Back to Journals » Drug Design, Development and Therapy » Volume 20
Prospects for Neuroprotective Therapies in Glaucoma: Drug Targets and Emerging Clinical Strategies
Authors Rho S ![]() , Williams PA
, Williams PA ![]()
Received 28 April 2026
Accepted for publication 11 June 2026
Published 16 June 2026 Volume 2026:20 612176
DOI https://doi.org/10.2147/DDDT.S612176
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tuo Deng
Seungsoo Rho,1 Pete A Williams2,3
1Department of Ophthalmology, CHA Bundang Medical Center, CHA University, Seongnam, Republic of Korea; 2Centre for Eye Research Australia, Royal Victorian Eye and Ear Hospital, Melbourne, Australia; 3Department of Clinical Neuroscience, Division of Eye and Vision, St. Erik Eye Hospital, Karolinska Institutet, Stockholm, Sweden
Correspondence: Pete A Williams, Centre for Eye Research Australia, Level 10, 200 Victoria Parade, East Melbourne, VIC, 3002, Australia, Email [email protected]
Abstract: Management of glaucoma is now at an inflection point with a new generation of therapeutic candidates, whilst targeting intraocular pressure-independent strategies is challenged by the landmark Phase III failure of memantine regarding trial design and endpoint sensitivity. Preclinical research has identified promising targets including glutamate excitotoxicity, neurotrophic factor deprivation, and mitochondrial dysfunction, with nicotinamide emerging as a leading candidate due to its ability to robustly protect RGCs by supporting NAD levels and bioenergetics. Current clinical efforts are expanding into metabolic repurposing with agents (eg metformin and semaglutide), sustained-delivery systems with neurotrophic factors (eg ciliary neurotrophic factor implant), and functional enhancers (eg citicoline). To bridge the translational gap, the field is integrating new endpoints with higher sensitivity (eg advanced assessment of photopic negative response), AI-guided endpoint selection (eg graph attention neural network), novel biomarkers (eg detection of apoptotic retinal cells and neurofilament light chain in aqueous humor), and precision medicine frameworks (eg polygenic risk scores and multi-omics analysis) to develop the first clinically validated neuroprotective treatments for glaucoma.
Keywords: glaucoma, neuroprotection, retinal ganglion cells, clinical trials
Introduction
Glaucoma is a progressive neurodegenerative disease characterized by the irreversible loss of retinal ganglion cells (RGCs) and their axons, leading to optic nerve damage and visual field loss. Whilst intraocular pressure (IOP) reduction remains the only evidence-based treatment strategy, disease progression commonly occurs despite adequate IOP control, indicating that IOP-independent mechanisms play critical roles in RGC degeneration. These include excitotoxicity, mitochondrial dysfunction, oxidative stress, neuroinflammation, neurotrophic factor deprivation, and apoptosis (independently or in concert). This unmet clinical need has spurred intense investigation into neuroprotective drugs that directly target the neurodegenerative component of glaucoma, independent of IOP lowering. This review synthesizes the current landscape of drug-based neuroprotective strategies, delineates preclinical from clinical evidence, and highlights emerging therapeutic targets. Clinical evidence is detailed in Table 1 and a summary including preclinical and clinical evidence is shown in Figure 1.
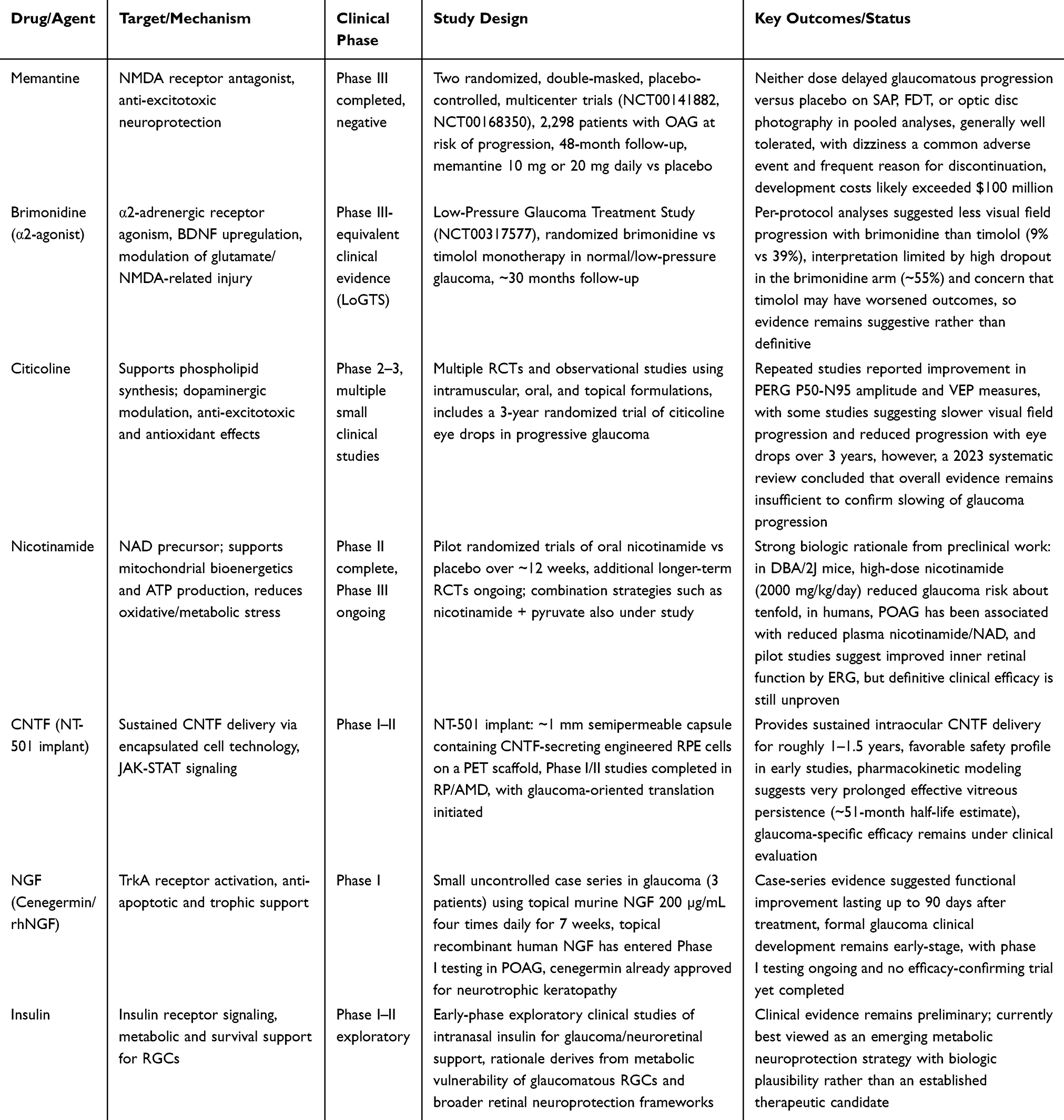 |
Table 1 Summary of the Main Clinical-Stage Drug Candidates for Glaucoma Neuroprotection |
 |
Figure 1 Drug-based neuroprotective strategies for glaucoma. Pathogenic mechanisms, pharmacological targets, and clinical development stage. |
Pre-Clinical Drug Targets and Evidence
Anti-Excitotoxic Strategies: NMDA and AMPA Receptor Modulation
Glutamate excitotoxicity is one of the best-characterized IOP-independent mechanisms of RGC death in glaucoma. Under physiological conditions, NMDA-type glutamate-gated ion channels are blocked at resting membrane potential by a magnesium ion occupying the channel pore. This channel thus functions as a coincidence detector, requiring both glutamate binding and sufficient depolarization to relieve the Mg2⁺ block and permit Na⁺ and Ca2⁺ conductance.1–3 In glaucomatous tissue, dysregulation of the glutamate–glutamine cycle is supported by findings of elevated glutamate levels and reduced expression of excitatory amino acid transporters (EAAT-1/GLAST, GLT-1) in human glaucomatous eyes, which together create conditions favoring excessive NMDA receptor activation and pathological calcium influx into RGCs.4–9
Memantine (1-amino-3,5-dimethyladamantane; a rigid tricyclic amine) is an uncompetitive, open-channel NMDA receptor antagonist that preferentially binds to activated (open) NMDA channels. Its mechanism is voltage-dependent with fast blocking and unblocking kinetics, ie it blocks excessive glutamatergic activity at pathological concentrations while sparing normal physiological neurotransmission. This is a critical pharmacological property that distinguishes it from competitive NMDA antagonists. Structurally, memantine interacts with critical asparagine residues (primarily N616) in the NR1 and NR2 subunits of the NMDA receptor channel pore.10 In electrophysiological preparations, the protective action of memantine has been directly demonstrated. In an ex vivo dark-adapted perfused rabbit retina single-unit RGC recording system, bath application of 200 µM NMDA produced progressive increases in tonic RGC spiking, reduced spike amplitude, and eventual failure of spike generation within 6 minutes. Co-perfusion with 10 µM memantine initiated recovery within 2 minutes and restored control spike frequency and amplitude by 4 minutes, demonstrating rapid and complete reversal of NMDA-induced excitotoxicity. For comparison, the competitive NMDA antagonist AP5 (30 µM) produced a similar complete reversal.11
In vivo, memantine has demonstrated neuroprotective efficacy in multiple preclinical model systems, including rodent models of experimental glaucoma and, critically, a non-human primate (NHP) chronic glaucoma model where functional measures and reductions in retinal injury were reported. The NHP data, reported by Hare et al, were particularly influential in building confidence for clinical translation, as they demonstrated that systemic NMDA antagonism could provide measurable RGC protection in a species with an eye and visual system closely homologous to humans.12,13 Supporting this, systemic NMDA antagonists have been shown to contribute to RGC injury reduction in rat models of experimental glaucoma. Beyond memantine, additional mediators of excitotoxicity have been identified. AMPA receptor-mediated excitotoxicity in purified RGCs is governed by receptor desensitization kinetics, and down-regulation of the RNA editing enzyme ADAR2 contributes to RGC death through non-cell-autonomous mechanisms, expanding the landscape of potential anti-excitotoxic targets. Novel preclinical compounds targeting excitotoxicity include the EP2 prostaglandin receptor agonist butaprost, which showed neuroprotective effects in retinal cultures, and ITH12657, which protected against excitotoxic RGC loss in rat models.12,14–16
Neurotrophic Factor Augmentation
Neurotrophic factor deprivation is a hallmark of glaucomatous neurodegeneration, arising from both interrupted retrograde axonal transport from the brain to the retina and reduced local retinal production. BDNF (brain-derived neurotrophic factor) is the most extensively studied neurotrophic factor in glaucoma. BDNF promotes RGC survival by binding to the TrkB receptor, activating downstream Erk1/2 signaling and c-jun pathways, and inhibiting caspase-2 to prevent apoptosis. BDNF also prevents amyloid-β-induced RGC apoptosis in rat models, suggesting a role in preventing/overcoming protein-aggregation-related toxicity in glaucoma. BDNF levels are significantly reduced in both the sera and ocular fluids of glaucoma patients, and interruption of retrograde BDNF transport has been detected in early stages of the disease, directly linking neurotrophic deprivation to clinical pathology.17–22
CNTF (ciliary neurotrophic factor), locally produced by RGCs and reduced in primary open-angle glaucoma (POAG), activates pro-survival JAK/STAT signaling. NGF (nerve growth factor) acts through TrkA receptors to activate the PI3K/Akt pathway, which raises Bcl-2 and lowers Bax. These directly modulate the apoptotic balance within RGCs. Repeated intravitreal injections of BDNF, CNTF, and GDNF have all demonstrated reduction in RGC loss in experimental glaucoma models, though the short vitreous half-life of these proteins remains a barrier to clinical translation.22–26 Sustained delivery strategies are therefore critical. PLGA microspheres have been engineered for extended neurotrophic factor release, and the NT-501 encapsulated cell technology device represents the most advanced delivery platform, providing continuous intravitreal CNTF secretion from implanted modified RPE cells. Despite strong biologic rationale, BDNF itself has not progressed beyond preclinical testing for glaucoma, primarily because of delivery challenges; intravitreal BDNF must be dosed repeatedly to sustain protection, and systemic administration does not achieve adequate retinal levels.22,27–30
Sigma-1 Receptor Agonists
The sigma-1 receptor (S1R) is an endoplasmic reticulum (ER) chaperone protein highly expressed in RGCs. S1R has emerged as an attractive neuroprotective target. Genetic studies demonstrate that S1R-deficient mice develop accelerated retinal ganglion cell death and retinal degeneration, establishing endogenous S1R as essential for retinal neuronal survival. Pridopidine, a highly selective and potent S1R agonist currently in clinical development for Huntington’s disease, has been tested in two distinct rat models of experimental glaucoma by Geva et al, with compelling results.31–33
In the Morrison hypertonic saline injection model using pigmented Brown Norway rats, where hypertonic saline is injected into episcleral veins on days 0 and 7 to produce sustained IOP elevation, pridopidine was administered daily by oral gavage at 3, 30, and 60 mg/kg from day 1 through day 41. Quantification of RGCs by Brn-3a immunostaining revealed dose-dependent neuroprotection. Importantly, a pilot study confirmed that 60 mg/kg pridopidine did not alter IOP in normotensive rats, establishing that the neuroprotective effect is IOP-independent. In a second model, laser photocoagulation model using albino Wistar rats, assessed at 14 days, the dose–response profile was inverted. Here, the low dose (3 mg/kg) provided maximal neuroprotection. This discrepancy was attributed to melanin binding: a radiolabel study using 14C-pridopidine (3 mg/kg) in Long Evans rats demonstrated that radioactivity accumulated dramatically in melanin-rich uveal tissues, peaking at 168 hours (1 week) post-dose and declining to only 7% of maximum by 672 hours (4 weeks), whereas non-pigmented tissues peaked at 1 hour and fell rapidly. Pigmented Brown Norway rats thus sequester pridopidine in melanin, reducing the free fraction available for S1R activation and necessitating higher systemic doses, while albino Wistar rats achieve sufficient retinal drug levels at low doses.31,34
Mechanistically, pridopidine’s neuroprotection is likely mediated through S1R-dependent mitochondrial rescue. In neuronal cell cultures, 1 µM pridopidine for 24 hours increased oxygen consumption rate (measured by Seahorse assay). Pretreatment with 5 µM pridopidine prevented H2O2-induced mitochondrial dysfunction and cell death in human lymphoblasts, preserving mitochondrial membrane potential as assessed by TMRE/TMRM and Rhodamine123 fluorescence. S1R knockdown abrogated these protective effects, confirming target specificity. Additional mechanistic pathways include stabilization of ER-mitochondria (MAM) contacts regulating Ca2⁺ flux, induction of the Nrf2-ARE antioxidant gene pathway to decrease ROS, modulation of autophagy, and upregulation of BDNF secretion and transport.31,35
Endocannabinoid System Modulation
The endocannabinoid system (ECS) is fully represented in retinal tissue, with both CB1 and CB2 receptors expressed on RGCs, Müller cells, and other retinal neurons across multiple species. Endocannabinoids (anandamide, 2-arachidonoylglycerol) and their metabolic machinery (synthesizing enzymes NAPE-PLD, DAGLα/β; degrading enzymes FAAH, MAGL) have been structurally and functionally characterized in the retina. The neuroprotective potential of ECS modulation in glaucoma operates through multiple complementary mechanisms.36–38
In preclinical excitotoxicity models, synthetic cannabinoids HU-210 and methanandamide injected intravitreally protected amacrine cells from AMPA-mediated excitotoxicity through CB1-dependent activation of PI3K/Akt and MEK/ERK signaling pathways. THC and CBD reduced NMDA-induced retinal neurotoxicity through both CB1/CB2-dependent and independent mechanisms, including direct antioxidant activity. This reduces peroxynitrite and oxidative stress across retinal layers. Anandamide exerted neuroprotective effects against high-IOP-induced retinal cell death via both CB1 and TRPV1 receptors, and the FAAH inhibitor URB597 promoted RGC neuroprotection in a rat optic nerve axotomy model by enhancing endocannabinoid tone, though its efficacy declined with age correlating with increased endogenous AEA levels.37,39,40
The interplay between cannabinoid receptors and non-CB targets adds further complexity. TRPV1 expression increased with elevated IOP, and TRPV1 activation can be protective. Genetic or pharmacologic TRPV1 blockade accelerates RGC degeneration under elevated IOP conditions, indicating that this ECS-related target plays a constitutive neuroprotective role. CB2 receptors have been specifically proposed as targets for controlling neural cell survival with less psychoactive liability than CB1, and strategies targeting FAAH/MAGL inhibition to elevate endogenous cannabinoid tone may circumvent the systemic psychoactive effects, tolerance, and short duration of action that limit clinical translation of phytocannabinoids. CBD, which lacks psychoactive properties, has demonstrated anti-inflammatory and neuroprotective effects in preclinical retinal studies through mechanisms including inhibition of equilibrative nucleoside transporter and engagement of A2A receptors.41–44
Anti-Apoptotic Peptides
Peptains are short, anti-apoptotic peptides derived from the core sequences of small heat-shock proteins HspB5 (αB-crystallin) and HspB6 (Hsp20), possessing both chaperone and anti-apoptotic activity. Peptain-3a is an acetylated derivative of peptain-3 designed to increase proteolytic resistance and extend its biological activity. Nam et al, 2022, systematically evaluated peptains across multiple glaucoma-relevant model systems. In primary rat RGCs subjected to neurotrophic factor deprivation for 48 hours, peptain-1 and peptain-3a inhibited apoptosis by 29% and 35%, respectively. In a retinal ischemia/reperfusion injury model intravitreal peptain-1 and peptain-3a dramatically restricted loss, representing ~75% and ~70% neuroprotection. In two distinct ocular hypertension models, microbead injection and silicone oil injection, peptain treatment was similarly effective. RGC loss was reduced to only 4% and 12% (peptain-1 and peptain-3a) in the MB model, and to 16% and 15% in the SO model. Beyond somal protection, peptains also ameliorated defective anterograde axonal transport, assessed by intravitreal injection of cholera toxin-B, which was substantially impaired in eyes with ocular hypertension but restored in peptain-treated eyes. Furthermore, peptains suppressed retinal glial activation in ocular hypertension models, indicating anti-inflammatory properties alongside their primary anti-apoptotic mechanism. Prior work had demonstrated that peptain-1 restores retinal mitochondrial cytochrome C oxidase subunit 6B2, suggesting a mitochondrial component to the protective mechanism.45–47
Rho Kinase Inhibitors
While ROCK inhibitors (ripasudil, fasudil, netarsudil) are clinically approved for IOP lowering through their direct action on the trabecular meshwork outflow pathway, accumulating preclinical evidence suggests they also possess IOP-independent neuroprotective properties. Fasudil, originally developed as a vasodilator for cerebral vasospasm, illustrates the vasorelaxant properties of ROCK inhibition that may benefit optic nerve perfusion. Proposed neuroprotective mechanisms include increased blood flow to the optic nerve, direct enhancement of RGC survival, and anti-inflammatory actions through inhibition of inflammatory cell activation and cytokine production.48,49
In a mouse optic nerve injury model, topical ripasudil suppressed expression of the pro-inflammatory mediators TNF-α, IL-1β, and MCP-1 and was associated with significant RGC rescue. Notably, the combination of ripasudil with brimonidine was more effective than either agent alone, suggesting additive or synergistic mechanisms through modulation of multiple signaling pathways, including suppression of proinflammatory mediators and upregulation of trophic factors. Clinically approved ROCK inhibitors for glaucoma include ripasudil (Japan, 2014) and netarsudil (USA, 2017), but the translation of neuroprotection to demonstrate human benefit remains unestablished and requires specifically designed clinical trials.50–53
Novel Emerging Preclinical Targets
Several innovative targets have emerged from recent preclinical work, expanding the target space beyond classic excitotoxicity and trophic support.
- The P2X7 purinergic receptor antagonist A740003 reduces microglial activation and neuroinflammation in glaucoma models, targeting the innate immune contribution to RGC degeneration.54,55
- The HDAC8 inhibitor H7E inhibits Müller glial activation and oxidative stress-related retinal cell death, representing a novel epigenetic approach to neuroprotection.56
- Irisin, a myokine released during exercise, promotes autophagy and reduces neuroinflammation in retinal models.57
- Dopamine D1 receptor agonism upregulates DRD1 signaling to improve both RGC survival and axon regeneration, potentially bridging neuroprotection and neuro-repair.58
- DHED, a bioprecursor that selectively generates retinal estradiol (E2), protected RGCs and axons in male rat models, offering a sex-hormone-based neuroprotective strategy with organ selectivity.59–61
- Omidenepag, an EP2 prostaglandin receptor agonist primarily developed for IOP lowering, has also been shown to suppress excitotoxic RGC death.62,63
These early-stage compounds are all at the discovery/validation phase and represent the expanding frontier of glaucoma neuroprotection research.
Metabolic and Mitochondrial Targets
Nicotinamide (the amide of vitamin B3) has emerged as arguably the most compelling neuroprotective candidate for glaucoma based on the depth and reproducibility of its preclinical evidence.64–66 The foundational work by Williams et al used the DBA/2J mouse model of hereditary glaucoma to demonstrate that age-related declines in retinal NAD render RGC mitochondria vulnerable to IOP-dependent stresses. Using RNA sequencing of isolated RGCs, unsupervised hierarchical clustering revealed progressive transcriptomic groups with increasing mitochondrial abnormalities: elevated mitochondrial-to-nuclear read ratios, altered oxidative phosphorylation gene expression, increased mitochondrial fission markers, and activation of the unfolded protein response. Pathway analysis highlighted eIF2 and mTOR-related stress/redox pathways. Electron microscopy confirmed reduced cristae volume in RGC dendritic mitochondria, and biochemical assays documented declining NAD⁺/NADH ratios and reduced glutathione levels, with elevated γ-H2AX staining and PARP activity indicating increased ROS and DNA damage. Oral nicotinamide supplementation was administered at two doses (550 and 2000 mg/kg/day). NAM was protective both prophylactically (beginning at ~6 months of age, before IOP elevation) and interventionally (beginning at ~9 months, after IOP elevation). The low dose robustly protected optic nerve axons, while the high dose resulted in 93% of eyes not developing glaucoma. This represents approximately a 10-fold reduction in glaucoma risk. NAM supplementation prevented the age-associated decline in retinal NAD levels through 12 months, preserved visual function, and prevented retina and optic nerve degeneration metrics. Importantly, NAM did not change IOP, confirming the neuroprotective mechanism is IOP-independent.67,68
Furthering this, a companion study demonstrated that combining NAM with the Wallerian degeneration slow allele (WldS), which increases retinal NAD levels through a different mechanism, produced additive protection. Now 94% of eyes were absent of glaucomatous neurodegeneration in the combination group, more than either treatment alone. This combinatorial approach protected somal, synaptic, and axonal compartments of RGCs, maintained anterograde axoplasmic transport, and preserved visual function.69 More recently, Cimaglia et al 2024 demonstrated that a nicotinamide-enriched diet provides robust dendritic protection in a rat model of experimental glaucoma, preserving dendritic structure that is characteristically lost early in glaucomatous degeneration. Metabolomic analysis of optic nerve samples from the same animals confirmed robust metabolic neuroprotection.70,71 The mechanistic understanding of NAM as a potential therapy that may enable visual recovery by preserving stressed but not yet fully degenerated RGC dendrites represents an important conceptual advance for the field.70,71 NAD as a target for neuroprotection in glaucoma is further supported by success in NAM overcoming RGC mitochondrial dysfunction, both Nmnat1 and Nmnat2 (the terminal enzymes for NAD synthesis from NAM) being successful as gene therapies in preclinical models, and new compounds targeting NMNAT2 demonstrating early preclinical success.67,70,72–74
Citicoline (cytidine 5′-diphosphocholine) is an endogenous mononucleotide that acts as an intermediary in the CDP-choline (Kennedy) pathway for phospholipid biosynthesis. Following oral or parenteral administration (bioavailability >90%), citicoline is rapidly metabolized to cytidine and choline. Cytidine is converted to uridine, which crosses the blood–brain barrier and is converted to CTP, while free choline is phosphorylated to phosphocholine. The combination of CTP and phosphocholine represents the rate-limiting step of phosphatidylcholine biosynthesis. Citicoline thereby supports synthesis of key neuronal membrane phospholipids including phosphatidylcholine, phosphatidylethanolamine, sphingomyelin, and cardiolipin (the latter critical for mitochondrial electron transport-chain function). It also prevents phospholipid degradation through inhibition of phospholipase A2 (PLA2), which protects against hydroxyl radical-induced loss of cardiolipin and phosphatidylcholine in the inner mitochondrial membrane.75–77
Beyond membrane stabilization, citicoline enhances neurotransmitter availability. It increases dopamine levels by enhancing tyrosine hydroxylase activity and inhibiting dopamine reuptake, and also raises noradrenaline, serotonin, and acetylcholine through its choline-donating function. Citicoline may reduce glutamate excitotoxicity by upregulating excitatory amino acid transporter-2 (EAAT2). In preclinical models, citicoline has demonstrated anti-apoptotic effects across multiple systems: in mouse retinal explant cultures, intraperitoneal citicoline markedly reduced TUNEL-positive cells in the ganglion cell layer; in vitro concentrations of 0.1–10 µmol/L lowered apoptotic markers, increased regenerating neurites, and 100 µM citicoline reduced apoptosis from both high-glucose and glutamate-induced excitotoxicity in cultured rat retinal neurons. In optic nerve crush, citicoline treatment upregulated the anti-apoptotic protein Bcl-2, providing molecular evidence for its anti-apoptotic mechanism. In a kainic acid-induced retinal damage model, citicoline (500 mg/kg twice daily for 7 days) partially preserved retinal layer thickness, and treatment significantly increased retinal dopamine levels. Topical 2% citicoline prevented diabetes-associated thinning of retinal layers in a mouse model. In vivo, citicoline attenuated reductions in RGC density, improved visual acuity, and preserved the integrity of pre-chiasmatic optic nerve white matter without affecting IOP.76–78
A review of all the preclinical targets in context is presented in Figure 1.
Clinical Evidence and Ongoing Trials
Memantine: The Landmark Phase III Failure
The memantine glaucoma program represents the most extensive and costly neuroprotection trial effort in ophthalmology to date. Two identically designed, randomized, double-masked, placebo-controlled, multicenter, parallel-group Phase III trials (NCT00141882 and NCT00168350) were conducted over 48 months with 20 scheduled visits each, enrolling a total of 2298 patients with bilateral open-angle glaucoma at risk of progression. Patients were randomized 3:2:2 to receive memantine 20 mg, memantine 10 mg, or placebo daily, with dose escalation from 5 mg permitted.79
The prespecified primary efficacy endpoint was glaucomatous visual field progression measured by standard automated perimetry (SAP; Humphrey 24–2 full-threshold). SAP progression required the same ≥5 visual field locations with significant reductions confirmed within 8 weeks; (frequency-doubling technology) FDT progression was declared if 2 adjacent test locations showed significant reductions on the FDT glaucoma change probability analysis without requiring confirmation. Planned enrollment was ~1050 patients per study to yield ~233 completers per treatment group at 48 months, with power calculations assuming 45% dropout in the 20-mg group and 25% in other groups, targeting 85–96% power to detect 10–15% differences. The second study protocol was notably amended (before unmasking) to exclude low tension (normal tension) glaucoma from the primary population, to make FDT the primary efficacy measure, and to focus on memantine 20 mg versus placebo as the primary comparison.
The results were unequivocally negative. Compared with placebo, neither memantine dose significantly delayed glaucomatous progression in either individual study or in pooled analyses over 48 months. FDT and optic disc photograph analyses showed no significant differences between memantine groups and placebo. In an unexpected (and paradoxical) finding, SAP analysis in the first study showed a statistically higher cumulative probability of visual field progression with memantine versus placebo at month 48. A post hoc subgroup analysis restricted to high tension glaucoma patients suggested a lower probability of progression with memantine 20 mg, but this was not a prespecified primary finding and did not reach significance in pooled analyses. Completion rates were similar across groups (~80–83%). Regarding safety, memantine was generally well tolerated. Dizziness was the most common adverse event and the most frequent reason for treatment discontinuation. A higher incidence of prostate cancer was noted in the memantine 20 mg group, though no treatment-related deaths were reported. The trials cost an estimated >$80–100 million and took nearly 5 years.
The failure generated critical lessons for future neuroprotection trial design. Identified limitations include: enrollment of patients with relatively advanced disease (mean cup-to-disc ratio ~0.8), potential insensitivity of the chosen functional endpoints (SAP and FDT) for detecting neuroprotection over feasible durations, variable IOP management across sites, and a protocol requirement that subjects stop study treatment once HFA-based progression was declared, limiting the ability of later assessments to detect post-treatment benefit. The memantine experience has profoundly influenced the design of subsequent neuroprotection trials, driving the field toward more sensitive structural and functional biomarkers (eg PhNR, OCT-based RNFL thinning rates) and enriched patient populations.
Brimonidine: The Low-Pressure Glaucoma Treatment Study (LoGTS)
The LoGTS (NCT00317577) is the most prominent clinical neuroprotection study for brimonidine. This randomized trial compared brimonidine 0.2% monotherapy versus timolol 0.5% in patients with low pressure (normal tension) glaucoma over approximately 30 months. Despite similar IOP lowering in both groups, visual field progression was observed in only 9% of brimonidine-treated patients who could tolerate treatment compared to 39% of timolol-treated patients. This striking fourfold difference in progression rates generated considerable enthusiasm for brimonidine’s putative neuroprotective properties.80–82
However, the study suffered from a high differential dropout rate, with substantially more brimonidine patients discontinuing treatment than timolol patients (primarily due to local adverse effects including allergic conjunctivitis). This per-protocol analysis bias means the brimonidine completers may have represented a self-selected group with more favorable disease characteristics. Additionally, the timolol arm showed worse visual field progression rates than control groups in other contemporaneous glaucoma trials, raising the possibility that the observed difference reflects timolol-associated worsening (potentially through nocturnal hypotension affecting optic nerve perfusion) rather than brimonidine neuroprotection per se. Additional clinical data have shown that topical brimonidine improved contrast sensitivity and may preserve RNFL independent of IOP reduction, but these findings remain inconsistent across studies, and overall clinical evidence for neuroprotection is mixed and inconclusive. Larger, well-designed randomized controlled trials with structural endpoints are needed to definitively establish brimonidine’s neuroprotective efficacy in humans.
Citicoline
Citicoline has been evaluated through multiple administration routes and in numerous clinical studies, generating a substantial body of evidence although with notable heterogeneity in design and endpoints. Intramuscular citicoline (commonly 1 g/day for 10–60 days, with repeated treatment courses) improved visual field performance, enhanced both PERG (pattern electroretinogram) and VEP (visual evoked potential) responses, with some studies reporting sustained or enhanced effects during long-term treatment up to 8 years of follow-up. A study by Parisi et al showed that topical citicoline eye drops, which reach the retina through vitreous bioavailability, produced positive trends in visual field indices in individual eyes, though group-level changes did not reach statistical significance; electrophysiologically, PERG showed reduced P50 latency, and increased P50-N95 amplitude, suggesting enhanced RGC function.83
Oral citicoline at 250 mg/day in a 54-patient study produced a significant increase in average RNFL thickness at 3 months (notably in average and inferior quadrants), though effects partially regressed after washout, with no significant macular ganglion cell-inner plexiform layer (mGCIPL) changes. A multicenter randomized placebo-controlled crossover trial of 500 mg/day oral citicoline reported improved vision-related quality of life as assessed by VFQ-25 and SF-36 questionnaires. Randomized, single-blind crossover trials of citicoline combined with homotaurine (CIT/HOMO) or with homotaurine plus vitamin E demonstrated significant improvements in transient PERG amplitudes (N35-P50 and P50-N95 components) during supplementation, with gains reversing after washout. Contrast sensitivity and patient-reported quality of life (NEI-VFQ-25 dependency subscale, GQL-15) also improved, while IOP, optic nerve head morphology, and OCT measures (cup-to-disc ratio) remained unchanged. No serious adverse events were reported.84–86
Despite these encouraging functional findings, a 2023 systematic review analyzing 10 studies encompassing 424 patients found no significant effects on IOP, visual field mean deviation, RNFL thickness, or PERG amplitude at the group level, highlighting substantial heterogeneity in dosing regimens, treatment durations, routes of administration, and the frequent absence of structural endpoints as major limitations of the current evidence base. Citicoline is available as a nutritional supplement in the EU and Italy and has been endorsed in some national guidelines for glaucoma adjunctive therapy, but definitive proof of disease modification through slowed structural progression is still lacking. Ongoing larger randomized trials (NCT05315206, NCT05710198) aim to address these gaps.
Nicotinamide: Potential for Rapid Clinical Translation
The dramatic preclinical efficacy of nicotinamide has driven rapid clinical translation. Nicotinamide’s clinical safety profile is well-established, with approximately 80 years of clinical use at therapeutic doses.87,88 Hui et al led a randomized, double-masked, placebo-controlled crossover trial which enrolled 49 glaucoma patients who received nicotinamide at 1.5 g/day escalating to 3.0 g/day.89 After 12 weeks, there was a significant improvement in the photopic negative response (PhNR) Vmax (a direct electrophysiological marker of inner retinal/RGC function) compared to placebo. A greater proportion of nicotinamide-treated participants exceeded test–retest repeatability limits, and there was a trend toward improved visual field mean deviation, though the study was not powered to detect visual field changes. Further supporting this, a Phase II trial by De Moraes et al demonstrated that combined nicotinamide and pyruvate (3 g/d for each agent) safely enhanced short-term visual function versus placebo in open-angle glaucoma patients.90 In the Korean normal tension nicotinamide study (NCT06078605), nicotinamide at a lower dose (2 g/d) also demonstrated similar short term protective effects.91 This supports nicotinamide as a treatment that could be beneficial across glaucoma subtypes. Multiple ongoing Phase III trials are now testing NAD augmentation for glaucoma neuroprotection, including the TGNT, TAMING, and NAMinG studies (NCT05275738, NCT06731582, NCT05405868; Australia, Singapore, Sweden, UK).
Nicotinamide riboside (NR), an alternative NAD precursor, has also been shown to be well tolerated in healthy volunteers, doubling circulating NAD levels by day 9 of supplementation.92–94 Notably, a 125-patient, 24-month randomized NR versus placebo trial (300 mg/day) uses RNFL thinning as the primary structural outcome to definitively test whether NAD augmentation can slow the rate of optic nerve degeneration.95 Lower systemic niacin intake has been associated with normal-tension glaucoma in epidemiological studies,96 and POAG patients have been reported to have lower blood nicotinamide concentrations and lower PMBC NAD,97,98 supporting the translational relevance of targeting the NAD pathway.
CNTF (NT-501 Encapsulated Cell Therapy)
The NT-501 device represents the most advanced sustained neurotrophic factor delivery system in clinical testing for glaucoma. The implant consists of encapsulated modified RPE cells engineered to continuously secrete CNTF into the vitreous, addressing the fundamental challenge of short neurotrophic factor half-life. A Phase 1 trial (NCT01408472, 11 patients) established safety and tolerability of the intravitreal implant. In other retinal diseases, the NT-501 device demonstrated a positive dose-response for visual acuity over 1 year (high dose > low dose > sham), providing proof of concept for the sustained-delivery platform.99 A randomized, sham-controlled Phase II trial (NCT02862938, 54 patients) is now assessing whether sustained intravitreal CNTF delivery preserves visual fields and retinal structure in glaucoma patients, with visual field and structural (OCT) endpoints.
rhNGF Eye Drops
Recombinant human NGF (rhNGF) eye drops, FDA-approved for neurotrophic keratitis, have been evaluated in a Phase Ib randomized, double-masked safety and tolerability study in patients with progressive POAG (NCT02855450).100 The trial completed recruitment and demonstrated safety and tolerability of topical rhNGF administration. Preclinical evidence showed that topical NGF preserved RGCs and supported retinal survival in multiple injury models, and the clinical development path benefits from the established safety profile of rhNGF in neurotrophic keratitis. However, definitive efficacy evidence in glaucoma awaits larger trials.
Fas Pathway Inhibitor (ONL1204)
ONL1204 is a first-in-class inhibitor of Fas (CD95/APO-1) receptor-mediated apoptosis that targets a fundamentally different mechanism than any other agent in the glaucoma neuroprotection pipeline. Preclinical studies demonstrated that ONL1204 protects multiple retinal cell types in numerous models of retinal disease by directly blocking the Fas death receptor signaling cascade that culminates in caspase activation and RGC apoptosis. A Phase 1b multicenter, randomized, single-masked, sham-controlled study (NCT05160805) was conducted enrolling 25 patients with progressing open-angle glaucoma despite controlled IOP (≤21 mmHg). Patients were randomized to two intravitreal dose levels of ONL1204 versus sham procedure (needle-less syringe touch to the eye surface). The primary endpoint was safety and tolerability assessed over 39 weeks through adverse event reporting, ophthalmic examination, BCVA, IOP, electroretinogram, and ophthalmic imaging. This completed Phase 1b trial represents the first clinical test of a direct apoptosis-blocking strategy in glaucoma, and its results will inform dose selection for subsequent efficacy trials.
Metformin and Semaglutide (Repurposing Metabolic Agents)
Metformin, widely used for type 2 diabetes, has garnered interest as a potential neuroprotective agent based on epidemiological data showing that diabetic patients taking metformin at >1,500 mg/day had a 25% reduced risk of developing open-angle glaucoma compared to non-users.101 Its proposed mechanisms include modulation of cellular energetics through AMPK/Nrf2 activation, reduction of oxidative stress, targeting of fibrotic signaling, and effects on NAD/mitochondrial pathways. A Phase II, double-blind, placebo-controlled, parallel-group randomized controlled trial (NCT05426044) is now recruiting 125 POAG patients who demonstrate progressive RNFL and/or GCIPL thinning by OCT trend-based or guided progression analysis. Patients are randomized 1:1 to metformin 1,500 mg/day (750 mg twice daily) or identical placebo for 24 months, with OCT RNFL/GCIPL imaging and visual field testing at 2-month intervals. The primary outcome is the rate of change of average RNFL thickness, with the secondary outcome being the rate of VF mean deviation decline. Randomization is stratified by age, gender, VF MD, spherical equivalent, mean IOP, and number of glaucoma medications. Importantly, patients with diabetes or renal impairment are excluded.
Semaglutide, a GLP-1 receptor agonist, is being tested in the ABSALON trial (NCT06792422), a Phase III, quadruple-masked (participant, care provider, investigator, outcomes assessor), randomized, placebo-controlled study. The trial is enrolling 126 patients with POAG with MD ≤16 dB and reliable visual fields, who are already receiving IOP-lowering treatment. Participants receive oral semaglutide (3 mg/day for 1 month to 7 mg/day for 1 month to 14 mg/day maintenance) or matched placebo for 6 months. The primary outcome is the change in the photopic negative response of the electroretinogram from baseline to month 6. Secondary outcomes include contrast sensitivity (Pelli-Robson), health-related quality of life (EQ-5D-3L and NEI VFQ-25), and safety/tolerability. Exploratory outcomes include OCT ring and macular scans for RNFL thickness and ganglion cell volume, standard automated perimetry, and multi-omics analyses (cytokine proteome, metabolome, lipidome). Patients with diabetes, renal impairment, pancreatitis history, or BMI <18.5 are excluded. This novel trial represents one of the newest entries in the glaucoma neuroprotection landscape.102
Insulin
Insulin activates PI3K/Akt and mTORC1/2 signaling pathways important for RGC metabolism, and it crosses the blood-brain and blood-retinal barriers. Preclinical work demonstrated that exogenous insulin preserves RGCs in injury models through protection of neurons, glia, and retinal vasculature from apoptosis and metabolic stress.66,103,104 Intranasal delivery may avoid systemic hypoglycemia while achieving adequate retinal concentrations. A Phase I study of topical insulin eye drops demonstrated safety in glaucoma patients (NCT05206877), providing the foundation for potential efficacy evaluation.105
A review of all the clinical targets in context is presented in Figure 1 and trial details are shown in Table 1.
Challenges and Future Directions
Lessons from Translational Failures
The history of glaucoma neuroprotection drug development is defined by a persistent translational gap ie numerous agents demonstrate robust efficacy in animal models, yet none have achieved clinically validated, IOP-independent neuroprotection in humans in Phase III trials. Understanding the reasons for this disconnect is essential for charting a productive path forward.
The memantine Phase III program, the most expensive and ambitious neuroprotection trial in ophthalmology to date, exemplifies the challenges. Two identically designed, randomized, double-masked, placebo-controlled, multicenter 48-month trials enrolling 2,298 patients failed to show any statistically significant benefit of oral memantine versus placebo on visual field progression.79 The trials consumed an estimated USD $100 million and nearly five years, yet the primary endpoint was not met in either individual study or pooled analyses. Post hoc analyses identified several critical limitations: preclinical data in non-human primates were not completely convincing with observed benefits occurring at very high IOP levels that may have reflected protection against ischemia rather than chronic glaucomatous neurodegeneration.13 The enrolled patient population was relatively advanced, potentially beyond the window where neuroprotective intervention could produce measurable benefit. The functional endpoints used (standard automated perimetry, frequency-doubling technology) may have lacked the sensitivity needed to detect neuroprotective effects over feasible trial durations.
More broadly, translational failures in glaucoma neuroprotection can be classified as type 1 problems (flawed preclinical or clinical data) or type 2 problems (valid data in each domain but failure to bridge between them). Animal models commonly rely on a single, abrupt insult (eg acute IOP elevation) and do not recapitulate the fluctuating, multifactorial, slowly progressive nature of human POAG. Neuroprotective agents in animal studies are frequently administered before or at the onset of damage, a timing that bears little resemblance to treating patients who present with established disease. Additionally, the inability to measure target engagement in human ocular tissues and uncertain drug bioavailability at the retina remain persistent barriers.
Reimagining Clinical Trial Design and Endpoints
The failure of SAP to detect neuroprotective effects in the memantine trials has spurred innovation in endpoint selection. Slow disease progression requiring follow-up exceeding five years, the absence of a consensus primary neuroprotection outcome measure accepted by regulators, and inadequate endpoint sensitivity have all been identified as structural barriers to successful trials.
Several promising new approaches are emerging. The photopic negative response (PhNR) of the electroretinogram has been adopted as a primary endpoint in multiple ongoing neuroprotection trials, including the semaglutide ABSALON trial (NCT06792422) and the nicotinamide Phase III trials (NCT05275738, NCT06731582, NCT05405868), because it provides a direct measure of inner retinal (RGC) function. Pattern electroretinography (PERG) similarly offers a sensitive electrophysiological readout and has been used extensively in citicoline and combination supplement trials.85 On the structural side, OCT-based probability maps (rather than summary thickness metrics) can detect localized progression earlier, and combining structural and functional measures yields higher sensitivity than either alone. The 10–2 visual field may be more appropriate for capturing macular/central glaucomatous damage that standard 24–2 testing misses.106–108
A particularly transformative development is AI-guided endpoint selection. A recent study developed a graph attention neural network (GNN) that uses baseline SAP data to predict the five visual field locations most likely to deteriorate for each eye (“High-5”).109 Trained on 6,996 eyes and externally validated across two independent cohorts (>7,000 eyes), the model identified high-risk locations that declined at approximately −2.1 dB/year among progressors (versus −1.0 dB/year for global mean deviation), achieved AUCs of 0.883–0.937 for discriminating progressors from non-progressors, and showed that nearly all progressing eyes exhibited a repeatable ≥7 dB loss at High-5 points (versus <30% using global MD). Critically, the authors estimated that using the High-5 endpoint could reduce required trial sample sizes by approximately 42% compared to mean deviation, while remaining aligned with FDA-accepted criteria. This data-driven approach could fundamentally improve the feasibility of neuroprotection trials.110,111
Trial design enrichment strategies (including recruiting patients with documented structural progression, using cluster-measurement designs that concentrate assessments at study start and end, and employing futility analysis for earlier go/no-go decisions) can further improve trial efficiency.
Novel Biomarkers for Stratification and Monitoring
Beyond trial endpoints, novel biomarkers hold promise for patient stratification and real-time monitoring of treatment response. DARC (Detection of Apoptosing Retinal Cells) enables direct, in vivo visualization of individual apoptotic retinal cells using intravenous fluorescently labeled annexin V (ANX776). A Phase I proof-of-concept study demonstrated increased DARC counts in glaucoma patients, with correlation to structural and functional progression (NCT02394613). While DARC does not yet specifically identify RGCs, it represents the only clinical-stage tool for imaging active apoptotic processes in the retina.112–115
Neurofilament light chain (NfL) in aqueous humor has been shown to be significantly elevated in glaucoma patients (mean 429 pg/mL vs. 3.1 pg/mL in controls) and is associated with markers of active disease (maximum IOP, number of medications) but not with conventional severity measures such as visual field MD or OCT RNFL thickness.116 This dissociation suggests that NfL may reflect ongoing neurodegeneration rather than historical damage, making it a potential pharmacodynamic biomarker. The authors propose that enrolling patients with elevated NfL could increase trial efficiency by targeting those with active disease.116,117 Serum neurofilaments have also been validated as prognostic and pharmacodynamic biomarkers in other neurodegenerative diseases, supporting translational relevance.118
Additional emerging biomarkers include flavoprotein fluorescence (FPF) as a measure of mitochondrial dysfunction at the optic nerve head, hyperspectral retinal imaging for metabolic assessment of RGC health, and molecular markers including microRNAs, inflammatory cytokines, and oxidative stress markers for multi-dimensional patient profiling.119,120
Epigenetic Reprogramming
An emerging and conceptually distinct strategy involves partial epigenetic reprogramming of RGCs using the Yamanaka transcription factors OCT4, SOX2, and KLF4 (collectively, OSK). Originally demonstrated by Lu et al, 2020,121 OSK expression delivered via an inducible AAV system restored youthful DNA methylation patterns and improved visual function in aged and glaucomatous mice without altering cell identity. Subsequent work demonstrated sustained vision recovery after only 2 months of OSK expression, with benefits persisting for up to 11 months post-induction, and no adverse effects observed over 21 months of continuous expression in glaucomatous mice.122 Efficacy has been replicated in non-human primates in a model of non-arteritic anterior ischemic optic neuropathy (NAION), with OSK treatment significantly restoring pattern ERG responses and increasing axon survival compared to controls. These data supported the first-in-human IND application for ER-100 (Life Biosciences), an AAV2-delivered OSK construct for intravitreal injection, which received FDA clearance in January 2026 (Phase 1; NCT07290244. Indications: open-angle glaucoma and NAION). While OSK represents a shift from neuroprotection toward neuro-rejuvenation, key mechanistic questions remain regarding the fidelity of epigenetic reprogramming in primate RGCs, and independent replication of efficacy data is needed before broad clinical conclusions can be drawn.
Multi-Target Combinations and Drug Delivery Innovation
The multifactorial pathophysiology of glaucomatous neurodegeneration which encompasses mitochondrial dysfunction, excitotoxicity, neuroinflammation, trophic factor deprivation, and apoptosis, provides a compelling rationale for multi-target combination strategies. Early clinical evidence supports this approach ie combinations of citicoline with homotaurine, vitamin E, CoQ10, or vitamin B3 have shown additive electrophysiological and functional benefits beyond monotherapy in randomized trials.85,123,124 Preclinical data suggest that nicotinamide combined with gene therapy-mediated NAD augmentation produces the strongest neuroprotective effect in glaucoma models.69 However, optimal compound ratios, dosing regimens, and long-term safety require systematic investigation through preclinical synergy testing and adequately powered clinical trials.
Drug delivery remains a critical bottleneck. Neurotrophic factors have short vitreous half-lives, many small molecules have limited retinal bioavailability from topical administration, and intravitreal injections carry procedural risks. Several innovations are being developed to address these challenges. Multifunctional nanocarriers (liposomes, PLGA microspheres, dendrimers) can co-encapsulate hydrophilic and hydrophobic agents, offering sustained release and targeted delivery. Chitosan-based mucoadhesive eye drops provide enhanced corneal penetration and prolonged retention. Encapsulated cell technology (eg the NT-501 CNTF implant) enables sustained intravitreal protein delivery for months. Intranasal delivery routes may bypass the blood-retinal barrier and rapidly deliver drugs to ocular tissues. Extracellular vesicle-based delivery (MSC-derived exosomes) represents a frontier approach for targeted RGC neuroprotection, though optimization is needed to mitigate potential gliosis and inflammatory responses.99,125–127
Precision Medicine and Patient Stratification
Glaucoma is a heterogeneous group of diseases, and a one-size-fits-all approach to neuroprotection is unlikely to succeed. Precision medicine approaches are beginning to mature: GWAS-identified loci and polygenic risk scores (PRS) can stratify patients by genetic susceptibility, while integration of genetic, environmental, and individual response data can identify subgroups most likely to benefit from specific interventions.128–130 Machine learning applied to multi-omics datasets (genomics, transcriptomics, proteomics, metabolomics) can discover disrupted pathways and categorize genetically similar patient subgroups.131 Biological insights into compartmentalized degeneration (where dendritic, synaptic, somatic, and axonal loss follow distinct molecular programs) and selective vulnerability of specific RGC subtypes (eg α OFF-transient cells) suggest that therapeutic strategies may need to be tailored to the predominant degenerative pattern in individual patients.66,132–136 Ensuring equity across diverse populations by accounting for ancestry-specific genetic variation is also essential for the validity and generalizability of precision approaches.
Preclinical Model Improvement and Regulatory Engagement
Improving animal models is a prerequisite for better translational outcomes. Current models predominantly employ acute, severe IOP elevation in young animals, which does not recapitulate the chronic, age-related, multifactorial nature of human POAG. Development of chronic progressive models with fluctuating IOP, use of IOP telemetry, and incorporation of longitudinal functional assessments that mirror human disease monitoring are needed. Pharmacodynamic validation of target engagement in animal models before advancing to human trials would reduce the risk of expensive clinical failures. Harmonizing preclinical outcome measures with clinical endpoints is also critical to reducing the translational disconnect.
At the regulatory level, no IOP-independent neuroprotective drug has ever been approved for glaucoma, and there is no FDA-accepted primary outcome measure for neuroprotection. Early engagement with regulatory agencies to define acceptable surrogate endpoints, potentially including RNFL thinning rate, PhNR change, or AI-selected visual field metrics, will be essential for providing a viable regulatory pathway. Establishment of consensus primary outcome measures through multi-stakeholder workshops (as initiated by NEI/FDA) could accelerate the field. Multicenter, international collaborations will be necessary to achieve adequate sample sizes and population diversity for definitive neuroprotection trials. This is already the case for the nicotinamide Phase III that will incorporate data from Australia, Singapore, Sweden, and the UK.
Concluding Perspective
The field of glaucoma neuroprotection stands at an inflection point. The lessons of the memantine failure (regarding endpoint sensitivity, patient selection, and preclinical model relevance) have been absorbed and are now informing a new generation of better-designed trials. The convergence of AI-guided endpoint innovation (potentially reducing required sample sizes), novel biomarkers that can detect active neurodegeneration in real time, multi-target combination strategies, advanced drug delivery systems, and precision medicine frameworks creates a realistic pathway toward clinically validated neuroprotective therapies. With multiple well-designed randomized controlled trials of nicotinamide, metformin, semaglutide, ONL1204, and CNTF now underway, the next five years may prove decisive in determining whether IOP-independent neuroprotection becomes a clinical reality in glaucoma management.
Search Strategy
This narrative review was conducted using systematic database searches of PubMed/MEDLINE and ClinicalTrials.gov (last searched: March 2026 for R#1, June 2026 for R#2). Search terms included, individually and in combination:
- glaucoma,
- retinal ganglion cell,
- neuroprotection,
- neuroprotective,
- optic nerve,
- clinical trial,
- nicotinamide,
- NAD,
- NMDA,
- excitotoxicity,
- neurotrophic factor,
- BDNF,
- CNTF,
- NGF,
- citicoline,
- metformin,
- semaglutide,
- brimonidine,
- gene therapy,
- epigenetic reprogramming.
Inclusion criteria were: peer-reviewed primary research articles, systematic reviews, and registered clinical trials reporting pharmacological neuroprotection in glaucoma or relevant preclinical models. No formal date restriction was applied; however, emphasis was placed on articles published from 2015 to 2025, with key foundational studies included irrespective of publication date. Non-English language articles without English-language abstracts were excluded.
Funding
SR is supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant number: RS-2025-25459803). PAW is supported by the The Ulla and Ingemar Dahlberg Foundation and Centre for Eye Research Australia philanthropic funds.
Disclosure
Both authors are currently leadership in Phase II and Phase III clinical trials for glaucoma including nicotinamide as a treatment for glaucoma. The authors report no conflicts of interest in this work.
References
1. Ishikawa M. Abnormalities in glutamate metabolism and excitotoxicity in the retinal diseases. Scientifica. 2013;2013:528940. doi:10.1155/2013/528940
2. Harada T, Harada C, Nakamura K, et al. The potential role of glutamate transporters in the pathogenesis of normal tension glaucoma. J Clin Invest. 2007;117(7):1763–19. doi:10.1172/JCI30178
3. Lucas DR, Newhouse JP. The toxic effect of sodium L-glutamate on the inner layers of the retina. AMA Arch Ophthalmol. 1957;58(2):193–201. doi:10.1001/archopht.1957.00940010205006
4. Dreyer EB, Zurakowski D, Schumer RA, Podos SM, Lipton SA. Elevated glutamate levels in the vitreous body of humans and monkeys with glaucoma. Arch Ophthalmol. 1996;114(3):299–305. doi:10.1001/archopht.1996.01100130295012
5. Dreyer EB, Grosskreutz CL. Excitatory mechanisms in retinal ganglion cell death in primary open angle glaucoma (POAG). Clin Neurosci. 1997;4(5):270–273.
6. Naskar R, Vorwerk CK, Dreyer EB. Saving the nerve from glaucoma: memantine to caspaces. Semin Ophthalmol. 1999;14(3):152–158. doi:10.3109/08820539909061468
7. Naskar R, Vorwerk CK, Dreyer EB. Concurrent downregulation of a glutamate transporter and receptor in glaucoma. Invest Ophthalmol Vis Sci. 2000;41(7):1940–1944.
8. Vorwerk CK, Gorla MS, Dreyer EB. An experimental basis for implicating excitotoxicity in glaucomatous optic neuropathy. Surv Ophthalmol. 1999;43 Suppl 1:S142-50.
9. Russo R, Cavaliere F, Varano GP, et al. Impairment of neuronal glutamate uptake and modulation of the glutamate transporter GLT-1 induced by retinal ischemia. PLoS One. 2013;8(8):e69250. doi:10.1371/journal.pone.0069250
10. Chen HS, Lipton SA. Pharmacological implications of two distinct mechanisms of interaction of memantine with N-methyl-D-aspartate-gated channels. J Pharmacol Exp Ther. 2005;314(3):961–971. doi:10.1124/jpet.105.085142
11. Hare WA, Wheeler L. Experimental glutamatergic excitotoxicity in rabbit retinal ganglion cells: block by memantine. Invest Ophthalmol Vis Sci. 2009;50(6):2940–2948. doi:10.1167/iovs.08-2103
12. Hare W, WoldeMussie E, Lai R, et al. Efficacy and safety of memantine, an NMDA-type open-channel blocker, for reduction of retinal injury associated with experimental glaucoma in rat and monkey. Surv Ophthalmol. 2001;45(Suppl 3):
13. Hare WA, WoldeMussie E, Lai RK, et al. Efficacy and safety of memantine treatment for reduction of changes associated with experimental glaucoma in monkey, I: functional measures. Invest Ophthalmol Vis Sci. 2004;45(8):2625–2639. doi:10.1167/iovs.03-0566
14. Park YH, Mueller BH, McGrady NR, Ma HY, Yorio T. AMPA receptor desensitization is the determinant of AMPA receptor mediated excitotoxicity in purified retinal ganglion cells. Exp Eye Res. 2015;132:136–150. doi:10.1016/j.exer.2015.01.026
15. Wang AL, Carroll RC, Nawy S. Down-regulation of the RNA editing enzyme ADAR2 contributes to RGC death in a mouse model of glaucoma. PLoS One. 2014;9(3):e91288. doi:10.1371/journal.pone.0091288
16. Di Pierdomenico J, Gallego-Ortega A, Norte-Muñoz M, et al. Evaluation of the neuroprotective efficacy of the gramine derivative ITH12657 against NMDA-induced excitotoxicity in the rat retina. Front Neuroanat. 2024;18:1335176. doi:10.3389/fnana.2024.1335176
17. Lambuk L, Mohd Lazaldin MA, Ahmad S, et al. Brain-derived neurotrophic factor-mediated neuroprotection in glaucoma: a review of current state of the art. Front Pharmacol. 2022;13:875662. doi:10.3389/fphar.2022.875662
18. Pease ME, McKinnon SJ, Quigley HA, Kerrigan-Baumrind LA, Zack DJ. Obstructed axonal transport of BDNF and its receptor TrkB in experimental glaucoma. Invest Ophthalmol Vis Sci. 2000;41(3):764–774.
19. Kurokawa T, Katai N, Shibuki H, et al. BDNF diminishes caspase-2 but not c-Jun immunoreactivity of neurons in retinal ganglion cell layer after transient ischemia. Invest Ophthalmol Vis Sci. 1999;40(12):3006–3011.
20. Lazaldin MAM, Iezhitsa I, Agarwal R, Agarwal P, Ismail NM. Neuroprotective effects of exogenous brain-derived neurotrophic factor on amyloid-beta 1-40-induced retinal degeneration. Neural Regen Res. 2023;18(2):382–388. doi:10.4103/1673-5374.346546
21. Binley KE, Ng WS, Barde YA, Song B, Morgan JE. Brain-derived neurotrophic factor prevents dendritic retraction of adult mouse retinal ganglion cells. Eur J Neurosci. 2016;44(3):2028–2039. doi:10.1111/ejn.13295
22. Johnson TV, DeKorver NW, Levasseur VA, et al. Identification of retinal ganglion cell neuroprotection conferred by platelet-derived growth factor through analysis of the mesenchymal stem cell secretome. Brain. 2014;137(Pt 2):503–519. doi:10.1093/brain/awt292
23. Watanabe M, Fukuda Y. Survival and axonal regeneration of retinal ganglion cells in adult cats. Prog Retin Eye Res. 2002;21(6):529–553.
24. Pease ME, Zack DJ, Berlinicke C, et al. Effect of CNTF on retinal ganglion cell survival in experimental glaucoma. Invest Ophthalmol Vis Sci. 2009;50(5):2194–2200. doi:10.1167/iovs.08-3013
25. Wang H, Wang R, Thrimawithana T, et al. The nerve growth factor signaling and its potential as therapeutic target for glaucoma. Biomed Res Int. 2014;2014:759473. doi:10.1155/2014/759473
26. Dulz S, Bassal M, Flachsbarth K, et al. Intravitreal Co-Administration of GDNF and CNTF confers synergistic and long-lasting protection against injury-induced cell death of retinal ganglion cells in mice. Cells. 2020;9(9). doi:10.3390/cells9092082
27. Jiang C, Moore MJ, Zhang X, Klassen H, Langer R, Young M. Intravitreal injections of GDNF-loaded biodegradable microspheres are neuroprotective in a rat model of glaucoma. Mol Vis. 2007;13:1783–1792.
28. Johnson TV, Bull ND, Martin KR. Neurotrophic factor delivery as a protective treatment for glaucoma. Exp Eye Res. 2011;93(2):196–203. doi:10.1016/j.exer.2010.05.016
29. Xiao JH, Zhang MN. Neuroprotection of retinal ganglion cells with GDNF-Loaded biodegradable microspheres in experimental glaucoma. Int J Ophthalmol. 2010;3(3):189–191. doi:10.3980/j.issn.2222-3959.2010.03.01
30. Checa-Casalengua P, Jiang C, Bravo-Osuna I, et al. Retinal ganglion cells survival in a glaucoma model by GDNF/Vit E PLGA microspheres prepared according to a novel microencapsulation procedure. J Control Release. 2011;156(1):92–100. doi:10.1016/j.jconrel.2011.06.023
31. Geva M, Gershoni-Emek N, Naia L, et al. Neuroprotection of retinal ganglion cells by the sigma-1 receptor agonist pridopidine in models of experimental glaucoma. Sci Rep. 2021;11(1):21975. doi:10.1038/s41598-021-01077-w
32. Mavlyutov TA, Nickells RW, Guo LW. Accelerated retinal ganglion cell death in mice deficient in the Sigma-1 receptor. Mol Vis. 2011;17:1034–1043.
33. Mavlyutov TA, Guo LW. Peeking into Sigma-1 receptor functions through the retina. Adv Exp Med Biol. 2017;964:285–297. doi:10.1007/978-3-319-50174-1_19
34. Li L, He S, Liu Y, Yorio T, Ellis DZ. Sigma-1R protects retinal ganglion cells in optic nerve crush model for glaucoma. Invest Ophthalmol Vis Sci. 2021;62(10):17. doi:10.1167/iovs.62.10.17
35. Naia L, Ly P, Mota SI, et al. The Sigma-1 receptor mediates pridopidine rescue of mitochondrial function in Huntington disease models. Neurotherapeutics. 2021;18(2):1017–1038. doi:10.1007/s13311-021-01022-9
36. Bouskila J, Javadi P, Elkrief L, Casanova C, Bouchard JF, Ptito M. A comparative analysis of the endocannabinoid system in the retina of mice, tree shrews, and monkeys. Neural Plast. 2016;2016:3127658. doi:10.1155/2016/3127658
37. Kokona D, Thermos K. Synthetic and endogenous cannabinoids protect retinal neurons from AMPA excitotoxicity in vivo, via activation of CB1 receptors: involvement of PI3K/Akt and MEK/ERK signaling pathways. Exp Eye Res. 2015;136:45–58. doi:10.1016/j.exer.2015.05.007
38. Micaelo-Fernandes C, Bouskila J, Bouchard JF, Ptito M. Presence of the endocannabinoid system in the inferior pulvinar of the vervet monkey. Brain Sci. 2021;11(6). doi:10.3390/brainsci11060770
39. Nucci C, Gasperi V, Tartaglione R, et al. Involvement of the endocannabinoid system in retinal damage after high intraocular pressure-induced ischemia in rats. Invest Ophthalmol Vis Sci. 2007;48(7):2997–3004. doi:10.1167/iovs.06-1355
40. Slusar JE, Cairns EA, Szczesniak AM, Bradshaw HB, Di Polo A, Kelly ME. The fatty acid amide hydrolase inhibitor, URB597, promotes retinal ganglion cell neuroprotection in a rat model of optic nerve axotomy. Neuropharmacology. 2013;72:116–125. doi:10.1016/j.neuropharm.2013.04.018
41. Sappington RM, Sidorova T, Long DJ, Calkins DJ. TRPV1: contribution to retinal ganglion cell apoptosis and increased intracellular Ca2+ with exposure to hydrostatic pressure. Invest Ophthalmol Vis Sci. 2009;50(2):717–728. doi:10.1167/iovs.08-2321
42. Sappington RM, Sidorova T, Ward NJ, Chakravarthy R, Ho KW, Calkins DJ. Activation of transient receptor potential vanilloid-1 (TRPV1) influences how retinal ganglion cell neurons respond to pressure-related stress. Channels. 2015;9(2):102–113. doi:10.1080/19336950.2015.1009272
43. Cairns EA, Baldridge WH, Kelly ME. The endocannabinoid system as a therapeutic target in glaucoma. Neural Plast. 2016;2016:9364091. doi:10.1155/2016/9364091
44. Liou GI, Auchampach JA, Hillard CJ, et al. Mediation of cannabidiol anti-inflammation in the retina by equilibrative nucleoside transporter and A2A adenosine receptor. Invest Ophthalmol Vis Sci. 2008;49(12):5526–5531. doi:10.1167/iovs.08-2196
45. Nam MH, Stankowska DL, Johnson GA, Nahomi RB, Pantcheva MB, Nagaraj RH. Peptains block retinal ganglion cell death in animal models of ocular hypertension: implications for neuroprotection in glaucoma. Cell Death Dis. 2022;13(11):958. doi:10.1038/s41419-022-05407-2
46. Rajeswaren V, Wong JO, Yabroudi D, et al. Small heat shock proteins in retinal diseases. Front Mol Biosci. 2022;9:860375. doi:10.3389/fmolb.2022.860375
47. Stankowska DL, Nam MH, Nahomi RB, et al. Systemically administered peptain-1 inhibits retinal ganglion cell death in animal models: implications for neuroprotection in glaucoma. Cell Death Discov. 2019;5:112. doi:10.1038/s41420-019-0194-2
48. Abbhi V, Piplani P. Rho-kinase (ROCK) inhibitors – a neuroprotective therapeutic paradigm with a focus on ocular utility. Curr Med Chem. 2020;27(14):2222–2256. doi:10.2174/0929867325666181031102829
49. Satoh S, Ikegaki I, Kawasaki K, Asano T, Shibuya M. Pleiotropic effects of the rho-kinase inhibitor fasudil after subarachnoid hemorrhage: a review of preclinical and clinical studies. Curr Vasc Pharmacol. 2014;12(5):758–765. doi:10.2174/1570161112666140613115813
50. Otsubo M, Sase K, Tsukahara C, et al. Axonal protection by combination of ripasudil and brimonidine with upregulation of p-AMPK in TNF-induced optic nerve degeneration. Int Ophthalmol. 2024;44(1):173. doi:10.1007/s10792-024-03095-9
51. Akaiwa K, Namekata K, Azuchi Y, et al. Topical Ripasudil suppresses retinal ganglion cell death in a mouse model of normal tension glaucoma. Invest Ophthalmol Vis Sci. 2018;59(5):2080–2089. doi:10.1167/iovs.17-23276
52. Nishijima E, Namekata K, Kimura A, et al. Topical ripasudil stimulates neuroprotection and axon regeneration in adult mice following optic nerve injury. Sci Rep. 2020;10(1):15709. doi:10.1038/s41598-020-72748-3
53. Chatzimichail E, Christodoulaki E, Konstas PAG, et al. Rho kinase inhibitors in glaucoma management: current perspectives and future directions. Drug Des Devel Ther. 2025;19:2519–2531. doi:10.2147/DDDT.S515166
54. Zhu Y, Li SY, Zhang LJ, Lei B, Wang YC, Wang Z. Neuroprotection of the P2X7 receptor antagonist A740003 on retinal ganglion cells in experimental glaucoma. Neuroreport. 2024;35(13):822–831. doi:10.1097/WNR.0000000000002071
55. Yang M, Qiu R, Wang W, et al. P2X7 receptor antagonist attenuates retinal inflammation and neovascularization induced by oxidized low-density lipoprotein. Oxid Med Cell Longev. 2021;2021:5520644. doi:10.1155/2021/5520644
56. Wu LH, Cheng YW, Lin FL, et al. A novel HDAC8 inhibitor H7E exerts retinoprotective effects against glaucomatous injury via ameliorating aberrant Müller glia activation and oxidative stress. Biomed Pharmacother. 2024;174:116538. doi:10.1016/j.biopha.2024.116538
57. Zhang Q, Xiang S, Chen X, et al. Irisin attenuates acute glaucoma-induced neuroinflammation by activating microglia-integrin αVβ5/AMPK and promoting autophagy. Int Immunopharmacol. 2024;138:112545. doi:10.1016/j.intimp.2024.112545
58. Zhang Q, Xue J, Tang J, et al. Modulating amacrine cell-derived dopamine signaling promotes optic nerve regeneration and preserves visual function. Sci Adv. 2024;10(31):eado0866. doi:10.1126/sciadv.ado0866
59. Kapic A, Zaman K, Nguyen V, et al. The prodrug DHED delivers 17β-Estradiol into the retina for protection of retinal ganglion cells and preservation of visual function in an animal model of glaucoma. Cells. 2024;13(13). doi:10.3390/cells13131126
60. Prokai-Tatrai K, Nguyen V, De La Cruz DL, et al. Retina-targeted delivery of 17β-Estradiol by the topically applied DHED prodrug. Pharmaceutics. 2020;12(5). doi:10.3390/pharmaceutics12050456
61. Prokai-Tatrai K, Zaman K, Kapic A, et al. Retina-targeted 17β-Estradiol by the DHED prodrug rescues visual function and actuates neuroprotective protein networks after optic nerve crush in a rat model of surgical menopause. Int J Mol Sci. 2025;26(5). doi:10.3390/ijms26051846
62. Nakamura N, Honjo M, Yamagishi R, Igarashi N, Sakata R, Aihara M. Effects of selective EP2 receptor agonist, omidenepag, on trabecular meshwork cells, Schlemm’s canal endothelial cells and ciliary muscle contraction. Sci Rep. 2021;11(1):16257. doi:10.1038/s41598-021-95768-z
63. Nakamura N, Honjo M, Yamagishi-Kimura R, Sakata R, Watanabe S, Aihara M. Neuroprotective effect of omidenepag on excitotoxic retinal ganglion cell death regulating COX-2-EP2-cAMP-PKA/Epac pathway via Neuron-Glia interaction. Neuroscience. 2024;553:145–159. doi:10.1016/j.neuroscience.2024.07.006
64. Hui F, Williams PA. Vitamins and nutraceuticals in glaucoma research. Eur J Ophthalmol. 2026;36(3):11206721261419640. doi:10.1177/11206721261419640
65. Tribble JR, Hui F, Jöe M, et al. Targeting Diet and Exercise for Neuroprotection and Neurorecovery in Glaucoma. Cells. 2021;10(2). doi:10.3390/cells10020295
66. Tribble JR, Hui F, Quintero H, et al. Neuroprotection in glaucoma: mechanisms beyond intraocular pressure lowering. Mol Aspects Med. 2023;92:101193. doi:10.1016/j.mam.2023.101193
67. Williams PA, Harder JM, Foxworth NE, et al. Vitamin B3 modulates mitochondrial vulnerability and prevents glaucoma in aged mice. Science. 2017;355(6326):756–760. doi:10.1126/science.aal0092
68. Williams PA, Harder JM, Cardozo BH, Foxworth NE, John SWM. Nicotinamide treatment robustly protects from inherited mouse glaucoma. Commun Integr Biol. 2018;11(1):e1356956. doi:10.1080/19420889.2017.1356956
69. Williams PA, Harder JM, Foxworth NE, Cardozo BH, Cochran KE, John SWM. Nicotinamide and WLDS act together to prevent neurodegeneration in glaucoma. Front Neurosci. 2017;11:232. doi:10.3389/fnins.2017.00232
70. Tribble JR, Otmani A, Sun S, et al. Nicotinamide provides neuroprotection in glaucoma by protecting against mitochondrial and metabolic dysfunction. Redox Biol. 2021;43:101988. doi:10.1016/j.redox.2021.101988
71. Cimaglia G, Tribble JR, Votruba M, Williams PA, Morgan JE. Oral nicotinamide provides robust, dose-dependent structural and metabolic neuroprotection of retinal ganglion cells in experimental glaucoma. Acta Neuropathol Commun. 2024;12(1):137. doi:10.1186/s40478-024-01850-8
72. Tribble JR, Jöe M, Varricchio C, et al. NMNAT2 is a druggable target to drive neuronal NAD production. Nat Commun. 2024;15(1):6256. doi:10.1038/s41467-024-50354-5
73. Otmani A, Jóhannesson G, Brautaset R, Tribble JR, Williams PA. Prophylactic nicotinamide treatment protects from rotenone-induced neurodegeneration by increasing mitochondrial content and volume. Acta Neuropathol Commun. 2024;12(1):37. doi:10.1186/s40478-024-01724-z
74. Fang F, Zhuang P, Feng X, et al. NMNAT2 is downregulated in glaucomatous RGCs, and RGC-specific gene therapy rescues neurodegeneration and visual function. Mol Ther. 2022;30(4):1421–1431. doi:10.1016/j.ymthe.2022.01.035
75. Grieb P, Rejdak R. Pharmacodynamics of citicoline relevant to the treatment of glaucoma. J Neurosci Res. 2002;67(2):143–148.
76. Han YS, Chung IY, Park JM, Yu JM. Neuroprotective effect of citicoline on retinal cell damage induced by kainic acid in rats. Korean J Ophthalmol. 2005;19(3):219–226. doi:10.3341/kjo.2005.19.3.219
77. Oshitari T, Fujimoto N, Adachi-Usami E. Citicoline has a protective effect on damaged retinal ganglion cells in mouse culture retina. Neuroreport. 2002;13(16):2109–2111. doi:10.1097/00001756-200211150-00023
78. Schuettauf F, Rejdak R, Thaler S, et al. Citicoline and lithium rescue retinal ganglion cells following partial optic nerve crush in the rat. Exp Eye Res. 2006;83(5):1128–1134. doi:10.1016/j.exer.2006.05.021
79. Weinreb RN, Liebmann JM, Cioffi GA, et al. Oral memantine for the treatment of glaucoma: design and results of 2 randomized, placebo-controlled, phase 3 studies. Ophthalmology. 2018;125(12):1874–1885. doi:10.1016/j.ophtha.2018.06.017
80. Evans DW, Hosking SL, Gherghel D, Bartlett JD. Contrast sensitivity improves after brimonidine therapy in primary open angle glaucoma: a case for neuroprotection. Br J Ophthalmol. 2003;87(12):1463–1465. doi:10.1136/bjo.87.12.1463
81. Garudadri CS, Choudhari NS, Rao HL, Senthil S. A randomized trial of brimonidine versus timolol in preserving visual function: results from the low-pressure glaucoma treatment study. Am J Ophthalmol. 2011;152(5):
82. Krupin T, Liebmann JM, Greenfield DS, Ritch R, Gardiner S; Group L-PGS. A randomized trial of brimonidine versus timolol in preserving visual function: results from the low-pressure glaucoma treatment study. Am J Ophthalmol. 2011;151(4):671–681. doi:10.1016/j.ajo.2010.09.026
83. Parisi V. Electrophysiological assessment of glaucomatous visual dysfunction during treatment with cytidine-5’-diphosphocholine (citicoline): a study of 8 years of follow-up. Doc Ophthalmol. 2005;110(1):91–102. doi:10.1007/s10633-005-7348-7
84. Lanza M, Gironi Carnevale UA, Mele L, Bifani Sconocchia M, Bartollino S, Costagliola C. Morphological and functional evaluation of Oral Citicoline therapy in chronic open-angle glaucoma patients: a pilot study with a 2-year follow-up. Front Pharmacol. 2019;10:1117. doi:10.3389/fphar.2019.01117
85. Rossi GCM, Rolle T, De Silvestri A, et al. Multicenter, prospective, randomized, single blind, cross-over study on the effect of a fixed combination of Citicoline 500 mg Plus Homotaurine 50 mg on Pattern Electroretinogram (PERG) in patients with open angle glaucoma on well controlled intraocular pressure. Front Med Lausanne. 2022;9:882335. doi:10.3389/fmed.2022.882335
86. Prinz J, Prokosch V, Liu H, Walter P, Fuest M, Migliorini F. Efficacy of citicoline as a supplement in glaucoma patients: a systematic review. PLoS One. 2023;18(9):e0291836. doi:10.1371/journal.pone.0291836
87. Knip M, Douek IF, Moore WP, et al. Safety of high-dose nicotinamide: a review. Diabetologia. 2000;43(11):1337–1345. doi:10.1007/s001250051536
88. Williams PA, Harder JM, John SWM. Glaucoma as a metabolic optic neuropathy: making the case for nicotinamide treatment in glaucoma. J Glaucoma. 2017. doi:10.1097/IJG.0000000000000767
89. Hui F, Tang J, Williams PA, et al. Improvement in inner retinal function in glaucoma with nicotinamide (vitamin B3) supplementation: a crossover randomized clinical trial. Clin Exp Ophthalmol. 2020. doi:10.1111/ceo.13818
90. De Moraes CG, John SWM, Williams PA, Blumberg DM, Cioffi GA, Liebmann JM. Nicotinamide and pyruvate for neuroenhancement in open-angle glaucoma: a phase 2 randomized clinical trial. JAMA Ophthalmol. 2022;140(1):11–18. doi:10.1001/jamaophthalmol.2021.4576
91. Ha A, Kim YK, Lee CK, et al. Effects of nicotinamide supplementation in normal-tension glaucoma: a crossover placebo-controlled randomised clinical trial. Br J Ophthalmol. 2025. doi:10.1136/bjo-2025-328096
92. Trammell SA, Schmidt MS, Weidemann BJ, et al. Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat Commun. 2016;7:12948. doi:10.1038/ncomms12948
93. Ratajczak J, Joffraud M, Trammell SA, et al. NRK1 controls nicotinamide mononucleotide and nicotinamide riboside metabolism in mammalian cells. Nat Commun. 2016;7:13103. doi:10.1038/ncomms13103
94. Martens CR, Denman BA, Mazzo MR, et al. Chronic nicotinamide riboside supplementation is well-tolerated and elevates NAD. Nat Commun. 2018;9(1):1286. doi:10.1038/s41467-018-03421-7
95. Leung CKS, Ren ST, Chan PPM, et al. Nicotinamide riboside as a neuroprotective therapy for glaucoma: study protocol for a randomized, double-blind, placebo-control trial. Trials. 2022;23(1):45. doi:10.1186/s13063-021-05968-1
96. Jung KI, Kim YC, Park CK. Dietary Niacin and Open-Angle Glaucoma: the Korean National Health and Nutrition Examination Survey. Nutrients. 2018;10(4). doi:10.3390/nu10040387
97. Petriti B, Rabiolo A, Chau KY, et al. Peripheral blood mononuclear cell respiratory function is associated with progressive glaucomatous vision loss. Nat Med. 2024. doi:10.1038/s41591-024-03068-6
98. Kouassi Nzoughet J, Chao de la Barca JM, Guehlouz K, et al. Nicotinamide deficiency in primary open-angle glaucoma. Invest Ophthalmol Vis Sci. 2019;60(7):2509–2514. doi:10.1167/iovs.19-27099
99. Goldberg JL, Beykin G, Satterfield KR, Nuñez M, Lam BL, Albini TA. Phase I NT-501 ciliary neurotrophic factor implant trial for primary open-angle glaucoma: safety, neuroprotection, and neuroenhancement. Ophthalmol Sci. 2023;3(3):100298. doi:10.1016/j.xops.2023.100298
100. Beykin G, Stell L, Halim MS, et al. Phase 1b randomized controlled study of short course topical Recombinant Human Nerve Growth Factor (rhNGF) for neuroenhancement in glaucoma: safety, tolerability, and efficacy measure outcomes. Am J Ophthalmol. 2022;234:223–234. doi:10.1016/j.ajo.2021.11.002
101. Lin HC, Stein JD, Nan B, et al. Association of geroprotective effects of metformin and risk of open-angle glaucoma in persons with diabetes mellitus. JAMA Ophthalmol. 2015;133(8):915–923. doi:10.1001/jamaophthalmol.2015.1440
102. Mouhammad ZA, Vohra R, Horwitz A, et al. Glucagon-like peptide 1 receptor agonists - potential game changers in the treatment of glaucoma? Front Neurosci. 2022;16:824054. doi:10.3389/fnins.2022.824054
103. Agostinone J, Alarcon-Martinez L, Gamlin C, Yu WQ, Wong ROL, Di Polo A. Insulin signalling promotes dendrite and synapse regeneration and restores circuit function after axonal injury. Brain. 2018;141(7):1963–1980. doi:10.1093/brain/awy142
104. El Hajji S, Shiga Y, Belforte N, et al. Insulin restores retinal ganglion cell functional connectivity and promotes visual recovery in glaucoma. Sci Adv. 2024;10(32):eadl5722. doi:10.1126/sciadv.adl5722
105. Saludares M, Wennberg-Smith Z, Beykin G, et al. A phase i randomized trial of topical insulin for glaucoma: safety and efficacy outcomes. Ophthalmol Sci. 2026;6(2):101032. doi:10.1016/j.xops.2025.101032
106. Wu Z, Medeiros FA, Weinreb RN, Girkin CA, Zangwill LM. Comparing 10-2 and 24-2 visual fields for detecting progressive central visual loss in glaucoma eyes with early central abnormalities. Ophthalmol Glaucoma. 2019;2(2):95–102. doi:10.1016/j.ogla.2019.01.003
107. Lee WJ, Kim TJ, Kim YK, Jeoung JW, Park KH. Serial combined wide-field optical coherence tomography maps for detection of early glaucomatous structural progression. JAMA Ophthalmol. 2018;136(10):1121–1127. doi:10.1001/jamaophthalmol.2018.3160
108. West ME, Sharpe GP, Hutchison DM, et al. Value of 10-2 visual field testing in glaucoma patients with early 24-2 visual field loss. Ophthalmology. 2021;128(4):545–553. doi:10.1016/j.ophtha.2020.08.033
109. da Costa DR, Scherer R, Swaminathan S, Tseng H, Medeiros FA. AI-guided endpoint selection for neuroprotection trials in glaucoma. medRxiv. 2025. doi:10.1101/2025.08.23.25334286
110. da Costa D R, Scherer R, Swaminathan SS, Tseng HA, Medeiros F. Artificial intelligence-guided endpoint selection for neuroprotection trials in glaucoma. Am J Ophthalmol. 2026;284:88–100. doi:10.1016/j.ajo.2025.12.038
111. De Moraes CG, Lane KJ, Wang X, Liebmann JM. A potential primary endpoint for clinical trials in glaucoma neuroprotection. Sci Rep. 2023;13(1):7098. doi:10.1038/s41598-023-34009-x
112. Cordeiro MF, Migdal C, Bloom P, Fitzke FW, Moss SE. Imaging apoptosis in the eye. Eye. 2011;25(5):545–553. doi:10.1038/eye.2011.64
113. Galvao J, Davis BM, Cordeiro MF. In vivo imaging of retinal ganglion cell apoptosis. Curr Opin Pharmacol. 2013;13(1):123–127. doi:10.1016/j.coph.2012.08.007
114. Guo L, Cordeiro MF. Assessment of neuroprotection in the retina with DARC. Prog Brain Res. 2008;173:437–450. doi:10.1016/S0079-6123(08)01130-8
115. Normando EM, Turner LA, Cordeiro MF. The potential of annexin-labelling for the diagnosis and follow-up of glaucoma. Cell Tissue Res. 2013;353(2):279–285. doi:10.1007/s00441-013-1554-5
116. Lin JB, Pitts KM, El Helwe H, et al. Neurofilament light chain in aqueous humor as a marker of neurodegeneration in glaucoma. Clin Ophthalmol. 2023;17:2209–2217. doi:10.2147/OPTH.S417664
117. Lin JB, El Helwe H, Falah H, et al. Evaluation of serum and aqueous humor neurofilament light chain as markers of neurodegeneration in glaucoma. Transl Vis Sci Technol. 2025;14(2):24. doi:10.1167/tvst.14.2.24
118. Benatar M, Wuu J, Turner MR. Neurofilament light chain in drug development for amyotrophic lateral sclerosis: a critical appraisal. Brain. 2023;146(7):2711–2716. doi:10.1093/brain/awac394
119. Zhou DB, Castanos MV, Geyman L, et al. Mitochondrial dysfunction in primary open-angle glaucoma characterized by flavoprotein fluorescence at the optic nerve head. Ophthalmol Glaucoma. 2022;5(4):413–420. doi:10.1016/j.ogla.2021.12.006
120. Dammak A, Sanchez Naves J, Huete-Toral F, Carracedo G. New biomarker combination related to oxidative stress and inflammation in primary open-angle glaucoma. Life. 2023;13(7). doi:10.3390/life13071455
121. Lu Y, Brommer B, Tian X, et al. Reprogramming to recover youthful epigenetic information and restore vision. Nature. 2020;588(7836):124–129. doi:10.1038/s41586-020-2975-4
122. Karg MM, Lu YR, Refaian N, et al. Sustained vision recovery by OSK gene therapy in a mouse model of glaucoma. Cell Reprogram. 2023;25(6):288–299. doi:10.1089/cell.2023.0074
123. Martucci A, Cesareo M, Pinazo-Durán MD, et al. Next-Gen neuroprotection in glaucoma: synergistic molecules for targeted therapy. J Clin Med. 2025;14(17). doi:10.3390/jcm14176145
124. Rossi GCM, Rinaldi M, Matarazzo F, et al. Randomized, cross over, multicenter, single-blind study comparing Citicoline 500 mg/Homotaurine 50 mg/Vitamin B3 54 mg/Pyrroloquinoline Quinone 5 mg (Neuprozin Mito. J Clin Med. 2025;14(11). doi:10.3390/jcm14113774
125. Kwon S, Kim SH, Khang D, Lee JY. Potential therapeutic usage of nanomedicine for glaucoma treatment. Int J Nanomed. 2020;15:5745–5765. doi:10.2147/IJN.S254792
126. Kim M, Kim JY, Rhim WK, et al. Extracellular vesicle encapsulated nicotinamide delivered via a trans-scleral route provides retinal ganglion cell neuroprotection. Acta Neuropathol Commun. 2024;12(1):65. doi:10.1186/s40478-024-01777-0
127. Pei K, Georgi M, Hill D, Lam CFJ, Wei W, Cordeiro MF. Review: neuroprotective nanocarriers in glaucoma. Pharmaceuticals. 2024;17(9). doi:10.3390/ph17091190
128. Craig JE, Han X, Qassim A, et al. Multitrait analysis of glaucoma identifies new risk loci and enables polygenic prediction of disease susceptibility and progression. Nat Genet. 2020;52(2):160–166. doi:10.1038/s41588-019-0556-y
129. Qassim A, Souzeau E, Siggs OM, et al. An intraocular pressure polygenic risk score stratifies multiple primary open-angle glaucoma parameters including treatment intensity. Ophthalmology. 2020;127(7):901–907. doi:10.1016/j.ophtha.2019.12.025
130. Kolovos A, Aung T, Khawaja AP, Wiggs JL, Craig JE. Polygenic risk scores for glaucoma: impact on diagnosis and disease course. Prog Retin Eye Res. 2026;112:101469. doi:10.1016/j.preteyeres.2026.101469
131. Fea AM, Ricardi F, Novarese C, Cimorosi F, Vallino V, Boscia G. Precision medicine in glaucoma: artificial intelligence, biomarkers, genetics and redox state. Int J Mol Sci. 2023;24(3). doi:10.3390/ijms24032814
132. Howell GR, Soto I, Libby RT, John SW. Intrinsic axonal degeneration pathways are critical for glaucomatous damage. Exp Neurol. 2012. doi:10.1016/j.expneurol.2012.01.014
133. Syc-Mazurek SB, Libby RT. Axon injury signaling and compartmentalized injury response in glaucoma. Prog Retin Eye Res. 2019;73:100769. doi:10.1016/j.preteyeres.2019.07.002
134. Whitmore AV, Libby RT, John SW. Glaucoma: thinking in new ways-a rôle for autonomous axonal self-destruction and other compartmentalised processes? Prog Retin Eye Res. 2005;24(6):639–662. doi:10.1016/j.preteyeres.2005.04.004
135. Risner ML, Pasini S, Cooper ML, Lambert WS, Calkins DJ. Axogenic mechanism enhances retinal ganglion cell excitability during early progression in glaucoma. Proc Natl Acad Sci U S A. 2018;115(10):E2393–E2402. doi:10.1073/pnas.1714888115
136. Tapia ML, Nascimento-Dos-Santos G, Park KK. Subtype-specific survival and regeneration of retinal ganglion cells in response to injury. Front Cell Dev Biol. 2022;10:956279. doi:10.3389/fcell.2022.956279
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.