Back to Journals » Drug Design, Development and Therapy » Volume 9
Protective effect of galangin in Concanavalin A- induced hepatitis in mice
Authors Luo Q ![]() , Zhu L, Ding J, Zhuang X, Xu L, Chen F
, Zhu L, Ding J, Zhuang X, Xu L, Chen F
Received 15 January 2015
Accepted for publication 14 April 2015
Published 10 June 2015 Volume 2015:9 Pages 2983—2992
DOI https://doi.org/10.2147/DDDT.S80979
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Shu-Feng Zhou
Qingqiong Luo,1,* Liping Zhu,1,* Jieying Ding,1 Xing Zhuang,1 Lili Xu,2 Fuxiang Chen1
1Department of Clinical Immunology, Shanghai Ninth People’s Hospital, Shanghai JiaoTong University School of Medicine, 2Division of Gastroenterology, Zhongshan Hospital, Fudan University, Shanghai, People’s Republic of China
*These authors contributed equally to this work
Abstract: Galangin is an active pharmacological ingredient from propolis and Alpinia officinarum Hance, and has been reported to have anti-inflammatory and antioxidative properties. The present study aims to reveal the effect of galangin on Concanavalin A (ConA)-induced hepatitis (CIH), a well-established animal model of immune-mediated liver injury, and to clarify the related mechanism. C57BL/6 mice were pretreated with galangin followed by ConA challenge. Results indicated that galangin inhibited ConA-induced liver damage. Mice pretreated with galangin showed more reduction of liver damage when compared with control mice pretreated with vehicle solution. In galangin-pretreated mice with induced CIH, increases in serum levels of several inflammatory cytokines, including tumor necrosis factor-α, interferon-γ, and interleukin-12 were dramatically attenuated, and chemokines and adhesion molecules like interferon inducible protein-10, macrophage inflammatory protein-1α, and intercellular adhesion molecule-1 messenger RNA expressions in liver were decreased. Moreover, CIH mice pretreated with galangin showed less leukocyte infiltration and T-cell activation in the liver. Further, the mechanism of the anti-inflammatory effects of galangin may be attributed to its modulation of crucial inflammatory signaling pathways, including nuclear factor kappa B and interferon-gamma/signal transducer and activator of transcription 1. Collectively, these findings suggest the preventive and therapeutic potential of galangin in immune-mediated liver injury in vivo.
Keywords: galangin, Concanavalin A-induced hepatitis, nuclear factor kappa B, STAT1
Introduction
Hepatitis, caused by virus infection, autoimmunity, alcoholism, and certain drugs, is a serious threat to human health and a worldwide problem. In the pathogenesis of various hepatitides, excessive or misdirected immune stimulation is considered to play a critical role.1–3 Concanavalin A (ConA)-induced hepatitis (CIH) in mice is a well-established experimental model for immune-mediated liver injury and has been widely used in studies of the pathogenesis and clinical treatment of immune hepatitis in humans. In CIH, inflammatory cells, such as T-cells, natural killer T-cells, macrophages, and neutrophils, infiltrate the liver and produce a variety of hepatotoxic inflammatory cytokines, including interferon (IFN)-γ, tumor necrosis factor (TNF)-α, interleukin (IL)-1β, and IL-12,4,5 which finally leads to massive apoptosis and necrosis of hepatocytes and marked elevation of serum transaminases.
Galangin (3,5,7-trihydroxyflavone; Figure 1A) is a naturally active flavonoid extracted from propolis and the root of Alpinia officinarum Hance, a plant that has been used as a spice and as a herbal medicine for various ailments in Asia for centuries.6 The biological activity of galangin includes antimutagenic, anticlastogenic, antioxidative, radical scavenging, metabolic enzyme-modulating, bactericidal, antifibrotic, and antitumor effects.6–8 There is also some recent evidence indicating that galangin has anti-inflammatory activity, which has attracted increasing attention. Galangin was shown to inhibit mast cell-derived allergic inflammation.9 Galangin also showed anti-inflammatory effects on collagen-induced arthritis via attenuation of RANKL-induced activation of Janus kinase, p38, and nuclear factor kappa B (NF-κB) pathways.10 Moreover, galangin dampened lipopolysaccharide-induced acute lung injury and inhibited atopic dermatitis-like skin lesions.11,12 However, the effect of galangin on CIH has not yet been studied.
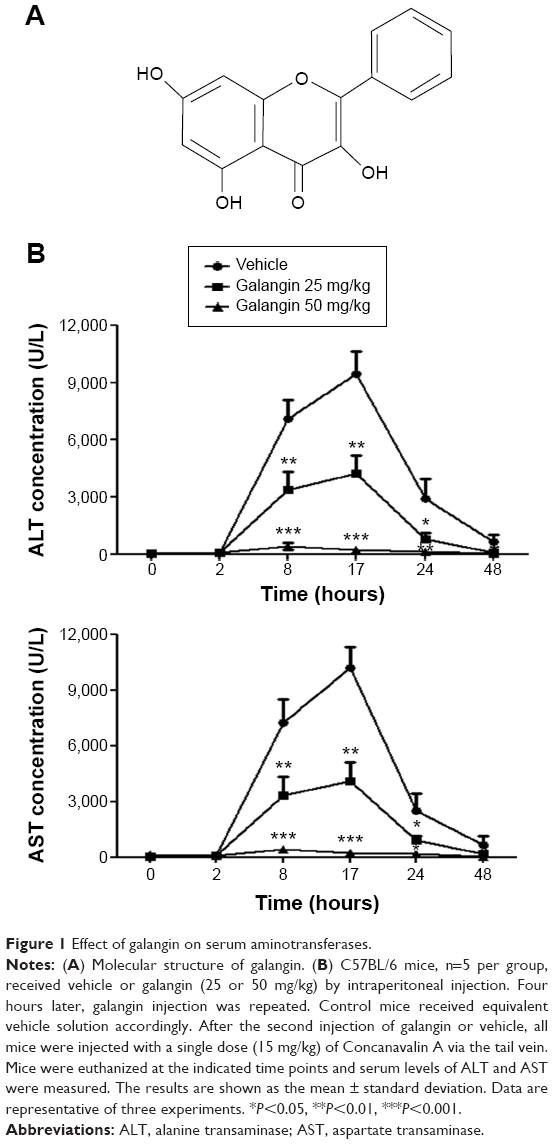 | Figure 1 Effect of galangin on serum aminotransferases. |
In this study, we demonstrated that pretreatment with galangin dramatically inhibited development of CIH in mice. In mice with CIH, the increases in serum aminotransferases, cytokine production, hepatic necroinflammation, and apoptosis were markedly attenuated by administration of galangin. Intrahepatic inflammatory cell infiltration and activation was also decreased by galangin. We also investigated the signaling pathways involved in the anti-inflammatory effects of galangin.
Materials and methods
Mice
Male C57BL/6 mice aged 8–10 weeks were purchased from Shanghai SLAC Laboratory Animal Co Ltd (Shanghai, People’s Republic of China). The mice were housed in the animal care facilities of the Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine, under pathogen-free conditions. Animal welfare and experimental procedures were carried out strictly in accordance with the Guide for the Care and Use of Laboratory Animals (Ministry of Science and Technology of China, 2006) and approved by the hospital’s laboratory animal care and use committee.
Galangin treatment and induction of CIH
Galangin (purity ≥95%, determined by high-performance liquid chromatography; Sigma-Aldrich, St Louis, MO, USA) was dissolved in dimethyl sulfoxide (Sigma-Aldrich) and diluted with phosphate-buffered saline (PBS) for use (the final concentration of dimethyl sulfoxide in the solution was 10%). Mice in the treated group received galangin by intraperitoneal injection at a dose of 25 mg/kg or 50 mg/kg. Four hours later, the galangin injection was repeated. Control mice received equivalent vehicle solution. After the second injection of galangin or vehicle, all mice were injected with ConA (Vector Laboratories, Burlingame, CA, USA) dissolved in PBS via the tail vein (15 mg/kg).
Measurement of serum aminotransferase activity
Blood samples were collected from the mice at different time points after ConA injection. Serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) activities were determined by the velocity method according to the manufacturer’s instructions using the Siemens Dimension RXL MAX Clinical Chemistry Analyzer (Dade Behring Inc., Newark, NJ, USA).
Liver histology and TUNEL staining
Eight hours after ConA injection, mice develop significant liver injury.5,13 Thus, we chose this time point for examination of liver pathology. Livers were removed after perfusion with PBS, fixed with 4% phosphate-buffered paraformaldehyde, and embedded in paraffin. Tissue sections (5 μm) were prepared and stained with hematoxylin-eosin. For TUNEL (terminal deoxynucleotidyl transferase (TdT)-mediated dUTP-biotin nick end labeling) staining, paraffin-embedded liver tissues were assayed for DNA fragmentation using a TUNEL reaction, according to the instructions provided by the manufacturer (Roche Molecular Biochemicals, Indianapolis, IN, USA). All sections were examined under light microscopy. A total of ten tissue sections were analyzed for each animal.
Analysis of serum cytokines
Serum concentrations of TNF-α, IFN-γ, and IL-12p70 were determined using a specific enzyme-linked immunosorbent assay kit (R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s instructions. As has been reported previously, cytokines reach their peak concentrations at different time points.14–16 Thus, TNF-α was measured 2 hours after ConA injection and the other cytokines were measured 8 hours after ConA injection.
RNA extraction and reverse transcription
Total RNA was isolated from the frozen liver tissues using TRIzol reagent (Life Technologies, Carlsbad, CA, USA) according to the protocol provided by the manufacturer. Genomic DNA was removed from total RNA prior to synthesis of complementary DNA using the RNase-free DNase set for DNase digestion during RNA purification (Qiagen, Hilden, Germany). RNA was stored at −80°C. First-strand complementary DNA synthesis was performed for each RNA sample using the Sensiscript RT kit (Qiagen). Random hexamers were used to prime complementary DNA synthesis.
Real-time polymerase chain reaction
Messenger (m)RNA expression of IFN-inducible protein (IP)-10, macrophage inflammatory protein (MIP)-1α, and intercellular adhesion molecule (ICAM)-1 was investigated by real-time polymerase chain reaction using SYBR Green master mix (Applied Biosystems, Foster City, CA, USA). Thermocycler conditions comprised an initial holding at 50°C for 2 minutes, then 95°C for 10 minutes. This was followed by a two-step polymerase chain reaction program consisting of 95°C for 15 seconds and 60°C for 60 seconds for 40 cycles. Data were collected and quantitatively analyzed on an ABI Prism 7500 sequence detection system (Applied Biosystems). The β−actin gene was used as an endogenous control to normalize for differences in the amount of total RNA in each sample. All quantities were expressed as number of folds relative to the expression of β−actin. Primer sequences were as follows: β−actin, sense 5′-TGTCCACCTTCCAGCAGATGT-3′, antisense 5′-AGCTCAGTAACAGTCCGCCTAGA-3′; MIP-1α, sense 5′-CACCCTCTGTCACCTGCTCAA-3′, antisense 5′-ATGGCGCTGAGAAGACTTGGT-3′; IP-10, sense 5′-GCCGTCATTTTCTG CCTC-3′, antisense 5′-ATGGCGCTGAGAAGACTTGGT-3′; ICAM-1, sense 5′-CCATCACCGTGTATTCGTTTCC-3′, antisense 5′-CTGGCGGCTCAGTATCTCCTC-3′.
Isolation of hepatic leukocytes
Isolation of intrahepatic leukocytes was performed as described previously.17 Briefly, the mice livers were removed and pressed through a 70 μm cell strainer. The liver cells were suspended in PBS and centrifuged at 50× g for 5 minutes. Supernatants containing mononuclear cells were collected, washed in PBS, and resuspended in 40% Percoll. The cell suspension was gently overlaid onto 70% Percoll and centrifuged for 30 minutes at 750× g. Mononuclear cells were collected from the interphase, washed twice in PBS, and resuspended in PBS for analysis by fluorescence-activated cell sorting. The intrahepatic leukocytes were also observed and counted under a light microscope.
Flow cytometry
Single-cell suspensions of liver or spleen mononuclear cells were washed in PBS containing 1% bovine serum albumin. The cells were stained with fluorochrome-conjugated monoclonal antibody for 30 minutes on ice. The following antibodies were used: anti-CD3e, anti-NK1.1, anti-Gr-1, anti-CD11b, and anti-CD69 (BD Pharmingen, San Diego, CA, USA). Samples were acquired on a FACSCalibur flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA) and the data were analyzed by FlowJo software.
Western blot analysis
Liver homogenates were lysed in RIPA buffer (150 mmol/L NaCl, 10 mmol/L Tris-HCl [pH 8.0], 1% Nonidet P-40, 0.5% deoxycholic acid, 0.1% sodium dodecyl sulfate, 5 mmol/L ethylenediaminetetraacetic acid) containing 0.7% phenylmethylsulfonyl fluoride, 0.2% aprotinin, and 0.2% leupeptin and sodium metavanadate. Samples containing 100 μg of protein were incubated at 100°C for 5 minutes, separated by 10% (w/v) sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and electrophoretically transferred onto a polyvinylidene difluoride transfer membrane (Bio-Rad Laboratories, Hercules, CA, USA). The nonspecific sites were blocked with a solution containing 5% non-fat milk powder in Tris-buffered saline/Tween 20 (TBST) for 2 hours at room temperature. The membrane was incubated with antibodies against β-actin (Sigma-Aldrich), IκBα (44D4), phospho-IκBα (pIκBα) (Ser32/36, 5A5), signal transducer and activator of transcription 1 (STAT1), and pSTAT1 (Ser727; Cell Signaling Technology, Danvers, MA, USA) in TBST containing 5% bovine serum albumin overnight at 4°C, then with horseradish peroxidase-conjugated second antibodies (anti-rabbit IgG [Sigma-Aldrich] and anti-mouse immunoglobulin G [Cell Signaling Technology], at a dilution of 1/5,000 and 1/2,000, respectively). Protein bands were visualized using chemiluminescence reagent (Thermo Scientific, Waltham, MA, USA).
Statistical analysis
All results are expressed as the mean ± standard deviation. The Student’s t-test was used for the statistical analysis. P<0.05 was considered to be statistically significant.
Results
Galangin inhibited ConA-induced aminotransferase release in a dose-dependent manner
It has been shown that galangin has anti-inflammatory activity;9,10 however, whether galangin plays a role in CIH is not clear. In order to clarify the effect of galangin in CIH, we pretreated mice with galangin at two different doses (25 mg/kg or 50 mg/kg) before injection of ConA. Mice were euthanized at different time points after induction of CIH, and serum levels of ALT and AST were measured for each mouse. As shown in Figure 1B, mice treated with galangin showed much lower serum ALT and AST activity in both the acute phase and the recovery stage, as compared with mice in the vehicle-injected control group. Moreover, galangin inhibited ConA-induced aminotransferase release in a dose-dependent manner. Mice receiving a 25 mg/kg dose of galangin showed more than half the reduction in ALT and AST release when compared with control mice, and in mice pretreated with 50 mg/kg galangin, the elevation of ALT and AST levels induced by ConA was almost completely suppressed. These data strongly suggest that the protective effect of galangin was permanent in the development of ConA-induced liver injury. We tried a higher dose of galangin (100 mg/kg), but did not observe further inhibition of release of ConA-induced aminotransferases (data not shown). Thus, the dose of galangin used in the following experiments was 50 mg/kg.
Galangin ameliorated ConA-induced liver damage
We then examined the effect of galangin on ConA-induced liver damage using hematoxylin-eosin and TUNEL staining. Mice administered ConA suffered severe liver damage, indicated by inflammatory infiltration around the central veins and large areas of centrilobular necrosis and apoptosis of hepatocytes (Figure 2A shows hematoxylin-eosin staining, Figure 2C shows TUNEL staining). However, mice pretreated with galangin (50 mg/kg) showed minimal liver damage: in these mice, fewer areas of intralobular necrosis or inflammatory infiltration were observed (Figure 2B, hematoxylin-eosin staining) and only a few hepatocytes exhibited TUNEL-positive nuclei (Figure 2D, TUNEL staining), indicating that apoptosis was markedly reduced in galangin-pretreated mice.
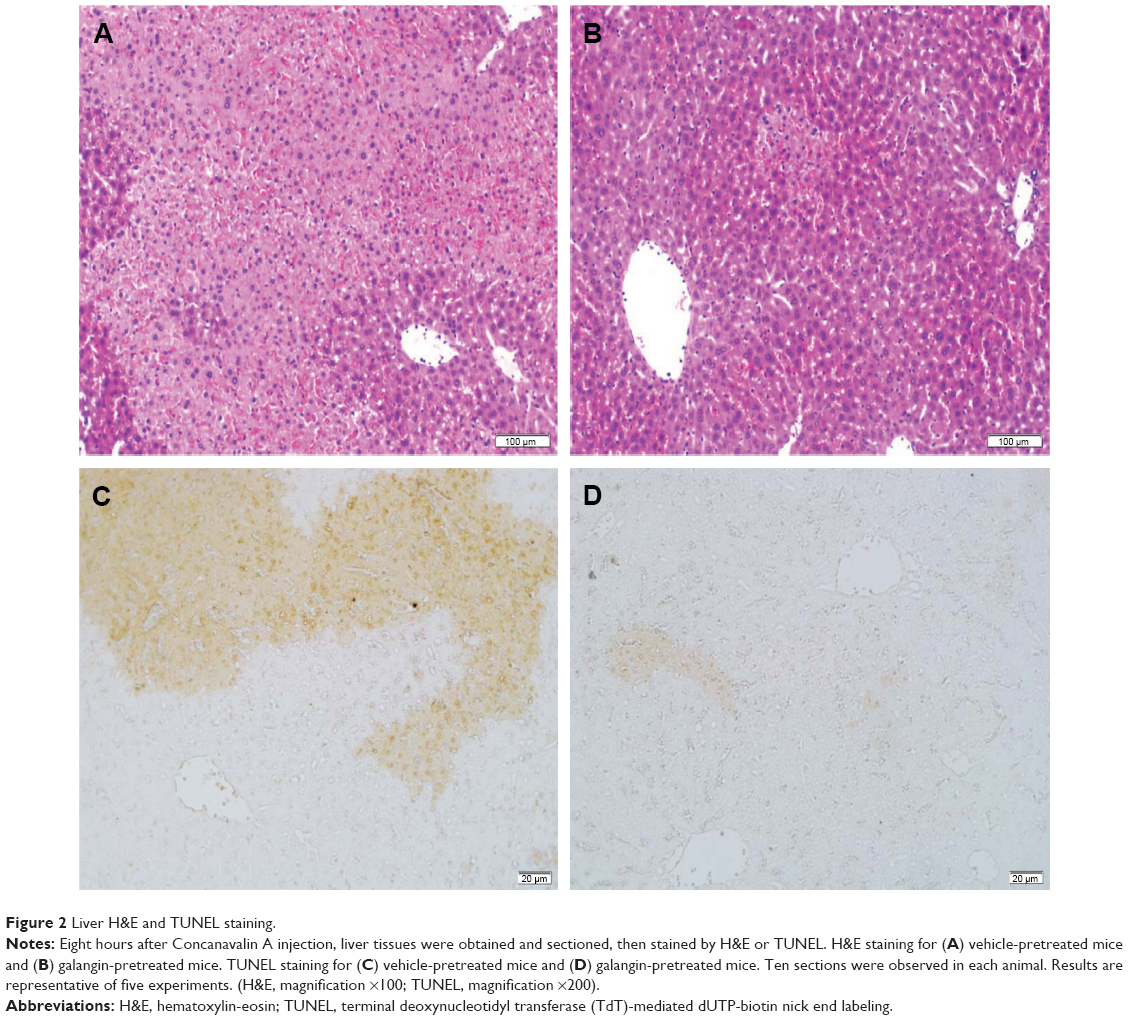 | Figure 2 Liver H&E and TUNEL staining. |
Galangin inhibited release of inflammatory cytokines in CIH mice
Previous studies indicate that immune cells are activated after ConA injection and that, as a result, a number of cytokines are released that aggravate liver injury.4,5 We therefore examined the influence of galangin on serum TNF-α, IFN-γ, and IL-12 levels, given that these cytokines are reported to play an important role in the development of ConA-induced liver injury.4,5 As shown in Figure 3, production of TNF-α, IFN-γ, and IL-12 was inhibited in galangin-pretreated mice when compared vehicle-pretreated mice (P<0.01 for TNF-α and IFN-γ, and P<0.001 for IL-12). These data demonstrate that galangin effectively suppressed production of various disease-related cytokines, thereby reducing the damage to liver tissues caused by these inflammatory mediators.
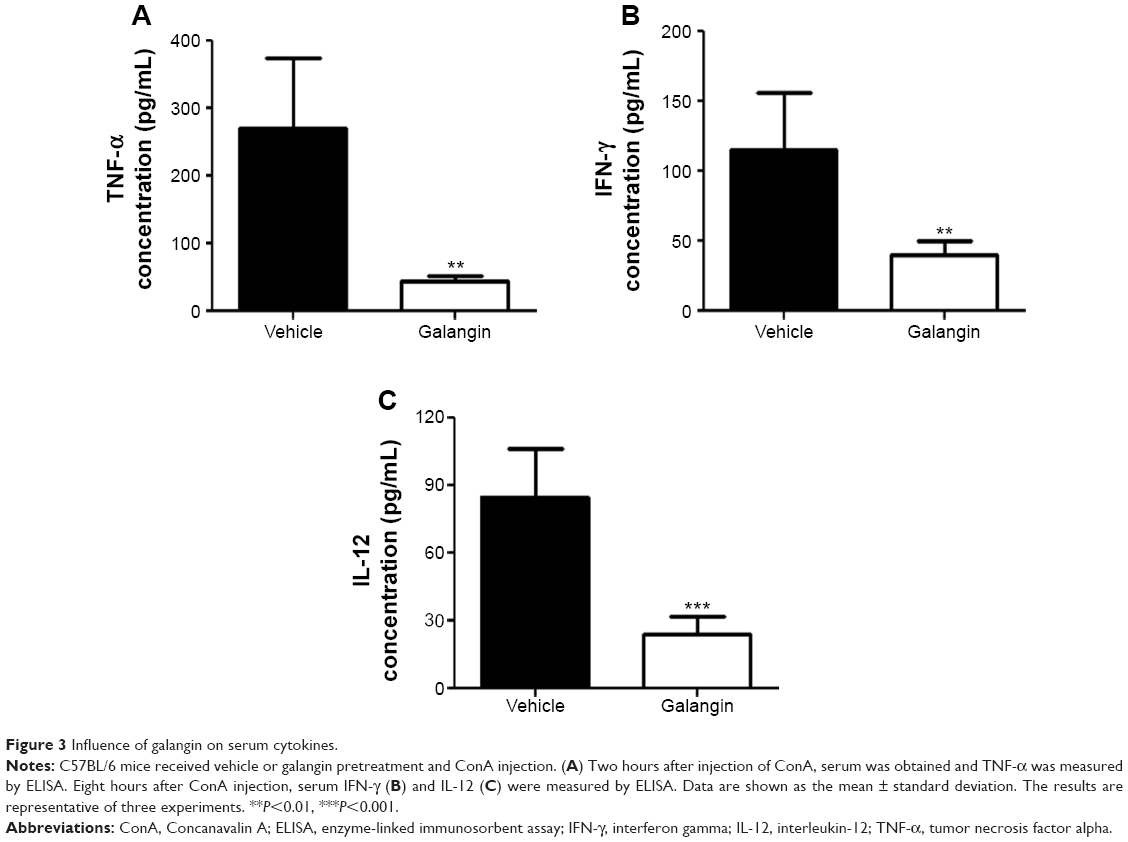 | Figure 3 Influence of galangin on serum cytokines. |
Galangin inhibited chemokine and adhesion molecule expression in the liver
Upregulated expression of chemokines and adhesion molecules such as IP-10, MIP-1α, and ICAM-1 has been reported to play a critical role in the development of CIH.18–20 Thus, we further studied the changes in IP-10, MIP-1α and ICAM-1 mRNA expression in the livers of mice with or without galangin pretreatment. Liver tissue RNA was obtained 8 hours after ConA injection, and mRNA expression of IP-10, MIP-1α, and ICAM-1 was measured. As shown in Figure 4, the mRNA levels of these three genes were all dramatically reduced by pretreatment with galangin (P<0.01 for IP-10, and P<0.05 for MIP-1α and ICAM-1).
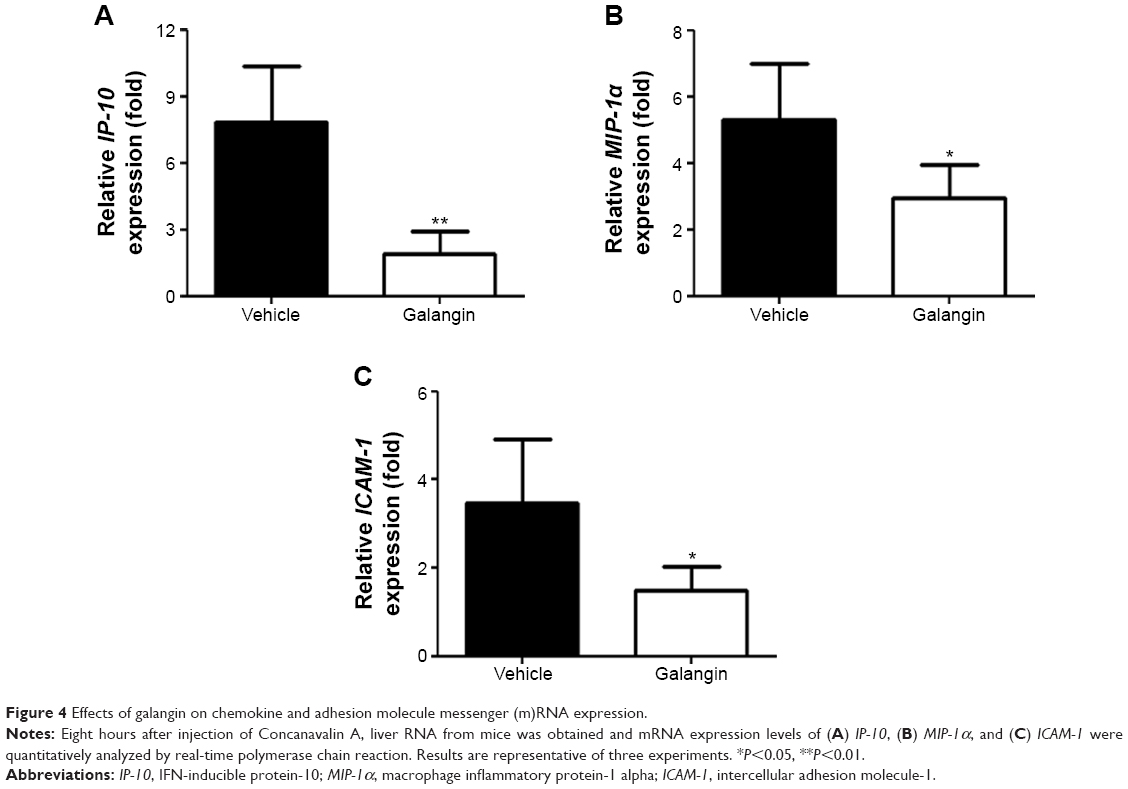 | Figure 4 Effects of galangin on chemokine and adhesion molecule messenger (m)RNA expression. |
Galangin prevented ConA-induced inflammatory cell recruitment and activation in the liver
To further study the profiles of the infiltrated inflammatory cells in the liver, we analyzed intrahepatic leukocytes by fluorescence-activated cell sorting. CIH mice with or without galangin pretreatment were euthanized 2 hours after ConA injection, and intrahepatic leukocytes in the liver were obtained. The total number of intrahepatic leukocytes in galangin pretreated mice was remarkably decreased compared with that in mice without galangin administration (Figure 5A, P<0.01). Neutrophils (Gr-1+CD11b+), macrophages (CD11b+Gr-1−), and T-cells (CD3+NK1.1−) recruited into the liver were significantly reduced by galangin, as shown in Figure 5A. Moreover, galangin significantly inhibited T-cell activation in CIH mice, as CD3+CD69+T cell percent decreased dramatically (Figure 5B). However, there was no significant change in natural killer cells (CD3−NK1.1+) or natural killer T-cells (CD3+NK1.1+). Taken together, these results indicate that pretreatment with galangin influenced ConA-induced intrahepatic inflammatory cell infiltration and activation.
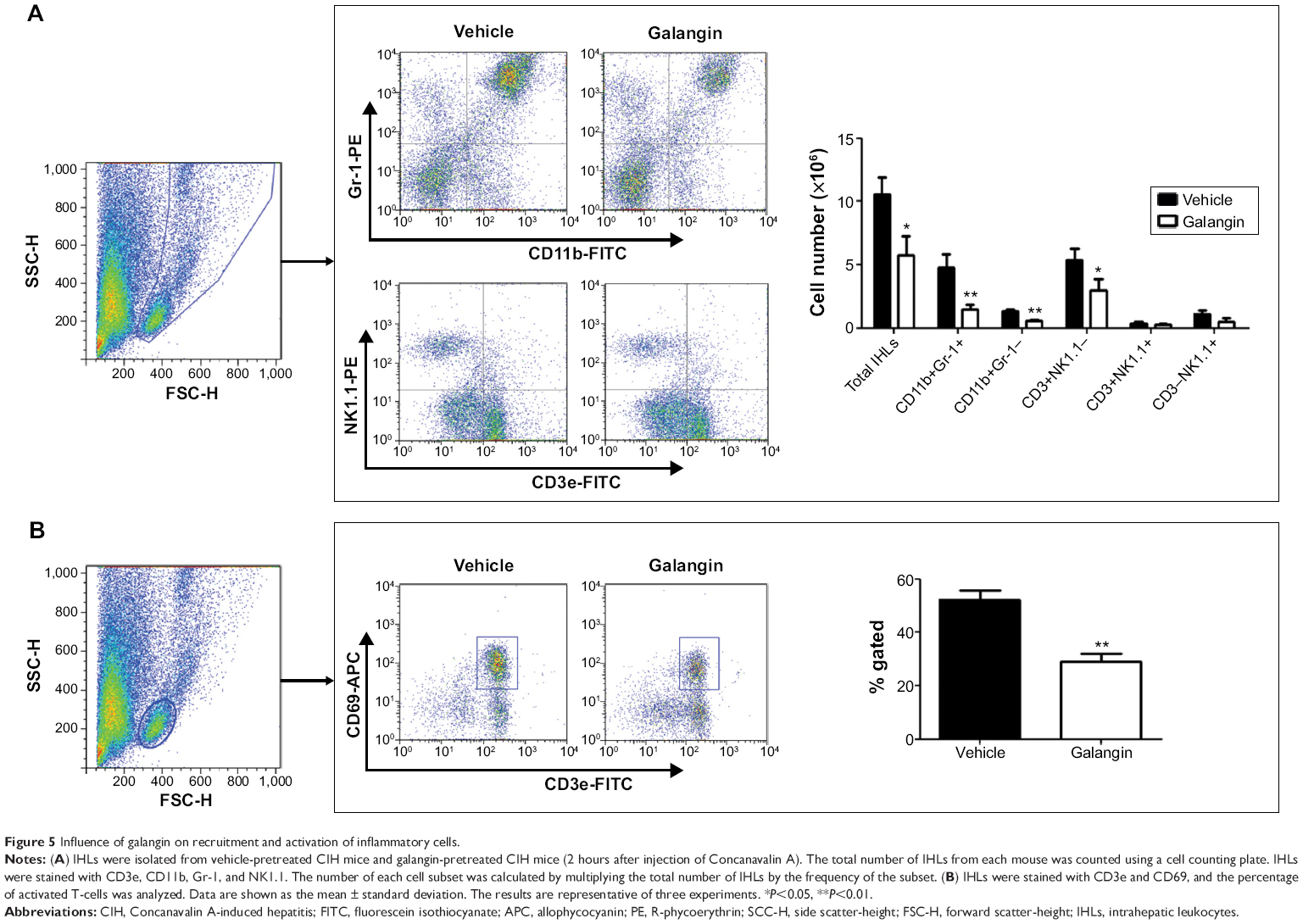 | Figure 5 Influence of galangin on recruitment and activation of inflammatory cells. |
Galangin suppressed ConA-induced NF-κB activation and STAT1 phosphorylation
Since the production of many inflammatory mediators, including the cytokines and chemokines mentioned above, depends on activation of the NF-κB signaling pathway, we analyzed the activation of NF-κB in mice received vehicle or galangin pretreatment and ConA injection. As shown in Figure 6, galangin significantly inhibited ConA-induced phosphorylation of IκBα, which is required for subsequent activation of NF-κB because it allows the NF-κB dimer to translocate into the nucleus and bind to the κB site. Therefore, more NF-κB complexes remained in an inactive state in the cytoplasm after pretreatment with galangin, leading to a weak response toward ConA stimulation. In addition, transcription factor STAT1, which is associated with the IFN-γ signaling pathway, is considered to be an important pathogenic mediator of T-cell activation in CIH.21,22 It has been reported that synthesis and activation of STAT1 is increased in the liver of the CIH mouse, and that STAT1 deficiency attenuates ConA-induced liver damage.21,22 Therefore, we also investigated the effect of galangin on activation of STAT1 in CIH. The data show that galangin effectively reduced phosphorylation of STAT1 (Figure 6A and B), suggesting that galangin decreased the release of IFN-γ and suppressed the STAT1 signaling pathway.
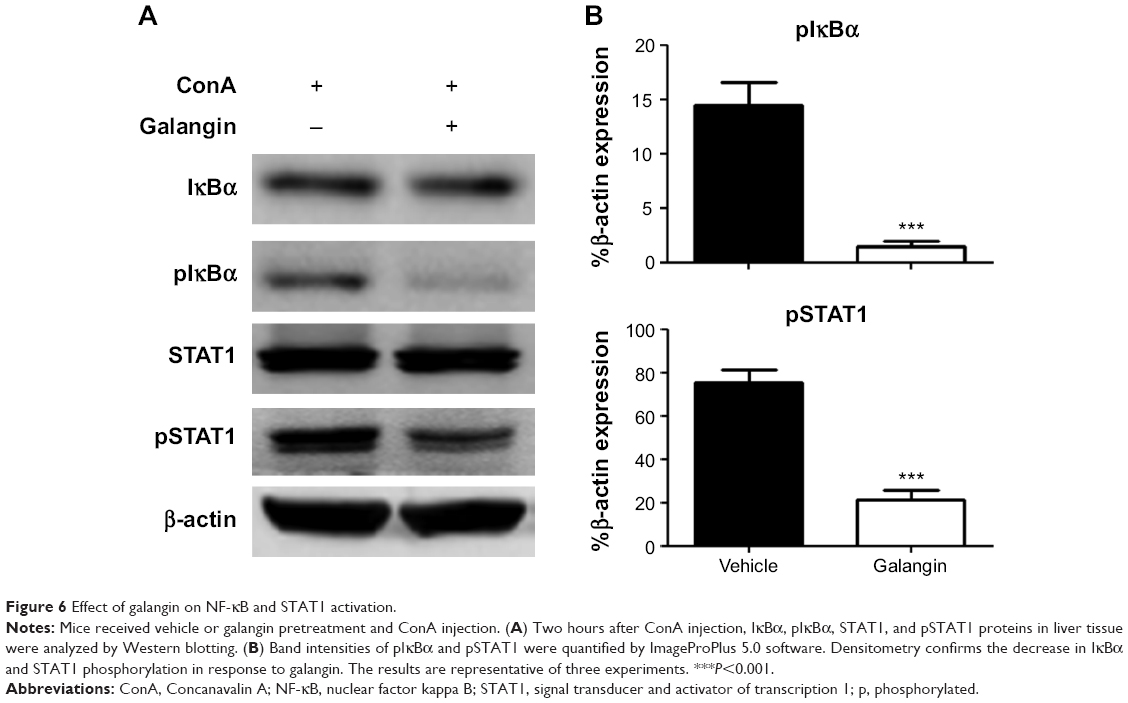 | Figure 6 Effect of galangin on NF-κB and STAT1 activation. |
Discussion
In the present study, we showed that galangin was able to protect against immune-mediated liver injury in mice with CIH. Histological staining indicated that liver inflammation and apoptosis of hepatocytes were clearly reduced in galangin-pretreated mice. Galangin also significantly reduced the expression and release of various disease-related cytokines and chemokines. Moreover, galangin inhibited infiltration and activation of intrahepatic inflammatory cells. The protective effects of galangin may be attributed to its suppression of activation of NF-κB and STAT1 signaling pathway.
Galangin, a member of the flavonol class of flavonoids, has multiple pharmacological activities, including anticancer,8 antioxidative,6 antiviral,23 antimicrobial,24 and vasorelaxant25 effects. Recently, the anti-inflammatory effect of galangin has been attracting increasing attention. Galangin has been reported to reduce IL-1β, TNF-α, and IL-17 concentrations and to attenuate activation of the Janus kinase, p38, and NF-κB pathways, thus reducing the severity of collagen-induced arthritis in mice.10 Galangin also abrogated ovalbumin-induced airway inflammation by modulating the expression of several related cytokines, including IL-4, IL-5, and IL-13, and chemokines such as monocyte chemoattractant protein-1, C-X-C motif chemokine 10, and vascular cell adhesion protein 1, thereby reducing infiltration and activation of eosinophil.26 In addition, several other groups have demonstrated that galangin inhibits atopic dermatitis-like lesions and dampens lipopolysaccharide-induced acute lung injury in mice.11,12 Our observations of the beneficial effects of galangin on CIH are consistent with previous reports. In particular, we demonstrated for the first time that galangin has marked protective effects against immune-mediated liver injury.
Previous studies have reported that TNF-α and IFN-γ play a critical role in the pathogenesis of CIH.14,27 TNF-α, secreted from activated macrophages and T-cells, binds to its receptors and induces an apoptotic cascade, thus acting as a major mediator in inflammation-induced hepatocyte death.27 Mice pretreated with anti-TNF-α antiserum or a TNF-α inhibitor and mice deficient in the TNF-α receptor exhibited obvious resistance to CIH. IFN-γ exerts pathogenic effects by synergizing with TNF-α to promote production of various chemokines and adhesion molecules.14,28 In addition, IFN-γ induces phosphorylation of STAT1, which leads to expression of proapoptotic genes and contributes to CIH.29 Besides TNF-α and IFN-γ, IL-12 has also been found to be important in the pathogenesis of CIH. Exogenous IL-12 administration prior to ConA injection exacerbated liver damage, and anti-IL-12 monoclonal antibody pretreatment prevented ConA-induced liver injury.15 In our study, galangin pretreatment reversed the increase in production of these pathogenic inflammatory cytokines in CIH, thereby suppressing activation of T-cells and alleviating the liver injury.
We found that galangin was able to inhibit IP-10, MIP-1α, and ICAM-1 mRNA expression in the livers of CIH mice. During the first few hours after injection of ConA, production of proinflammatory cytokines followed by expression of chemotactic factors such as IP-10, MIP-1α, and ICAM-1, which attract leukocytes to the liver, amplifies the local inflammatory damage.20,29 IP-10 has been found to play an important role in the hepatic neutrophil influx in CIH.19 MIP-1α is reported to promote recruitment of various leukocyte subtypes, including T-cells, macrophages, and neutrophils. In mice with MIP-1α deficiency, hepatic injury induced by ConA was markedly attenuated.18 Further, ICAM-1 has been shown to mediate adhesion and to support transendothelial migration of lymphocytes in the liver.20 A previous study demonstrated that ICAM-1 deficiency significantly inhibited liver injury and T-cell recruitment in CIH. In the present study, we report, for the first time, that galangin is able to downregulate IP-10, MIP-1α, and ICAM-1 mRNA expression in the liver. Such downregulation, in turn, inhibited infiltration of leukocytes into the liver, thus reducing the severity of liver damage in CIH mice.
We further investigated the mechanisms involved in the protective effect of galangin in CIH. One important signaling pathway in the regulation of these inflammatory mediators is activation of NF-κB. In resting cells, the NF-κB complex is retained in the cytoplasm by the inhibitory protein IκBα, and nuclear accumulation of NF-κB depends mainly on phosphorylation of IκBα and its subsequent degradation. Without the inhibitory effect of IκBα, NF-κB translocates from the cytoplasm to the nucleus to bind the κB site of target genes.30 In the present study, we found that galangin decreased phosphorylation of IκBα, leading to less translocalization of NF-κB. Moreover, the pathogenic inflammatory cytokine IFN-γ exerted its effects via activation of the Janus kinase-STAT signaling pathway. With the ligation of IFN-γ to its receptor, STAT1 was phosphorylated and dimerized, and then translocated into the nuclei to activate transcription of a number of genes, including those for IFN regulatory factor-1, Fas receptor, caspases, inducible nitric oxide synthase, cyclo-oxygenase, and intercellular cell adhesion molecules, all of which are critically involved in the pathogenesis of CIH.29,31 We observed an obvious decrease of STAT1 phosphorylation after pretreatment with galangin. Therefore, we speculate that the protective effect of galangin in CIH is mediated by inhibition of the NF-κB and IFN-γ/STAT1 signaling pathways.
Conclusion
Galangin attenuated ConA-induced liver injury in mice. The suppressive effect was attributed to inhibition of NF-κB and STAT1 signaling leading to the reduced expression and secretion of several inflammatory mediators. The results of this study suggest new opportunities to use galangin in the prevention and treatment of immune-mediated liver disease.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81200299 and 81472514) and the Medical Guide Program of Shanghai Municipal Science and Technology Commission (124119a9302).
Disclosure
The authors report no conflicts of interest in this work.
References
Knolle PA, Thimme R. Hepatic immune regulation and its involvement in viral hepatitis infection. Gastroenterology. 2014;146(5):1193–1207. | ||
Heneghan MA, Yeoman AD, Verma S, Smith AD, Longhi MS. Autoimmune hepatitis. Lancet. 2013;382(9902):1433–1444. | ||
Bieghs V, Trautwein C. The innate immune response during liver inflammation and metabolic disease. Trends Immunol. 2013;34(9):446–452. | ||
Wang HX, Liu M, Weng SY, et al. Immune mechanisms of Concanavalin A model of autoimmune hepatitis. World J Gastroenterol. 2012;18(2):119–125. | ||
Tiegs G, Hentschel J, Wendel A. A T cell-dependent experimental liver injury in mice inducible by concanavalin A. J Clin Invest. 1992;90(1):196–203. | ||
Heo MY, Sohn SJ, Au WW. Anti-genotoxicity of galangin as a cancer chemopreventive agent candidate. Mutat Res. 2001;488(2):135–150. | ||
Wang X, Gong G, Yang W, Li Y, Jiang M, Li L. Antifibrotic activity of galangin, a novel function evaluated in animal liver fibrosis model. Environ Toxicol Pharmacol. 2013;36(2):288–295. | ||
Su L, Chen X, Wu J, et al. Galangin inhibits proliferation of hepatocellular carcinoma cells by inducing endoplasmic reticulum stress. Food Chem Toxicol. 2013;62:810–816. | ||
Kim HH, Bae Y, Kim SH. Galangin attenuates mast cell-mediated allergic inflammation. Food Chem Toxicol. 2013;57:209–216. | ||
Huh JE, Jung IT, Choi J, et al. The natural flavonoid galangin inhibits osteoclastic bone destruction and osteoclastogenesis by suppressing NF-kappaB in collagen-induced arthritis and bone marrow-derived macrophages. Eur J Pharmacol. 2013;698(1–3):57–66. | ||
Shu YS, Tao W, Miao QB, Lu SC, Zhu YB. Galangin dampens mice lipopolysaccharide-induced acute lung injury. Inflammation. 2014;37(5):1661–1668. | ||
Choi JK, Kim SH. Inhibitory effect of galangin on atopic dermatitis-like skin lesions. Food Chem Toxicol. 2014;68:135–141. | ||
Shen M, Lu J, Cheng P, et al. Ethyl pyruvate pretreatment attenuates concanavalin a-induced autoimmune hepatitis in mice. PLoS One. 2014;9(2):e87977. | ||
Kusters S, Gantner F, Kunstle G, Tiegs G. Interferon gamma plays a critical role in T cell-dependent liver injury in mice initiated by concanavalin A. Gastroenterology. 1996;111(2):462–471. | ||
Nicoletti F, Di Marco R, Zaccone P, et al. Murine concanavalin A-induced hepatitis is prevented by interleukin 12 (IL-12) antibody and exacerbated by exogenous IL-12 through an interferon-gamma-dependent mechanism. Hepatology. 2000;32(4 Pt 1):728–733. | ||
Lv K, Zhang Y, Zhang M, Zhong M, Suo Q. Galectin-9 ameliorates Con A-induced hepatitis by inducing CD4(+)CD25(low/int) effector T-Cell apoptosis and increasing regulatory T cell number. PLoS One. 2012;7(10):e48379. | ||
Wang Y, Feng D, Wang H, et al. STAT4 knockout mice are more susceptible to concanavalin A-induced T-cell hepatitis. Am J Pathol. 2014;184(6):1785–1794. | ||
Ajuebor MN, Hogaboam CM, Le T, Proudfoot AE, Swain MG. CCL3/MIP-1alpha is pro-inflammatory in murine T cell-mediated hepatitis by recruiting CCR1-expressing CD4(+) T cells to the liver. Eur J Immunol. 2004;34(10):2907–2918. | ||
Tamaru M, Nishioji K, Kobayashi Y, et al. Liver-infiltrating T lymphocytes are attracted selectively by IFN-inducible protein-10. Cytokine. 2000;12(4):299–308. | ||
Kawasuji A, Hasegawa M, Horikawa M, et al. L-selectin and intercellular adhesion molecule-1 regulate the development of concanavalin A-induced liver injury. J Leukoc Biol. 2006;79(4):696–705. | ||
Siebler J, Wirtz S, Klein S, et al. A key pathogenic role for the STAT1/T-bet signaling pathway in T-cell-mediated liver inflammation. Hepatology. 2003;38(6):1573–1580. | ||
Jaruga B, Hong F, Kim WH, Gao B. IFN-gamma/STAT1 acts as a proinflammatory signal in T cell-mediated hepatitis via induction of multiple chemokines and adhesion molecules: a critical role of IRF-1. Am J Physiol Gastrointest Liver Physiol. 2004;287(5):G1044–G1052. | ||
Schnitzler P, Neuner A, Nolkemper S, et al. Antiviral activity and mode of action of propolis extracts and selected compounds. Phytother Res. 2010;24 Suppl 1:S20–S28. | ||
Cushnie TP, Hamilton VE, Chapman DG, Taylor PW, Lamb AJ. Aggregation of Staphylococcus aureus following treatment with the antibacterial flavonol galangin. J Appl Microbiol. 2007;103(5):1562–1567. | ||
Morello S, Vellecco V, Alfieri A, Mascolo N, Cicala C. Vasorelaxant effect of the flavonoid galangin on isolated rat thoracic aorta. Life Sci. 2006;78(8):825–830. | ||
Zha WJ, Qian Y, Shen Y, et al. Galangin abrogates ovalbumin-induced airway inflammation via negative regulation of NF-kappaB. Evid Based Complement Alternat Med. 2013;2013:767689. | ||
Gantner F, Leist M, Lohse AW, Germann PG, Tiegs G. Concanavalin A-induced T-cell-mediated hepatic injury in mice: the role of tumor necrosis factor. Hepatology. 1995;21(1):190–198. | ||
Mizuhara H, Uno M, Seki N, et al. Critical involvement of interferon gamma in the pathogenesis of T-cell activation-associated hepatitis and regulatory mechanisms of interleukin-6 for the manifestations of hepatitis. Hepatology. 1996;23(6):1608–1615. | ||
Hong F, Jaruga B, Kim WH, et al. Opposing roles of STAT1 and STAT3 in T cell-mediated hepatitis: regulation by SOCS. J Clin Invest. 2002;110(10):1503–1513. | ||
Hoesel B, Schmid JA. The complexity of NF-kappaB signaling in inflammation and cancer. Mol Cancer. 2013;12:86. | ||
Wu XX, Sun Y, Guo WJ, et al. Rebuilding the balance of STAT1 and STAT3 signalings by fusaruside, a cerebroside compound, for the treatment of T-cell-mediated fulminant hepatitis in mice. Biochem Pharmacol. 2012;84(9):1164–1173. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.