.88
Inhibition of Apoptosis
The liver has a high mitochondrial density and hepatocytes are rich to synthesize large amounts of secretory proteins. After I/R, mitochondrial damage, endoplasmic reticulum stress, and inflammatory responses intertwine, predisposing the liver to extensive apoptosis. Salidroside enhances mitochondrial resistance to damage by inhibiting mitochondrial permeability transition pore opening, reducing cytochrome c release, and downregulating caspase-3 activity.89 Administration of salidroside also upregulates the anti-apoptotic protein Bcl-2 and downregulates the pro-apoptotic protein Bax, thereby restoring the Bcl-2/Bax balance.90 Additionally, salidroside inhibits the IRE1α/JNK pathway, downregulates the expression of CHOP and caspase-12, and alleviates endoplasmic reticulum stress-induced apoptosis.91 Meanwhile, salidroside could significantly inhibit the phosphorylation of key MAPK family members, such as p38, JNK, and ERK, ameliorate inflammatory responses and apoptosis, and repair hepatocyte I/R injury.92
Salidroside and Renal I/R Injury
Renal I/R injury, which frequently occurs during procedures such as complex kidney stone extraction, extracorporeal shockwave lithotripsy, major vascular surgery, shock, and organ transplantation, often leads to acute kidney injury. Blood flow within the kidney is inhomogeneous, with the renal medulla, particularly the outer medulla, receiving only approximately 15% of the total renal blood flow, resulting in relatively low baseline perfusion. Owing to its high metabolic demand for oxygen, the medulla is highly vulnerable to ischemic injury.93 Notably, reperfusion-induced increases in renal blood flow elevate the glomerular filtration rate, which in turn further augments oxygen demand and predisposes the kidney to an imbalance between oxygen supply and demand.94 Following prolonged ischemia, reperfusion paradoxically intensifies renal dysfunction and structural injury instead of promoting recovery, which resulting in compromised metabolic waste clearance and disrupted water-electrolyte homeostasis.95
Salidroside acts on glomerular endothelial cells, podocytes, renal tubular cells, glomerular mesangial cells, and renal cancer cells, demonstrating significant protective effects against various renal diseases.96 As shown in Figure 4, research indicates that salidroside primarily acts on the renal tubules to alleviate I/R injury by enhancing tubular antioxidant capacity, suppressing inflammatory responses, inhibiting tubular epithelial cell apoptosis, suppressing ferroptosis, and restoring renal function.
 |
Figure 4 The protective mechanism of salidroside against renal ischemia-reperfusion injury. The mechanism is divided into five parts: (A) Enhancement of tubular antioxidant capacity. Salidroside enhances the antioxidant capacity primarily by increasing the activities of SOD and CAT, and reducing MDA levels. It exerts its effects by promoting the nuclear translocation of Nf2 and upregulating HO-1 and NQO-1. Additionally, it can inhibit EPO degradation by upregulating HIF-1α. (B) Alleviation of inflammatory responses. Salidroside alleviates inflammatory responses mainly by suppressing the NF-κB signaling pathway. Meanwhile, in macrophages, salidroside inhibits hypoxia-reoxygenation-induced stabilization of HIF-1α, thereby reducing NLRP3 inflammasome activation. (C) Inhibition of ferroptosis. Salidroside can suppress ferroptosis induced by high levels of oxidative stress through activation of the PI3K/Akt pathway. (D) Repairment of renal function. Salidroside reduces the level of serum creatinine, BUN, NGAL, and KIM-1. (E) Inhibition of cell apoptosis. Salidroside inhibits cell apoptosis by regulating the Bcl-2/Bax balance and suppressing the activity of caspase-3. Symbol definitions: A standard arrow (→) indicates activation or positive regulation. A flat-ended arrow (―|) indicates inhibition or negative regulation. An upward arrow (↑) indicates increase, activation, or upregulation. A downward arrow (↓) indicates decrease, inhibition, or downregulation. |
Enhancement of Tubular Antioxidant Capacity
Salidroside is primarily active by enhancing the activity of SOD and catalase in renal tubular epithelial cells and serum, while reducing the MDA and ROS levels, thereby boosting antioxidant activity.97,98 Nrf2 is an important regulatory factor of antioxidant defense in I/R injury, and notably, Nrf2 exhibits circadian rhythmic expression in the kidney. By promoting Nrf2 nuclear translocation and upregulating antioxidant genes (eg., HO-1 and NQO-1), salidroside enhances the cellular antioxidant defense capacity.99 Moreover, the rapid stabilization of HIF-1α protein after renal I/R is a key adaptive response that initiates endogenous repair. Salidroside could promote the HIF-1α protein accumulation by inhibiting EPO degradation, thereby enhancing the body’s adaptability to hypoxia.100
Alleviation of Inflammatory Responses
The activation of NF-κB in renal I/R injury occurs mainly in renal tubular epithelial cells and the NF-κB signaling pathway exhibits a sustained activation pattern after renal I/R.101 By inhibiting its activation and reducing p-NF-κB p65 levels, salidroside consequently diminishes the production of pro-inflammatory cytokines (eg., IL-1β, IL-6, TNF-α), thus alleviating the inflammatory response.97,98 Salidroside has opposite regulatory effects on HIF-1α in different cell types, in macrophages, salidroside inhibits hypoxia-reoxygenation-induced HIF-1α in macrophages, reduces NLRP3 inflammasome activation, inhibits gasdermin D cleavage, and suppresses IL-1β and IL-18 secretion.102 The immune cell balance is crucial for suppressing inflammatory responses in vivo. Salidroside could lower the Th17/Treg ratio in the peripheral blood, maintain immune homeostasis, and mitigate inflammation.97
Inhibition of Tubular Epithelial Cell Apoptosis
Salidroside reduces apoptosis in renal tubular epithelial cells by tipping the Bcl-2/Bax balance toward survival (upregulating Bcl-2, downregulating Bax) and consequently suppressing the executioner caspase-3 activity.98
Suppression of Ferroptosis
In renal I/R injury, after a tubular epithelial cell undergoes ferroptosis, it secretes specific signals that induce adjacent cells to enter a ferroptosis-prone state, thereby triggering ferroptosis in a relay manner.103 Salidroside could suppress ferroptosis induced by high levels of oxidative stress and protect renal tubular epithelial cells through activation of the PI3K/Akt pathway.104
Repairment of Renal Function
I/R injury not only manifests as damage to the kidney’s tissue structure but is also accompanied by a decline in the kidney’s ability to regulate metabolic waste. Salidroside treatment mitigated renal injury, as evidenced by reduced serum levels of creatinine, blood urea nitrogen, neutrophil gelatinase-associated lipocalin, and kidney injury molecule-1, along with alleviated histopathological damage.99
Salidroside and Intestinal I/R Injury
I/R injury of the intestines is commonly triggered by severe trauma, burns, shock, intestinal volvulus, mesenteric thromboembolism, and transplantation of the small intestines.105
Following I/R injury, intestinal capillary permeability is frequently increased, resulting in impaired intestinal barrier function. This leads to bacterial infiltration of the intestinal mucosa and submucosa.106 I/R-induced alterations in the intestinal microenvironment inevitably cause changes in the quantity and composition of gut microbiota.107 Intestinal I/R injury is driven by the dual mechanisms of oxidative stress and inflammation. The burst of ROS production within the tissue triggers intestinal oxidative stress,108,109 thereby promoting the synthesis and release of various cytokines and inflammatory mediators. Oxidative stress and inflammatory responses further trigger a series of programmed or non-programmed cell death processes.110 These mediators cause vasodilation, infiltration of white blood cells (particularly neutrophils), and platelet activation, markedly amplifying both local and systemic inflammatory responses and exacerbating damage to the intestinal mucosal barrier and further exacerbates intestinal inflammation.111,112 The translocation of bacteria and toxins further activates the systemic immune system and pro-inflammatory immune cells, leading to the release of even more inflammatory factors. Intestinal I/R injury has a high propensity to initiate systemic complications, as it can induce SIRS and MODS. Mortality rates significantly increase when the disease progresses to shock, MODS, or sepsis.111,112
Research on salidroside in intestinal I/R injury is still in its infancy, and current evidence merely preliminarily explores and confirms the effect of salidroside in regulating the thioredoxin-interacting protein (TXNIP) pathway, among them, TXNIP acts as a crucial molecular switch between oxidative stress and inflammatory response. Using an in vivo model of superior mesenteric artery ligation in mice to simulate intestinal I/R, a study found that salidroside alleviates intestinal I/R injury by modulating TXNIP and AMPK expression and reducing serum and intestinal tissue levels of TNF-α, IL-6, and IL-1β.113 Additionally, salidroside repairs intestinal mucosal damage by mitigating oxidative stress and suppressing the TXNIP/NLRP3 signaling pathway and its downstream inflammatory factors.114
Salidroside and Testicular I/R Injury
Ischemic reperfusion of the testicle often occurs following testicular torsion. Hypoxia in tissues caused by the ischemic period regulates the expression of prostaglandin E1 and vascular endothelial growth factor, leading to abnormal collagen deposition and vascular dysfunction, thereby laying the groundwork for tissue fibrosis and resulting in arrested spermatogenesis.115 After testicular I/R injury, the explosive generation of ROS, along with the release of pro-inflammatory cytokines and neutrophil infiltration, dramatically exacerbates tissue inflammation. ROS attack membrane lipids rich in polyunsaturated fatty acids, triggering a chain reaction of lipid peroxidation and inducing various forms of cell death.116 Moreover, ATP depletion leads to dysfunction of ion pumps, causing potassium efflux and intracellular calcium overload. These disturbances subsequently impair the metabolic functions of testicular cells, setting off a cascade of biological alterations.117 I/R injury directly results in testicular edema, tissue collapse, cytopenia, hemorrhage, and necrosis. These irreversible pathological changes ultimately disrupt spermatogenesis and lead to male infertility.118
Because the clinical scenarios are relatively limited, and the core protective goal is long-term fertility, both of which restrict drug development for testicular I/R. Research on salidroside in testicular I/R injury is still in its early stages. These preliminary studies indicate that salidroside may enhance the activity of antioxidant enzymes, thereby alleviate oxidative damage and maintain intracellular redox balance.119 Meanwhile, salidroside inhibits ferroptosis in the testis by reducing iron availability and limiting lipid peroxidation substrates. Meanwhile, salidroside also can activate the Nrf2 pathway, reduce the expression levels of ACSL4 and TRF1, and elevate the expression of ACSL3, FTMT, and GPX4 to precisely inhibit ferroptosis.120 However, it is undeniable that the current mechanistic research of salidroside against testicular I/R injury remains very superficial and still awaits validation.
Discussion and Conclusion
Nowadays, salidroside started relatively late in the field of I/R injury research, but it has shown significant protective effects in multiple organs. Pharmacokinetically, under normoxic conditions, salidroside exhibits moderate-to-high oral bioavailability and rapid clearance.33 When I/R injury occurs, the tissue is in an acute hypoxic state, the pharmacokinetic behavior of salidroside changes favorably showing exposure increases, clearance decreases, and bioavailability increases by approximately 108% compared with normoxia.121 Salidroside accumulates preferentially in the heart, lung, and spleen and is mainly excreted renally as Tyrosol, a process that is slowed by hypoxia.121 Although salidroside itself poorly crosses the blood-brain barrier, its metabolite Tyrosol does so far more efficiently, enabling salidroside to act indirectly via Tyrosol. Moreover, no obvious adverse reactions have been observed in acute, chronic, or genotoxicity studies of salidroside. Overall, the favorable pharmacokinetic properties and low toxicity of salidroside make it a promising candidate for further development.
Due to the unique physiological functions, cell types, metabolic requirements, and microvascular structures of different organs, the responses and vulnerability of various organs to I/R injuries vary significantly. By summarizing the above research, the common mechanism underlying salidroside’s protection against I/R injury has been elucidated that involving inhibiting oxidative stress, reducing inflammatory responses, regulating cell apoptosis, and suppressing ferroptosis. These mechanisms directly target the three core pathological processes of I/R injury: oxidative stress, inflammatory storm, and cell death, and exhibit cross-organ universal applicability. However, it is also important to note that, beneath these commonalities, the physiological and pathological characteristics of different organs determine their distinct regulatory priorities, which may guide future drug development. In terms of oxidative stress, the heart and brain have an extremely high demand for oxygen, and salidroside focuses on ameliorating energy metabolism disorders while regulating oxidative stress. Hepatocytes exhibit extremely high metabolic activity, making the maintenance of their endogenous antioxidant defense system crucial. In the treatment of the kidney, salidroside emphasizes enhancing the specific antioxidant capacity of renal tubular epithelial cells. Regarding inflammation, in brain, overactivation of microglia releases abundant pro-inflammatory cytokines, creating a neurotoxic microenvironment. Meanwhile, the liver is rich in Kupffer cells, which are activated after reperfusion and serve as the core trigger of the inflammatory storm. Therefore, the regulation of inflammatory responses by salidroside in the brain and liver is of great significance for the recovery from I/R injury. With respect to cell death, the temporal sequences of apoptosis and ferroptosis differ across organs. In the heart, apoptosis is triggered in the early phase of reperfusion, whereas ferroptosis predominates in the later stage. In contrast, in the brain, ferroptosis is triggered within the first few hours after reperfusion. Notably, in renal I/R injury, recent studies suggest that renal tubular epithelial cells undergoing ferroptosis may secrete specific signals to induce neighboring cells to undergo ferroptosis in a relay manner. Therefore, when treating I/R injury in different organs, attention should also be paid to the timing of drug administration. Of course, the current summary and understanding of these mechanisms are still at a preliminary stage. In the future, modern scientific techniques will be needed to further dissect how organ-specific physiology finely regulates the multi-target protective effects of salidroside.
The Nrf2 pathway serves as a primary mechanism through which salidroside inhibits oxidative stress and its protective effect is notably observed in the brain, lung, liver, kidney, and testis. Furthermore, salidroside could protect the heart, brain, liver, and kidney from I/R injury by inhibiting the NF-κB pathway and regulating the downstream NLRP3 inflammasome. Regulated by Bcl-2/Bax and Caspase family, salidroside can regulate cell apoptosis in myocardium, neurons, lung epithelial cells, liver cells, and renal tubules. The mechanisms by which salidroside regulates I/R injury in different organs are highly complex and closely related to the specific characteristics of damage in different organs. Salidroside can also exert organ-specific effects according to the physiological characteristics of different organs, such as improving mitochondrial function in cardiomyocytes, regulating synaptic plasticity, and modulating metabolic function in the kidneys, among others. The common and specific mechanism by which salidroside regulates I/R injury in different organs are shown in Figure 5.
 |
Figure 5 The common and special mechanisms by which salidroside regulates ischemia-reperfusion injury in different organs. The common mechanism includes inhibition of oxidative stress, reduction of inflammatory responses, regulation of cell apoptosis, and suppression of ferroptosis. The Nrf2 pathway is the primary pathway through which salidroside inhibits oxidative stress. Salidroside can suppress inflammatory responses by inhibiting the NF-κB pathway and regulating the downstream NLRP3 inflammasome. Under the coordinated regulation of the Bcl-2/Bax and Caspase families, salidroside inhibits cell apoptosis. Additionally, salidroside primarily suppresses ferroptosis by regulating the GSH/GPX4 axis. Symbol definitions: A double-ended arrow (↔) indicates an interaction. A standard arrow (→) indicates activation or positive regulation. A flat-ended arrow (―|) indicates inhibition or negative regulation. An upward arrow (↑) indicates increase, activation, or upregulation. A downward arrow (↓) indicates decrease, inhibition, or downregulation. |
Nowadays, the development of salidroside has become a hot topic and the nature product salidroside has been formulated in various forms, such as capsules, oral liquids, injection solutions, raw powders, nanoliposomes, and skincare gels, making its way into the health food, pharmaceutical, and cosmetic markets with promising development prospects. With salidroside as its primary component, a large-dose Rhodiola extract injection promotes blood circulation and removes blood stasis, has been used to treat coronary heart disease with angina pectoris caused by qi deficiency and blood stasis, as well as coronary heart disease complicated by heart failure.122,123 Rhodiola granules possibly through anti-inflammation, regulation of energy metabolism, and oxidative stress to alleviate fatigue, enhance antioxidant capacity to be used to improve I/R injury.124,125 Moreover, a capsule made from Rhodiola rosea, American ginseng, and Astragalus membranaceus, have been confirmed to help relieve physical fatigue.126 Nevertheless, the above-mentioned products are all multi-component formulations rather than single salidroside components. Meanwhile, the existing studies have obvious limitations: first, most of the evidence comes from small-sample preclinical in vivo and in vitro experiments and lacks independent validation. Second, the administered doses and treatment durations vary considerably across different studies, and systematic dose-effect studies are lacking. Third, the evidence across different organs is unbalanced, with fewer studies on organs such as the lung, intestine and testis; moreover, the mechanistic studies are relatively simplistic, lacking in-depth explorations such as gene knockout/overexpression or tissue-specific interventions. Fourth, pharmacokinetic data and toxicological evidence on salidroside for the treatment of I/R injury are scarce. Fifth, no direct comparison has been made between salidroside and other natural products, and it remains unknown whether its protective effects are superior to those of better-established compounds such as resveratrol and curcumin. Therefore, future research should focus on improving experimental quality, establishing standardized dose-effect relationships, filling the evidence gaps for under-studied organs, and conducting randomized controlled clinical trials to verify its therapeutic efficacy, thereby providing a solid foundation for the clinical translation of salidroside.
This review aims to systematically summarize the common and organ-specific protective mechanisms of salidroside against ischemia-reperfusion injury in different organs, thereby providing research directions for further investigation of its pharmacodynamic mechanisms, as well as for the development of related pharmaceutical formulations and their clinical translation. Currently, existing studies have limitations such as low quality of evidence, lack of dose standardization, and insufficient clinical translation evidence. Further in-depth investigation is still needed in order to provide a more reliable basis for clinical practice.
Abbreviations
Akt, protein kinase B; AMPK, Adenosine 5’-monophosphate-activated protein kinase; ASK1, apoptosis signal-regulating kinase 1; ARC, activity-regulated cytoskeleton-associated protein; ATP, adenosine triphosphate; BDNF, brain-derived neurotrophic factor; Drp1, dynamin-related protein 1; EGR, early growth response; ERK, extracellular signal-regulated kinase; EPO, Erythropoietin; GFAP, glial fibrillary acidic protein; GPX, glutathione peroxidase; GSK-3β, glycogen synthase kinase 3β; HO-1, heme oxygenase-1; I/R, Ischemia-reperfusion; JNK, c-Jun N-terminal kinase; MAPK, mitogen-activated protein kinase; MCP-1, monocyte chemotactic protein-1; MDA, malondialdehyde; NGF, nerve growth factor; PI3K, phosphoinositide 3-kinase; PGC-1α, peroxisome proliferator-activated receptor gamma coactivator 1-alpha; PPAR-α, peroxisome proliferator-activated receptor alpha; SIRS, systemic inflammatory response syndrome; STAT3, signal transducer and activator of transcription 3; Trx1, thioredoxin 1; TXNIP, Thioredoxin-interacting protein.
Acknowledgments
All figures were drawn by BioRender and have received the publication license.
Author Contributions
All authors make a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was supported by the National Natural Science Foundation of China (82205093), Fundamental Research Funds for the Central Public Welfare Research Institutes (ZZ16-YQ-003), Scientific and Technological Innovation Project of China Academy of Chinese Medical Sciences (CI2021B006), the Science and Technology Innovation Project for Graduate Students at China Academy of Chinese Medical Sciences (KC2025021), Hospital Capability Enhancement Project of Xiyuan Hospital, China Academy of Chinese Medical Sciences (XYZX0204-05), Talent Support Project of Xiyuan Hospital, China Academy of Chinese Medical Sciences (XYZXRC01-01), Clinical Collaboration Project of Traditional Chinese and Western Medicine for Major Difficult Diseases (ZDYN-2024-A-008).
Disclosure
The authors declare no competing interests in this work.
References
1. Zhang M, Liu Q, Meng H, et al. Ischemia-reperfusion injury: molecular mechanisms and therapeutic targets. Signal Transduc Target Ther. 2024;9(1):12. doi:10.1038/s41392-023-01688-x
2. Zang X, Zhou J, Zhang X, Han Y, Chen X. Ischemia reperfusion injury: opportunities for nanoparticles. ACS Biomater Sci Eng. 2020;6(12):6528–20. doi:10.1021/acsbiomaterials.0c01197
3. Li W, Liao Y, Chen J, et al. Ischemia - Reperfusion injury: a roadmap to precision therapies. Mol Aspects Med. 2025;104:101382. doi:10.1016/j.mam.2025.101382
4. Kassab AA, Aboregela AM, Shalaby AM. Edaravone attenuates lung injury in a hind limb ischemia-reperfusion rat model: a histological, immunohistochemical and biochemical study. Ann Anat. 2020;228:151433. doi:10.1016/j.aanat.2019.151433
5. Kashef SM, Abo Elnasr SE. Effect of peripheral blood mononuclear cells on ischemia-reperfusion injury of sciatic nerve of adult male albino rat: histological, immunohistochemical, and ultrastructural study. Ultrastruct Pathol. 2024;48(3):172–191. doi:10.1080/01913123.2024.2321144
6. Pang QY, Meng XM, Zhou ZF, et al. Temporal regulation of genetic programs governing multiple cell death during myocardial ischemia-reperfusion injury. Front Genet. 2025;16:1632867. doi:10.3389/fgene.2025.1632867
7. Du B, Deng ZJ, Chen K, et al. Iron promotes both ferroptosis and necroptosis in the early stage of reperfusion in ischemic stroke. Genes Dis. 2024;11(6):101262. doi:10.1016/j.gendis.2024.101262
8. Nakamura E, Aoki T, Endo Y, et al. Organ-Specific Mitochondrial Alterations Following Ischemia-Reperfusion Injury in Post-Cardiac Arrest Syndrome: a Comprehensive Review. Life. 2024;14(4):477. doi:10.3390/life14040477
9. Gheitasi I, Akbari G, Savari F. Physiological and cellular mechanisms of ischemic preconditioning microRNAs-mediated in underlying of ischemia/reperfusion injury in different organs. Mol Cell Biochem. 2025;480(2):855–868. doi:10.1007/s11010-024-05052-7
10. Peng J, Deng T, Wang XK, et al. Advances in the treatment of lower-extremity ischemia-reperfusion injury. Front Pharmacol. 2025;16:1576091. doi:10.3389/fphar.2025.1576091.
11. Monassier L, Ayme-Dietrich E, Aubertin-Kirch G, Pathak A. Targeting myocardial reperfusion injuries with cyclosporine in the CIRCUS Trial - pharmacological reasons for failure. Fundam Clin Pharmacol. 2016;30(2):191–193. doi:10.1111/fcp.12177
12. Atar D, Arheden H, Berdeaux A, et al. Effect of intravenous TRO40303 as an adjunct to primary percutaneous coronary intervention for acute ST-elevation myocardial infarction: MITOCARE study results. Eur Heart J. 2015;36(2):112–119. doi:10.1093/eurheartj/ehu331
13. Zhang WH, Guo SY, Dou JJ, et al. Berberine and its derivatives: mechanisms of action in myocardial vascular endothelial injury - a review. Front Pharmacol. 2025;16:1543697. doi:10.3389/fphar.2025.1543697
14. Abe M, Takiguchi Y, Ichimaru S, Tsuchiya K, Wada K. Comparison of the protective effect of N-acetylcysteine by different treatments on rat myocardial ischemia-reperfusion injury. J Pharmacol Sci. 2008;106(4):571–577. doi:10.1254/jphs.fp0071664
15. Prag HA, Aksentijevic D, Dannhorn A, et al. Ischemia-Selective Cardioprotection by Malonate for Ischemia/Reperfusion Injury. Circ Res. 2022;131(6):528–541. doi:10.1161/CIRCRESAHA.121.320717
16. Chahardehi AM, Arefnezhad R, Rafei S, et al. The Effect of Malonate as a Succinate Dehydrogenase Inhibitor on Myocardial Ischemia/Reperfusion Injury. Cell Biol Int. 2025;49(12):1545–1563. doi:10.1002/cbin.70079
17. Zhang S, Yan F, Luan F, et al. The pathological mechanisms and potential therapeutic drugs for myocardial ischemia reperfusion injury. Phytomedicine. 2024;129:155649. doi:10.1016/j.phymed.2024.155649
18. Malhotra A, Bath S, Elbarbry F. An Organ System Approach to Explore the Antioxidative, Anti-Inflammatory, and Cytoprotective Actions of Resveratrol. Oxid Med Cell Longev. 2015;2015:803971. doi:10.1155/2015/803971
19. Xie YZ, Gong XC, Jin Z, Xu W, Zhao K. Curcumin encapsulation in self-assembled nanoparticles based on amphiphilic palmitic acid-grafted-quaternized chitosan with enhanced cytotoxic, antimicrobial and antioxidant properties. Int J Biol Macromol. 2022;222(Pt B):2855–2867. doi:10.1016/j.ijbiomac.2022.10.064
20. Li H, Sureda A, Devkota HP, et al. Curcumin, the golden spice in treating cardiovascular diseases. Biotechnol Adv. 2020;38:107343. doi:10.1016/j.biotechadv.2019.01.010
21. Mohamadian M, Parsamanesh N, Chiti H, Sathyapalan T, Sahebkar A. Protective effects of curcumin on ischemia/reperfusion injury. Phytother Res. 2022;36(12):4299–4324. doi:10.1002/ptr.7620
22. Liu DQ, Chen SP, Sun J, et al. Berberine protects against ischemia-reperfusion injury: a review of evidence from animal models and clinical studies. Pharmacol Res. 2019;148:104385. doi:10.1016/j.phrs.2019.104385
23. Bi XJ, Wang Z, He JT. Recent advances in biomimetic nanodelivery systems for the treatment of myocardial ischemia reperfusion injury. Colloids Surf B Biointerfaces. 2025;247:114414. doi:10.1016/j.colsurfb.2024.114414
24. El Farissi M, Keulards DCJ, Zelis JM, et al. Hypothermia for Reduction of Myocardial Reperfusion Injury in Acute Myocardial Infarction: closing the Translational Gap. Circ Cardiovasc Interv. 2021;14(8):e010326. doi:10.1161/CIRCINTERVENTIONS.120.010326
25. Tinsley GM, Jagim AR, Potter GDM, Garner D, Galpin AJ. Rhodiola rosea as an adaptogen to enhance exercise performance: a review of the literature. Br J Nutr. 2024;131(3):461–473. doi:10.1017/S0007114523001988
26. Bernatoniene J, Jakstas V, Kopustinskiene DM. Phenolic compounds of rhodiola rosea L. as the potential alternative therapy in the treatment of chronic diseases. Int J Mol Sci. 2023;24(15):12293. doi:10.3390/ijms241512293
27. Panossian A, Wikman G, Sarris J. Rosenroot (rhodiola rosea): traditional use, chemical composition, pharmacology and clinical efficacy. Phytomed: Int J Phytother Phytopharm. 2010;17(7):481–493. doi:10.1016/j.phymed.2010.02.002
28. Tao H, Wu X, Cao J, et al. Rhodiola species: a comprehensive review of traditional use, phytochemistry, pharmacology, toxicity, and clinical study. Med Res Rev. 2019;39(5):1779–1850. doi:10.1002/med.21564
29. Ivanova Stojcheva E, Quintela JC. The Effectiveness of Rhodiola rosea L. Preparations in Alleviating Various Aspects of Life-Stress Symptoms and Stress-Induced Conditions-Encouraging Clinical Evidence. Molecules. 2022;27(12):3902. doi:10.3390/molecules27123902
30. Polumackanycz M, Konieczynski P, Orhan IE, Abaci N, Viapiana A. Chemical composition, antioxidant and anti-enzymatic activity of golden root (rhodiola rosea L.) commercial samples. Antioxid. 2022;11(5):919. doi:10.3390/antiox11050919
31. Liu Y, Weng W, Gao R, Liu Y. New insights for cellular and molecular mechanisms of aging and aging-related diseases: herbal medicine as potential therapeutic approach. Oxid Med Cell Longev. 2019;2019:4598167. doi:10.1155/2019/4598167
32. Xu F, Xu J, Xiong X, Deng Y. Salidroside inhibits MAPK, NF-κB, and STAT3 pathways in psoriasis-associated oxidative stress via SIRT1 activation. Redox Rep: Commun Free Radic Res. 2019;24(1):70–74. doi:10.1080/13510002.2019.1658377
33. Zhang X, Xie L, Long J, et al. Salidroside: a review of its recent advances in synthetic pathways and pharmacological properties. Chem Biol Interact. 2021;339:109268. doi:10.1016/j.cbi.2020.109268
34. Qu B, Liu X, Liang Y, Zheng K, Zhang C, Lu L. Salidroside in the treatment of NAFLD/NASH. Chem Biodivers. 2022;19(12):e202200401. doi:10.1002/cbdv.202200401
35. Juricic S, Klac J, Stojkovic S, et al. Molecular and Cellular Mechanisms of Myocardial Ischemia and Reperfusion Injury: a Narrative Review. Cells. 2026;15(3):265. doi:10.3390/cells15030265
36. Xiang Q, Yi X, Zhu XH, Wei X, Jiang DS. Regulated cell death in myocardial ischemia-reperfusion injury. Trends Endocrinol Metab TEM. 2024;35(3):219–234. doi:10.1016/j.tem.2023.10.010
37. Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol. 2018;19(2):121–135. doi:10.1038/nrm.2017.95
38. Chang X, Zhang K, Zhou R, et al. Cardioprotective effects of salidroside on myocardial ischemia-reperfusion injury in coronary artery occlusion-induced rats and langendorff-perfused rat hearts. Int J Cardiol. 2016;215:532–544. doi:10.1016/j.ijcard.2016.04.108
39. Tian X, Huang Y, Zhang X, et al. Salidroside attenuates myocardial ischemia/reperfusion injury via AMPK-induced suppression of endoplasmic reticulum stress and mitochondrial fission. Toxicol Appl Pharmacol. 2022;448:116093. doi:10.1016/j.taap.2022.116093
40. Yang B, Xu Y, Yu J, et al. Salidroside pretreatment alleviates ferroptosis induced by myocardial ischemia/reperfusion through mitochondrial superoxide-dependent AMPKα2 activation. Phytomed: int J Phytother. Phytopharm. 2024;128:155365. doi:10.1016/j.phymed.2024.155365
41. Shvedova M, Anfinogenova Y, Atochina-Vasserman EN, Schepetkin IA, Atochin DN. c-jun N-terminal kinases (JNKs) in myocardial and cerebral ischemia/reperfusion injury. Front Pharmacol. 2018;9:715. doi:10.3389/fphar.2018.00715
42. Jin P, Li LH, Shi Y, Hu NB. Salidroside inhibits apoptosis and autophagy of cardiomyocyte by regulation of circular RNA hsa_circ_0000064 in cardiac ischemia-reperfusion injury. Gene. 2021;767:145075. doi:10.1016/j.gene.2020.145075
43. Liu B, Wei H, Lan M, Jia N, Liu J, Zhang M. MicroRNA-21 mediates the protective effects of salidroside against hypoxia/reoxygenation-induced myocardial oxidative stress and inflammatory response. Exp Ther Med. 2020;19(3):1655–1664. doi:10.3892/etm.2020.8421
44. Shan H, Li XH, Ouyang C, et al. Salidroside prevents PM2.5-induced BEAS-2B cell apoptosis via SIRT1-dependent regulation of ROS and mitochondrial function. Ecotoxicol Environ Saf. 2022;231:113170. doi:10.1016/j.ecoenv.2022.113170
45. Zhou PY, Ji JQ, Zhang Z, et al. Salidroside-loaded metal-organic frameworks hydrogel to improve cardiac allograft function. J Heart Lung. Transplant. 2025;44(10):1621–1634 doi:10.1016/j.healun.2025.05.015.
46. Yu H, Lin L, Zhang Z, Zhang H, Hu H. Targeting NF-κB pathway for the therapy of diseases: mechanism and clinical study. Signal Transduc Target Ther. 2020;5(1):209. doi:10.1038/s41392-020-00312-6
47. Zhu L, Wei T, Gao J, et al. The cardioprotective effect of salidroside against myocardial ischemia reperfusion injury in rats by inhibiting apoptosis and inflammation. Apoptosis Int J Program Cell Death. 2015;20(11):1433–1443. doi:10.1007/s10495-015-1174-5
48. Bertheloot D, Latz E, Franklin B. Necroptosis, pyroptosis and apoptosis: an intricate game of cell death. Cell Mol Immunol. 2021;18(5):1106–1121. doi:10.1038/s41423-020-00630-3
49. Xu MC, Shi HM, Gao XF, Wang H. Salidroside attenuates myocardial ischemia-reperfusion injury via PI3K/akt signaling pathway. J Asian Nat Prod Res. 2013;15(3):244–252. doi:10.1080/10286020.2012.762358
50. Sun L, Isaak CK, Zhou Y, et al. Salidroside and tyrosol from rhodiola protect H9c2 cells from ischemia/reperfusion-induced apoptosis. Life Sci. 2012;91(5–6):151–158. doi:10.1016/j.lfs.2012.06.026
51. Jurcau A, Ardelean IA. Molecular pathophysiological mechanisms of ischemia/reperfusion injuries after recanalization therapy for acute ischemic stroke. J Integr Neurosci. 2021;20(3):727–744. doi:10.31083/j.jin2003078
52. Xiong XY, Liu L, Yang QW. Refocusing neuroprotection in cerebral reperfusion era: new challenges and strategies. Front Neurol. 2018;9:249. doi:10.3389/fneur.2018.00249
53. Wu L, Xiong X, Wu X, et al. Targeting oxidative stress and inflammation to prevent ischemia-reperfusion injury. Front Mol Neurosci. 2020;13:28. doi:10.3389/fnmol.2020.00028
54. Zhong ZF, Han J, Zhang JZ, et al. Neuroprotective effects of salidroside on cerebral ischemia/reperfusion-induced behavioral impairment involves the dopaminergic system. Front Pharmacol. 2019;10:1433. doi:10.3389/fphar.2019.01433
55. Semwal P, Rauf A, Simal-Gandara J. Editorial: high altitude medicinal plants and their bioactive compounds for the prevention of oxidative stress-induced diseases and disorders. Front Pharmacol. 2023;14:1257018. doi:10.3389/fphar.2023.1257018
56. Lai W, Xie X, Zhang X, et al. Inhibition of complement drives increase in early growth response proteins and neuroprotection mediated by salidroside after cerebral ischemia. Inflammation. 2018;41(2):449–463. doi:10.1007/s10753-017-0701-7
57. Jiang SN, Fan FF, Yang L, et al. Salidroside attenuates high altitude hypobaric hypoxia-induced brain injury in mice via inhibiting NF-κB/NLRP3 pathway. Eur J Pharmacol. 2022;925:175015. doi:10.1016/j.ejphar.2022.175015
58. Li F, Mao Q, Wang J, et al. Salidroside inhibited cerebral ischemia/reperfusion-induced oxidative stress and apoptosis via Nrf2/Trx1 signaling pathway. Metab Brain Dis. 2022;37(8):2965–2978. doi:10.1007/s11011-022-01061-x
59. Han J, Xiao Q, Lin YH, et al. Neuroprotective effects of salidroside on focal cerebral ischemia/reperfusion injury involve the nuclear erythroid 2-related factor 2 pathway. Neural Regen Res. 2015;10(12):1989–1996. doi:10.4103/1673-5374.172317
60. Song WT, Cao H, Zhang YH, Zheng XY, Liu JX. Protection of salidroside on endothelial cell barrier in cerebral ischemia-reperfusion model rats. Chin J Chin Mater Med. 2022;47(19):5284–5291. doi:10.19540/j.cnki.cjcmm.20220613.703
61. Atochin DN, Chernysheva GA, Smolyakova VI, et al. Neuroprotective effects of p-tyrosol after the global cerebral ischemia in rats. Phytomed: int J Phytother. Phytopharm. 2016;23(7):784–792. doi:10.1016/j.phymed.2016.03.015
62. Wu QW, Shan XH, Li XM, et al. Salidroside ameliorates neuroinflammation in autistic rats by inhibiting NLRP3/Caspase-1/GSDMD signal pathway. Brain Res Bull. 2025;220:111132. doi:10.1016/j.brainresbull.2024.11113
63. Chen T, Ma Z, Zhu L, et al. Suppressing receptor-interacting protein 140: a new sight for salidroside to treat cerebral ischemia. Mol Neurobiol. 2016;53(9):6240–6250. doi:10.1007/s12035-015-9521-7
64. Liu J, Ma W, Zang CH, et al. Salidroside inhibits NLRP3 inflammasome activation and apoptosis in microglia induced by cerebral ischemia/reperfusion injury by inhibiting the TLR4/NF-κB signaling pathway. Ann Transl Med. 2021;9(22):1694. doi:10.21037/atm-21-5752
65. Yin L, Ouyang D, Lin L, Xin X, Ji Y. Salidroside regulates imbalance of Th17/treg and promotes ischemic tolerance by targeting STAT-3 in cerebral ischemia-reperfusion injury. Arch Med Sci AMS. 2021;17(2):523–534. doi:10.5114/aoms.2019.85349
66. Dong T, Wang HY, Li YW, Tan J. Based on the PINK1/Parkin signaling pathway, the protective effect of salidroside on cerebral ischemia-reperfusion injury in male rats was investigated. J Stroke Cerebrovasc Dis. 2026;35(1):108519. doi:10.1016/j.jstrokecerebrovasdis.2025.108519
67. DU QS, Liao J. Salidroside suppresses ferroptosis in HT22 cells following oxygen-glucose deprivation/reoxygenation through regulation of non-ubiquitinated FUNDC1-dependent mitophagy pathway. Zhongguo ZhongYao ZaZhi. 2026;51(1):191–201. doi:10.19540/j.cnki.cjcmm.20250902.401
68. Xu L, Jia L, Wang Q, Hou J, Li S, Teng J. Salidroside attenuates hypoxia/reoxygenation-induced human brain vascular smooth muscle cell injury by activating the SIRT1/FOXO3α pathway. Exp Ther Med. 2018;15(1):822–830. doi:10.3892/etm.2017.5446
69. Zhang X, Du Q, Yang Y, et al. Salidroside alleviates ischemic brain injury in mice with ischemic stroke through regulating BDNK mediated PI3K/akt pathway. Biochem Pharmacol. 2018;156:99–108. doi:10.1016/j.bcp.2018.08.015
70. Lai W, Zheng Z, Zhang X, et al. Salidroside-mediated neuroprotection is associated with induction of early growth response genes (egrs) across a wide therapeutic window. Neurotoxic Res. 2015;28(2):108–121. doi:10.1007/s12640-015-9529-9
71. Zhang Y, Guo X, Wang G, et al. Effects of rhodioloside on the neurological functions of rats with total cerebral ischemia/reperfusion and cone neuron injury in the hippocampal CA1 region. PeerJ. 2020;8:e10056. doi:10.7717/peerj.10056
72. Li C, Chi J, Dai H, et al. Salidroside attenuates cerebral ischemia/reperfusion injury by regulating TSC2-induced autophagy. Exp Brain Res. 2023;241(1):113–125. doi:10.1007/s00221-022-06493-6
73. Rong X, Lu P, Li Y, et al. Salidroside attenuates cerebral ischemia-reperfusion injury via ERβ/BNIP3-mediated mitochondrial autophagy activation in a rat model. Neurochem Res. 2025;50(5):297. doi:10.1007/s11064-025-04535-3
74. Zheng J, Zhang J, Han J, Zhao Z, Lin K. The effect of salidroside in promoting endogenous neural regeneration after cerebral ischemia/reperfusion involves notch signaling pathway and neurotrophic factors. BMC Complementary Med Ther. 2024;24(1):293. doi:10.1186/s12906-024-04597-w
75. Yang Z, Huang X, Lai W, et al. Synthesis and identification of a novel derivative of salidroside as a selective, competitive inhibitor of monoamine oxidase B with enhanced neuroprotective properties. Eur J Med Chem. 2021;209:112935. doi:10.1016/j.ejmech.2020.112935
76. Besnier E, Clavier T, Bellien J, Selim J. Key role of the endothelium in lung ischemia-reperfusion injury: what the clinician needs to know. JHLT Open. 2026;12:100503. doi:10.1016/j.jhlto.2026.100503
77. Rancan L, Paredes SD, Huerta L, et al. Chemokine involvement in lung injury secondary to ischaemia/reperfusion. Lung. 2017;195(3):333–340. doi:10.1007/s00408-017-0001-x
78. Ta HQ, Kuppusamy M, Sonkusare SK, Roeser ME, Laubach VE. The endothelium: gatekeeper to lung ischemia-reperfusion injury. Respir Res. 2024;25(1):172. doi:10.1186/s12931-024-02776-4
79. Rizzo AN, Schmidt EP. The role of the alveolar epithelial glycocalyx in acute respiratory distress syndromE. Am J Physiol Cell Physiol. 2023;324(4):C799–C806. doi:10.1152/ajpcell.00555.2022
80. Wang Y, Chen Z, Luo J, et al. Salidroside postconditioning attenuates ferroptosis-mediated lung ischemia-reperfusion injury by activating the Nrf2/SLC7A11 signaling axis. Int Immunopharmacol. 2023;115:109731. doi:10.1016/j.intimp.2023.109731
81. Yu X, Xu B, Zhang M, Yao X, Xu K, Gao F. Salidroside inhibits the ferroptosis to alleviate lung ischemia reperfusion injury via the JAK2/STAT3 signalling pathway. Biochem Biophys Res Commun. 2024;722:150132. doi:10.1016/j.bbrc.2024.150132
82. Yu XB, Xu BB, Zhang MD, et al. Salidroside Alleviates Lung Ischemia-Reperfusion Injury by Inhibiting Ferroptosis Through the MAFK/lncRNA PTOV1-AS2/miR-525-5p/ACE2 Axis. FASEB J. 2025;39(23):e71291. doi:10.1096/fj.202502335R
83. Liu J, Man K. Mechanistic Insight and Clinical Implications of Ischemia/Reperfusion Injury Post Liver Transplantation. Cell Mol Gastroenterol Hepatol. 2023;15(6):1463–1474. doi:10.1016/j.jcmgh.2023.03.003
84. Ighodaro OM, Akinloye OA. First line defence antioxidants-superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPX): their fundamental role in the entire antioxidant defence grid. Alexandria J Med. 2018;54(4):287–293. doi:10.1016/j.ajme.2017.09.001
85. Choi EK, Lim DG. Hepatic ischemia-reperfusion injury with respect to oxidative stress and inflammatory response: a narrative review. J Yeungnam Med Sci. 2023;40(2):115–122. doi:10.12701/jyms.2022.00017
86. de Oliveira THC, Gonçalves GKN. Liver ischemia reperfusion injury: mechanisms, cellular pathways, and therapeutic approaches. Int Immunopharmacol. 2025;150:114299. doi:10.1016/j.intimp.2025.114299
87. Liu Y, Lei Z, Chai H, Kang Q, Qin X. Salidroside alleviates hepatic ischemia-reperfusion injury during liver transplant in rat through regulating TLR-4/NF-κB/NLRP3 inflammatory pathway. Sci Rep. 2022;12(1):13973. doi:10.1038/s41598-022-18369-4
88. Xiong Y, Wang Y, Xiong Y, Teng L. Protective effect of salidroside on hypoxia-related liver oxidative stress and inflammation via Nrf2 and JAK2/STAT3 signaling pathways. Food Sci Nutr. 2021;9(9):5060–5069. doi:10.1002/fsn3.2459
89. Cai L, Li Y, Zhang Q, et al. Salidroside protects rat liver against ischemia/reperfusion injury by regulating the GSK-3β/Nrf2-dependent antioxidant response and mitochondrial permeability transition. Eur J Pharmacol. 2017;806:32–42. doi:10.1016/j.ejphar.2017.04.011
90. Zhang H, Gao WZ. Effect of salidroside on liver injury and Bcl-2, Bax in rats with hepatic ischemia reperfusion. J Mod Med Health. 2018;34(5):690–692 doi:10.3969/j.issn.1009-5519.2018.05.014.
91. Xiong Y, Wang Y, Xiong Y, Gao W, Teng L. Salidroside alleviated hypoxia-induced liver injury by inhibiting endoplasmic reticulum stress-mediated apoptosis via IRE1α/JNK pathway. Biochem Biophys Res Commun. 2020;529(2):335–340. doi:10.1016/j.bbrc.2020.06.036
92. Feng J, Zhang Q, Mo W, et al. Salidroside pretreatment attenuates apoptosis and autophagy during hepatic ischemia-reperfusion injury by inhibiting the mitogen-activated protein kinase pathway in mice. Drug Des Devel Ther. 2017;11:1989–2006. doi:10.2147/DDDT.S136792
93. Ray SC, Mason J, O’Connor PM. Ischemic Renal Injury: can Renal Anatomy and Associated Vascular Congestion Explain Why the Medulla and Not the Cortex Is Where the Trouble Starts? Semin Nephrol. 2019;39(6):520–529. doi:10.1016/j.semnephrol.2019.10.002
94. Hosszu A, Fekete A, Szabo AJ. Sex differences in renal ischemia-reperfusion injury. Am J Physiol Renal Physiol. 2020;319(2):F149–F154. doi:10.1152/ajprenal.00099.2020
95. Pefanis A, Ierino FL, Murphy JM, Cowan PJ. Regulated necrosis in kidney ischemia-reperfusion injury. Kidney Int. 2019;96(2):291–301. doi:10.1016/j.kint.2019.02.009
96. Liu Q, Chen J, Zeng A, Song L. Pharmacological functions of salidroside in renal diseases: facts and perspectives. Front Pharmacol. 2023;14:1309598. doi:10.3389/fphar.2023.1309598
97. Zhang PJ, Fan XB, Peng D, Bin D, Tong D, Chen R. Prevention of salidroside pretreatment against ischemia-resperfusion injury in rats after kidney transplantation. J Clin Nephrol. 2019;19(3):191–196 doi:10.3969/j.issn.1671-2390.2019.03.008.
98. Sun Y, Xun L, Jin G, Shi L. Salidroside protects renal tubular epithelial cells from hypoxia/reoxygenation injury in vitro. J Pharmacol Sci. 2018;137(2):170–176. doi:10.1016/j.jphs.2018.05.011
99. Xu QB. Study on the Protective Effect and Mechanism of Rhodiola-Derived Salidro side Against Exhaustive Exercise-Induced Acute Kidney Injury. Molecular Plant Breeding. 2025;23(16):5546–5553. doi:10.13271/j.mpb.023.005546
100. Zheng KYZ, Zhang ZX, Guo AJY, et al. Salidroside stimulates the accumulation of HIF-1α protein resulted in the induction of EPO expression: a signaling via blocking the degradation pathway in kidney and liver cells. Eur J Pharmacol. 2012;679(1–3):34–39. doi:10.1016/j.ejphar.2012.01.027
101. Zhang SW, Luo HS, Wei MT, et al. Temporal Proteomic and Phosphoproteomic Profiling Deciphers Molecular Dynamics of Acute-to-Chronic Kidney Disease After Ischemia-Reperfusion Injury, With Dock2 Emerging as a Key Regulator. Mol Cell Proteomics. 2026;25(2):101509. doi:10.1016/j.mcpro.2026.101509
102. Zhan XQ, Zhang Y, Lin WD, Fu SY, Huang J. Salidroside alleviates renal ischemia-reperfusion injury by inhibiting macrophage pyroptosis through HIF signaling. Inflammation. 2025;48(6):4318–4329. doi:10.1007/s10753-025-02328-y
103. Yu HT, Yin HB, Mei YH, et al. Nucleophosmin 1 lactylation in graft kidney induces ferroptotic trigger waves that exacerbate delayed graft function. Nat Commun. 2025;17(1):277. doi:10.1038/s41467-025-66991-3
104. Tang Z, Wang Y, Liu Y, Li C. Salidroside inhibits renal ischemia/reperfusion injury-induced ferroptosis by the PI3K/AKT signaling pathway. Exp Ther Med. 2023;26(5):507. doi:10.3892/etm.2023.12206
105. Wen J, Xu B, Sun Y, et al. Paeoniflorin protects against intestinal ischemia/reperfusion by activating LKB1/AMPK and promoting autophagy. Pharmacol Res. 2019;146:104308. doi:10.1016/j.phrs.2019.104308
106. Nadatani Y, Watanabe T, Shimada S, Otani K, Tanigawa T, Fujiwara Y. Microbiome and intestinal ischemia/reperfusion injury. J Clin Biochem Nutr. 2018;63(1):26–32. doi:10.3164/jcbn.17-137
107. Chen J, Wang Y, Shi Y, Liu Y, Wu C, Luo Y. Association of gut microbiota with intestinal ischemia/reperfusion injury. Front Cell Infect Microbiol. 2022;12:962782. doi:10.3389/fcimb.2022.962782
108. Collange O, Charles AL, Lavaux T, et al. Compartmentalization of inflammatory response following gut ischemia reperfusion. Eur J Vasc Endovasc Surg. 2015;49(1):60–65. doi:10.1016/j.ejvs.2014.10.022
109. de Barboza G D, Guizzardi S, Moine L, de Talamoni N T. Oxidative stress, antioxidants and intestinal calcium absorption. World J Gastroenterol. 2017;23(16):2841–2853. doi:10.3748/wjg.v23.i16.2841
110. Wang F, Huang HM, Wei XJ, Tan P, Wang ZG, Hu ZD. Targeting cell death pathways in intestinal ischemia-reperfusion injury: a comprehensive review. Cell Death Discov. 2024;10(1):112. doi:10.1038/s41420-024-01891-x
111. Tang J, Zhuang S. Histone acetylation and DNA methylation in ischemia/reperfusion injury. Clin Sci. 2019;133(4):597–609. doi:10.1042/CS20180465
112. Isozaki S, Konishi H, Fujiya M, et al. Probiotic-Derived Polyphosphate Accelerates Intestinal Epithelia Wound Healing through Inducing Platelet-Derived Mediators. Mediators Inflamm. 2021;2021:5582943. doi:10.1155/2021/5582943
113. Chen F, Chai YH, Zhang F, et al. Network pharmacology analysis combined with experimental validation to explore the therapeutic mechanism of salidroside on intestine ischemia reperfusion. Biosci Rep. 2023;43(8):BSR20230539. doi:10.1042/BSR20230539
114. Zhong X, Tang GQ, Liu Q, Huang Z, Xu T. The effect and mechanism of salidroside on intestinal ischemia-reperfusion injury in rats via TXNIP/NLRP3 signaling pathway. J Guangxi Med Univ. 2021;38(8):1518–1523. doi:10.16190/j.cnki.45-1211/r.2021.08.009
115. Oyedokun PA, Akhigbe RE, Ajayi LO, Ajayi AF. Impact of hypoxia on Male reproductive functions. Mol Cell Biochem. 2023;478(4):875–885. doi:10.1007/s11010-022-04559-1
116. Shimizu S, Tsounapi P, Dimitriadis F, Higashi Y, Shimizu T, Saito M. Testicular torsion-detorsion and potential therapeutic treatments: a possible role for ischemic postconditioning. Int J Urol. 2016;23(6):454–463. doi:10.1111/iju.13110
117. Akhigbe RE, Odetayo AF, Akhigbe TM, Hamed MA, Ashonibare PJ. Pathophysiology and management of testicular ischemia/reperfusion injury: lessons from animal models. Heliyon. 2024;10(9):e27760. doi:10.1016/j.heliyon.2024.e27760
118. Li ZM. Role of antioxidants in preventing testicular ischemia-reperfusion injury: a narrative review. Eur Rev Med Pharmacol Sci. 2022;26(24):9126–9143. doi:10.26355/eurrev_202212_30663
119. Wei SM, Huang YM. Attenuation effect of salvianolic acid B on testicular ischemia-reperfusion injury in rats. Oxid Med Cell Longevity. 2022;2022:7680182. doi:10.1155/2022/7680182
120. Jiang YP, Liu BG, Dang Y, et al. Integrative analysis of transcriptomics and metabolomics reveals the protective effect and mechanism of salidroside on testicular ischemia-reperfusion injury. Front Pharmacol. 2024;15:1377836. doi:10.3389/fphar.2024.1377836
121. Weng JD, Shen X, Wang R, et al. Pharmacokinetic changes and mechanisms of salidroside in hypobaric hypoxic environment: a LC-MS and proteomics study. J Ethnopharmacol. 2026;360:121205. doi:10.1016/j.jep.2026.121205
122. Xue L, Dong X, Zhao CX, et al. Study on the Treatment of Qi Deficiency and Blood Stasis Syndrome of Coronary Heart Disease Angina Pectoris with Sofren Injection Based on Disease Module Analysis. J Nanjing Univ Tradit Chin Med. 2024;40(12):1430–1440. doi:10.14148/j.issn.1672-0482.2024.1430
123. Shi Y, Zeng L, Deng JJ. Efficacy study on Dazhu Hongjingtian Injection combined with sacubitril valsartan sodium tablets in treatment of coronary heart disease complicated with heart failure. J Hubei Univers Chin Medi. 2024;26(4):22–25. doi:10.3969/j.issn.1008987x.2024.04.05
124. Wang YM, Xue CM, Zhang SS, Yan PP, Zhao HY, Cao JL. Study on the anti-anoxia ability of Rhodiola granule in mice. World J Integr Trad West Med. 2016;11(9):1226–1227. doi:10.13935/j.cnki.sjzx.160912
125. Xing N, Qin J, Ren DS, et al. Integrating UPLC-Q-Exactive Orbitrap/MS, network pharmacology and experimental validation to reveal the potential mechanism of Tibetan medicine Rhodiola granules in improving myocardial ischemia-reperfusion injury. J Ethnopharmacol. 2023;314:116572. doi:10.1016/j.jep.2023.116572
126. Zhang RH, Chao M, Bai GJ, Wen QW, Wei BH. Thin-Layer Identification Study of Rhodiola rosea American Ginseng and Ophiopogon japonicus Capsules. Food Ind. 2025;46(2):101–105.
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Potential of Polydatin Against Ischemia-Reperfusion Injury: New Insights from Pharmacological-Pathological Mechanism Associations
Sun Z, Wang X, Pang X
Drug Design, Development and Therapy 2025, 19:1585-1594
Published Date: 6 March 2025