Back to Journals » Clinical Optometry » Volume 13
Progressive External Ophthalmoplegia Diagnosed in the Glaucoma Clinic: The Importance of a Complete Clinical Examination
Authors Karagiannis D, Kontomichos L, Tzimis V, Parikakis E, Batsos G ![]() , Karampelas M
, Karampelas M
Received 20 October 2021
Accepted for publication 14 December 2021
Published 24 December 2021 Volume 2021:13 Pages 335—339
DOI https://doi.org/10.2147/OPTO.S342972
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Mr Simon Berry
Dimitrios Karagiannis,1 Loukas Kontomichos,1 Vasileios Tzimis,1 Efstratios Parikakis,1 Georgios Batsos,1 Michael Karampelas2
1Second Department of Ophthalmology, Ophthalmiatreio Eye Hospital of Athens, Athens, Greece; 2Ophthalmology Department, Hippokration General Hospital, Athens, Greece
Correspondence: Loukas Kontomichos Email [email protected]
Purpose: To present a case of chronic progressive external ophthalmoplegia (CPEO) diagnosis in the setting of a busy glaucoma clinic.
Materials and Methods: Case report.
Results: A 70-year-old-female presented to our glaucoma clinic concerned on her glaucoma status. She was previously followed-up elsewhere. Her past ophthalmic history included a diagnosis of normal tension glaucoma 10 years earlier for which she was on topical anti-glaucoma medication. On inspection, symmetrical ptosis was noted and when she was asked about her family history, she reported that 4 members of her family had “problems with moving their eyes” as she also had from a young age. Her best corrected visual acuity was 20/100 in her right eye (RE) and 20/80 in her left eye (LE). Optic discs had normal margins with peripapillary atrophy and cup/disc ratio was 0.7 RE and 0.8 LE. Intraocular pressure was 10mmHg bilaterally. Ocular motility examination revealed severe restriction in all directions of gaze. She had mild alternating exotropia but she did not report any diplopia. Based on her family history, bilateral ptosis, ophthalmoplegia and reported symptoms, the diagnosis of CPEO was made.
Conclusion: In the setting of a busy subspecialty clinic, it is common for clinicians to focus on a specific pathology and overlook additional signs and symptoms that represent a second often unrelated condition. Our case highlights the importance of a thorough assessment including history taking (general, ophthalmic and family), good documentation of symptoms and an adequate clinical examination.
Keywords: chronic progressive external ophthalmoplegia, normal tension glaucoma, visual field defects
Introduction
Subspecialization in ophthalmology emerged around 1950 with the development of the first structured fellowship training programs.1 Nowadays, the majority of ophthalmic trainees choose to follow a subspecialization path at the end of their training and most eye units worldwide are organized in separate subspecialty clinics. There is no doubt that subspecialization has advanced the level of patient care we provide and enhanced our clinical, surgical and research skills. However, in the setting of a subspecialty clinic, it is common to focus on the specific field of study and overlook other ophthalmic pathology. This can be greatly exacerbated on a busy environment when there is often limited time for each consultation.
The case presented herein describes the diagnosis of Chronic progressive external ophthalmoplegia (CPEO) in the setting of a glaucoma clinic.
Case Report
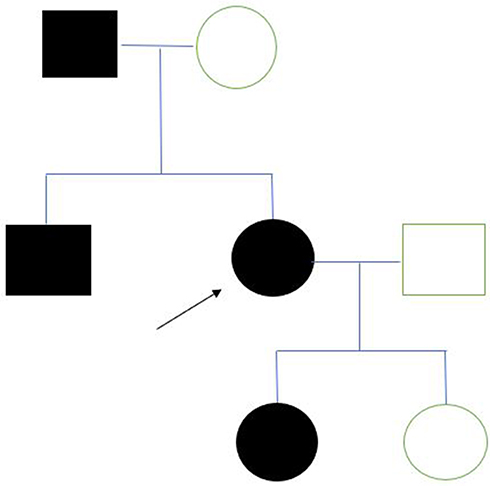
A 70-year-old-female presented to our glaucoma clinic as a new patient concerned on her glaucoma status. She was previously followed-up elsewhere. Her past medical history included hypertension and hyperlipidemia, both well controlled with oral medication. Her past ophthalmic history included a previous diagnosis of normal tension glaucoma 10 years earlier for which she was on topical anti-glaucoma medication in both eyes (dorzolamide twice per day, tafluprost once per day). On inspection, symmetrical ptosis was noted, as well as facial atrophy and when she was asked about her family history, after a thorough review, she reported that 4 members of her family (her father, her brother, her daughter and herself) had “problems with moving their eyes” as she also had from a young age (Figure 1). The patient denied to have genetic testing, but we managed to establish the pedigree of her family (Figure 2). After carefully studying this pedigree, we assumed that the inheritance pattern of this disease is probably autosomal dominant. Her best corrected visual acuity was 20/100 in her right eye (RE) and 20/80 in her left eye (LE). Pupillary reflexes were normal. The Ishihara colour test was normal. Slit-lamp examination revealed bilateral cataracts RE≫LE. Fundus examination revealed only minor pigmentary changes on both maculae. Optic discs had normal margins with peripapillary atrophy and cup/disc ratio was 0.7 RE and 0.8 LE. Intraocular pressure was 10mmHg bilaterally. Ocular motility examination revealed severe restriction in all directions of gaze. She had mild alternating exotropia but she did not report any diplopia. She did not mention hearing loss, or dysarthria. In addition, she did not have any symptoms of weakness in her limbs or heart problems. Humphrey visual fields revealed a significant general reduction of sensitivity in the right eye and a milder general reduction of sensitivity with a small central island of vision of about 5 degrees in the left eye (Figure 3). Based on her family history, bilateral ptosis, ophthalmoplegia and reported symptoms, the diagnosis of CPEO was made.
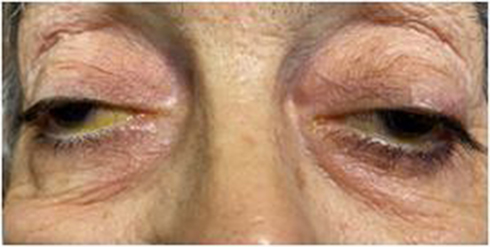 |
Figure 1 Bilateral ptosis and facial atrophy. |
 |

 |
Figure 3 Visual fields of the right eye showing a significant general reduction of sensitivity and left eye showing a mild general reduction of sensitivity with a small central island of vision of about 5 degrees. |
Discussion
We present a case of a 70-year-old female who was seen in the glaucoma clinic with a history of normal tension glaucoma. She was noted to have bilateral ptosis as well as ophthalmoplegia. These signs elicited a thorough review and clinical examination that eventually led to the diagnosis of CPEO. This is something not common at this age. The disease most commonly begins in the third or fourth decade.1 Due to her positive family history, we recommended her to have genetic testing but she did not accept it. The absence of non-ocular symptoms like limb weakness, dysphagia, dysarthria, or hearing loss, guide us to the conclusion that here is an isolated form of CPEO because it is not associated with other systemic findings (CPEO-plus). The patient was referred to glaucoma clinic due to vision deterioration. There were no previous visual field test to compare with our baseline test. Our report highlights the fact that in the setting of a busy subspecialty clinic, it is common to focus on a specific pathology potentially missing other unrelated signs or symptoms.
CPEO is the most frequent manifestation of mitochondrial myopathies.2 It is a bilateral, typically symmetric, and external (ie, spares the pupil) ophthalmoplegia commonly manifesting as bilateral ptosis followed by ophthalmoparesis months to years later. Other less common ophthalmic manifestations include pigmentary retinopathy and optic atrophy.2–4 CPEO may present with isolated oculomotor symptoms (isolated CPEO) or as part of a systemic myopathic or neurologic syndrome (“CPEO-plus syndrome”).3 Up to 60% cases are due to mitochondrial DNA (mtDNA) deletions. Sporadic cases of CPEO suggest de novo mutations in mtDNA while autosomal dominant or recessive inheritance patterns point to nuclear DNA mutations.4
CPEO most commonly begins in the third or fourth decade of life2,3 and it follows a slow progressive course, making it distinct from other etiologies that cause acute/subacute forms of ophthalmoplegia. There are reports of patients who develop ophthalmoplegia but not ptosis and unilateral or asymmetric ptosis may develop.5,6 CPEO is a clinical diagnosis, but laboratory studies can aid in confirming the diagnosis.
Unlike isolated CPEO that commonly presents at the third or fourth decade of life, patients with Kearns-Sayre Syndrome (KSS) typically have symptom onset before age 20.7 Patients with KSS also have an interesting bilateral pigmentary retinopathy, and other associated findings include cardiovascular conduction defects, elevated protein in the cerebrospinal fluid (CSF), and cerebellar ataxia.3,7 In Myotonic Dystrophy, distal muscle weakness is common. Myotonia, or delayed relaxation of muscles following contraction, most consistently involves the hand muscles.3,8 Congenital Fibrosis of the Extraocular Muscles (CFEOM) is a severe form of strabismus with deficits in ocular motility. In this congenital, non-progressive disorder, deficits of vertical eye movements, especially upgaze, are a hallmark of the condition, and patients’ eyes are often stuck in infraduction.9 Muscle biopsy is the definitive test for mitochondrial disorders, but polymerase chain reaction testing is also of great value.10 Currently, there is no defined treatment for CPEO.
In a study by Zhu et al, ophthalmological findings were identified in 74 patients with mitochondrial diseases.11 One out of 74 patients had associated visual field defects. Ten per cent of patients (8/74) were diagnosed with partial or total optic nerve atrophy. Optic neuropathy due to mitochondrial dysfunction may have an insidious or acute onset and tends to involve the papillomacular bundle at an earlier stage.12,13 As a consequence, central vision is mostly affected and patients have the characteristic temporal pallor. In our patient, we noticed peripapillary atrophy but no signs of optic nerve atrophy such as neuroretinal rim discoloration. In addition, the presence of an increased cup to disc ratio led us to the assumption that optic nerve neuropathy was related to normal tension glaucoma rather than primary or secondary optic disc atrophy.
Kalenak et al were the first to describe the association of primary open angle glaucoma with Kearns-Sayre syndrome in a 40-year-old woman.14 Zarnowski et al published a case report in which they studied the progression of normal tension glaucoma in Kearns-Sayre syndrome over a 10-year period.15 They presented a patient with established glaucomatous damage and progression who underwent trabeculectomy in both eyes. However, despite the fact that more than 50% reduction of initial intraocular pressure was achieved, visual fields continued to deteriorate. An earlier publication by Frezzotti et al presented an 82-year-old patient previously diagnosed with Kearns-Sayre syndrome with intraocular pressures in the mid 20s, increased cupping in both eyes and arcuate scotomas on visual fields.16
Our study presents a case suffering from an isolated CPEO syndrome. The association between CPEO and glaucoma is not well established. Unlike other mitochondrial inherited eye disorders such as autosomal dominant optic atrophy (ADOA) and Leber hereditary optic neuropathy (LHON) optic nerve function is normal in CPEO patients. In our case, the severe compromise in optic nerve function as illustrated in visual field testing could only be attributed to a longstanding glaucoma rather than any other optic neuropathy.
Conclusion
The recent trend towards subspecialization has greatly benefited ophthalmology practice and research. Nevertheless, in the setting of a busy subspecialty clinic, it is common for clinicians to focus on a specific pathology and overlook additional signs and symptoms that represent a second often unrelated condition. We present a patient originally seen for normal tension glaucoma, who was eventually diagnosed with CPEO, highlighting the importance of a thorough assessment including history taking (general, ophthalmic and family), good documentation of symptoms and an adequate clinical examination. Considering the fact that CPEO patients are at risk of losing vision throughout their life, clinicians should bear in mind that concurrent pathology such as glaucoma must be identified and treated promptly.
Ethics Statement
Institutional approval was required to publish the case details. We proceeded with the study after having received the approval from the Ethical Committee of our Hospital, Ophthalmiatreio Eye Hospital of Athens.
Consent Statement
The patient provided written informed consent to publish her case details and any accompanying images.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Spivey BE. Education in ophthalmology over the past 60 years. Indian J Ophthalmol. 2018;66(6):746–751. doi:10.4103/ijo.IJO_738_18
2. Yu-Wai-Man P. Chronic progressive external ophthalmoplegia secondary to nuclear-encoded mitochondrial genes. In: Mitochondrial Case Studies. Elsevier; 2016:159–169.
3. Lee AG, Brazis PW. Chronic progressive external ophthalmoplegia. Curr Neurol Neurosci Rep. 2002;2:413–417. doi:10.1007/s11910-002-0067-5
4. DiMauro S, Schon EA, Carelli V, Hirano M. The clinical maze of mitochondrial neurology. Nat Rev Neurol. 2013;9:429–444. doi:10.1038/nrneurol.2013.126
5. Finsterer J, Zarrouk-Mahjoub S. Unilateral ptosis and homolateral hemifacial weakness in chronic progressive external ophthalmoplegia. Neuroophthalmology. 2017;41(3):165–166. doi:10.1080/01658107.2017.1294187
6. Murdock J, Thyparampil PJ, Yen MT. Late-onset development of eyelid ptosis in chronic progressive external ophthalmoplegia: a 30-year follow-up. Neuroophthalmology. 2016;40(1):44–46. doi:10.3109/01658107.2015.1122815
7. Moraes CT, DiMauro S, Zeviani M, et al. Mitochondrial DNA deletions in progressive external ophthalmoplegia and Kearns-Sayre syndrome. N Engl J Med. 1989;320:1293–1299. doi:10.1056/NEJM198905183202001
8. Machuca‐Tzili L, Brook D, Hilton‐Jones D. Clinical and molecular aspects of the myotonic dystrophies: a review. Muscle Nerve. 2005;32:1–18. doi:10.1002/mus.20301
9. Whitman MC, Hunter DG. Congenital fibrosis of the extraocular muscles (CFEOM). Clinical Education/Education/Disease Reviews; October 14, 2015.
10. De Coo IF, Gussinklo T, Arts PJ, et al. A PCR test for progressive external ophthalmoplegia and Kearns-Sayre syndrome on DNA from blood samples. J Neurol Sci. 1997;149(1):37–40. doi:10.1016/S0022-510X(97)05366-5
11. Zhu -C-C, Traboulsi EI, Parikh S. Ophthalmological findings in 74 patients with mitochondrial disease. Ophthalmic Genet. 2017;38(1):67–69. doi:10.3109/13816810.2015.1130153
12. Yu-Wai-Man P, Griffiths PG, Chinnery PF. Mitochondrial optic neuropathies - disease mechanisms and therapeutic strategies. Prog Retin Eye Res. 2011;30:81–114. doi:10.1016/j.preteyeres.2010.11.002
13. Pan BX, Ross-Cisneros FN, Carelli V, et al. Mathematically modeling the involvement of axons in Leber’s hereditary optic neuropathy. Invest Ophthalmol Vis Sci. 2012;53:7608–7617. doi:10.1167/iovs.12-10452
14. Kalenak JW, Kolker AE. Kearns - Sayre syndrome and primary open angle glaucoma. Am J Ophthalmol. 1989;108:35–36. doi:10.1016/0002-9394(89)90134-7
15. Zarnowski T, Kosior-Jarecka E. Progression of normal tension glaucoma in Kearns-Sayre syndrome over 10 years. Case Rep Clin Exp Ophthalmol. 2012;40(2):218–220. doi:10.1111/j.1442-9071.2011.02721.x
16. Frezzoti P, Frezzotti R. Primary open angle glaucoma in a case of mitochondrial encephalomyopathy (Kearns - Sayre syndrome). Eur J Ophthalmol. 2005;15(6):809–810. doi:10.1177/112067210501500625
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.