Back to Journals » International Medical Case Reports Journal » Volume 18
Progressive Deterioration of the Right Ventricular Function in a Patient with Non-Obstructive HCM Complicated by Atrial Fibrillation: A Case Presentation
Authors Xu J, Tan C, Chen J, Hu J ![]()
Received 26 March 2025
Accepted for publication 28 June 2025
Published 7 July 2025 Volume 2025:18 Pages 837—843
DOI https://doi.org/10.2147/IMCRJ.S529591
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Vinay Kumar
Jiamin Xu,1,2,* Changshou Tan,3,* Jianzhou Chen,4 Jiaxin Hu1,2
1Cardiovascular Disease Center, The Central Hospital of Enshi Tujia and Miao Autonomous Prefecture, Enshi Clinical College of Wuhan University, Enshi, People’s Republic of China; 2Hubei Selenium and Human Health Institute, The Central Hospital of Enshi Tujia and Miao Autonomous Prefecture, Enshi, People’s Republic of China; 3Department of Interventional Radiology, the First Affiliated Hospital of Wannan Medical College, Wuhu, People’s Republic of China; 4Department of Cardiology, Nanjing Drum Tower Hospital, The Affiliated Hospital of Nanjing University Medical School, Nanjing, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jiaxin Hu, Email [email protected] Jianzhou Chen, Email [email protected]
Background: Hypertrophic cardiomyopathy (HCM) is a prevalent monogenic hereditary cardiovascular disorder caused by a mutation in one of the several sarcomere genes encoding components of the cardiac contractile system, distinguished by varied phenotypic presentation and fluctuating clinical advancement. HCM typically affects the left ventricle, resulting in impaired left ventricular function, instances of impaired right ventricular function are uncommon. This case study presents a rare occurrence of HCM accompanied by deteriorating right ventricular function, providing valuable insights for clinicians.
Case Presentation: Herein, we present a case study of an individual diagnosed with HCM utilizing next-generation sequencing (NGS). Over the disease course, the patient with hypertrophic cardiomyopathy also presented with sustained atrial fibrillation, characterized by progressive right ventricular dysfunction, resulting in the development of peripheral edema and ascites.
Conclusion: We report the case of a patient with non-obstructive HCM exhibiting progressive decline in right heart function, confirmed to have the c.470C>T (p.Ala157Val) mutation in exon 7 of the TNNI3 gene.
Keywords: hypertrophic cardiomyopathy, right heart failure, next generation sequencing, atrial fibrillation
Introduction
Hypertrophic cardiomyopathy (HCM) is a prevalent, heritable cardiac condition with worldwide occurrence, typified by hypertrophy of the left ventricle. This ailment stands as a primary contributor to instances of sudden cardiac demise, heart failure, and atrial fibrillation.1,2 HCM predominantly adheres to an autosomal dominant mode of inheritance, with pathogenic genetic alterations detected in around 60% of individuals affected by the condition.3 Molecular genetic investigations have unveiled 27 pathogenic genes linked to HCM, with particular emphasis on MYBPC3, MYH7 and TNNT2, pivotal in encoding myofilament proteins within the cardiac sarcomere and crucial for contractile functionality.4
Heart failure is a prevalent complication of HCM and constitutes an independent risk factor for mortality in affected individuals.5 The majority of heart failure cases in HCM are classified as heart failure with preserved ejection fraction (HFpEF), primarily due to impaired ventricular diastolic function.6 Advanced heart failure characterized by predominantly non-obstructive HCM is present in 5–7% of patients and is distinguished by resistance to pharmaceutical interventions and severe functional impairment.7 Right heart failure occurs in approximately 5–15% of cases of HCM8 and is most commonly seen in patients with end-stage HCM, ventricular remodelling leading to biventricular failure and an increased incidence of right heart failure. Other conditions associated with right heart failure include combined pulmonary hypertension and right ventricular hypertrophic HCM. However, left ventricular hypertrophy with right ventricular insufficiency is rare among HCM patients, and its incidence is unknown.
Atrial fibrillation is the most prevalent arrhythmia associated with HCM and constitutes a critical complication, significantly linked to an elevated risk of thromboembolic stroke as well as increased morbidity and mortality.9 The impaired atrial contractile function in atrial fibrillation commonly leads to a significant decrease in left ventricular (LV) diastolic filling, further worsening heart failure symptoms in individuals with HCM.10
We report a case of HCM coexisting with atrial fibrillation in a patient exhibiting preserved left ventricular function, yet experiencing progressive deterioration of right ventricular function. This relatively uncommon manifestation highlights the necessity for further exploration into the underlying mechanisms of disease progression and the development of potential therapeutic strategies.
Case Report
In January 2021, a 53-year-old female patient sought medical care due to complaints of chest tightness, palpitations, and peripheral edema. These symptoms had intermittently surfaced and deteriorated over the preceding decade. The patient had been diagnosed with hypertrophic cardiomyopathy (HCM) for 13 years and had also experienced atrial fibrillation for 5 years. Furthermore, in 2014, she underwent significant gastrectomy for gastric cancer. Notably, there was no pertinent travel history or contact with epidemic regions. She did not engage in smoking or indulge in excessive alcohol consumption. Her mother had a confirmed medical record of cardiac hypertrophy. Upon hospital admission, her vital signs were recorded as follows: temperature 36.9°C, blood pressure 106/64 mmHg, pulse rate 74 beats per minute, and respiratory rate 20 breaths per minute. Physical examination unveiled an enlarged cardiac silhouette, markedly irregular heart rhythm, and the absence of pathological murmurs within the cardiac valve regions. While jugular venous distention was not observed, mild edema in the lower extremities was noted. Additionally, no adventitious lung sounds, such as moist rales, were auscultated.
Laboratory investigations revealed elevated levels of brain natriuretic peptide (829 pg/mL) and D-dimer (1.04 mg/L). Hemoglobin was notably low at 84 g/L, while electrolyte assessments indicated hyponatremia (131 mmol/L) and hypokalemia (3.0 mmol/L). Tests for liver function, renal function, thyroid function, and markers of infectious disease exhibited no significant abnormalities. An electrocardiogram performed upon admission showed atrial fibrillation with ST-segment depression and T-wave inversion (Figure 1).
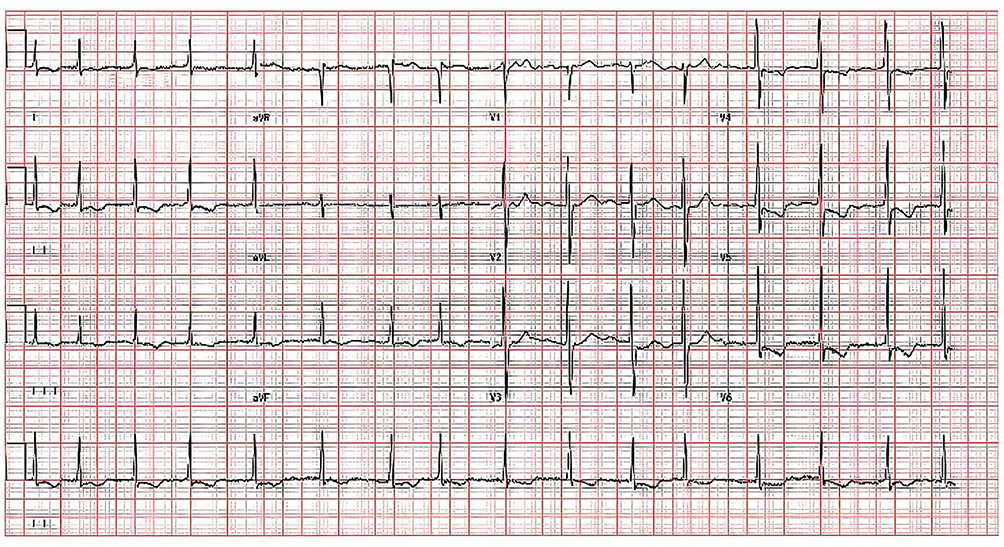 |
Figure 1 Electrocardiogram (ECG) of the patient at admission. 12-lead electrocardiogram indicated atrial fibrillation, ST segment depression, and T wave inversion in leads II, III, avF, V4, V5 and V6. |
Transthoracic echocardiography (Figure 2) demonstrated pronounced interventricular septal hypertrophy measuring 19 mm, along with preserved left ventricular systolic function (ejection fraction of 55%) and an absence of left ventricular outflow tract obstruction, both at rest and following the Valsalva maneuver. Additionally, right ventricular dilation with impaired systolic function was observed, as indicated by a TAPSE measurement of 14.5 mm, alongside notable tricuspid regurgitation. The pulmonary artery systolic pressure (PASP) was recorded at 53 mmHg.
 |
Figure 2 Echocardiograms of the patient at admission in January 2021 (A and B) and at outpatient review in August 2024 (C and D). Parasternal long axis view (A–C) shows significant thickness of interventricular septum (red arrow). Apical four chamber section (B and D) shows right heart dilatation with significant tricuspid regurgitation (red arrow). |
Through next-generation sequencing (NGS) analysis, a c.470C>T (p.Ala157Val) mutation in the TNNI3 gene was identified (refer to Table 1), which was also detected in two family members (specifically, her sister and daughter). Cardiac magnetic resonance imaging (CMR) is the gold standard for evaluating the structure and function of the right heart. Right heart catheterization allows direct measurement of intracardiac pressure, pulmonary artery pressure and cardiac output and assessment of right ventricular preload and afterload. It is recommended in refractory right heart failure or when non-invasive tests fail to make a definitive diagnosis. Despite recommendations for CMR and right heart catheterization, these diagnostic procedures were declined by the patient. The diagnosis rendered was non-obstructive HCM concomitant with atrial fibrillation, and treatment commenced with furosemide, spironolactone, rivaroxaban, and metoprolol succinate. Based on the 2024 AHA/ACC guideline for the management of HCM,8 and given the absence of pertinent family history involving sudden death, unexplained instances of syncope, abnormal left ventricular hypertrophy (maximum left ventricular wall thickness ≥30mm), apical ventricular wall tumor of the left ventricle, or non-sustained ventricular tachycardia during extended electrocardiogram monitoring, the implantation of an implantable cardioverter defibrillator (ICD) was deemed unnecessary.
 |
Table 1 Mutation Profile via Next-Generation Sequencing |
Upon discharge, the patient maintained regular follow-up appointments at our outpatient clinic. In September 2021, an abdominal ultrasound disclosed indications of congestive hepatopathy. Subsequent evaluations in March 2022 revealed the development of ascites. An echocardiogram performed in August 2024 displayed a modest reduction in left ventricular systolic function (ejection fraction of 52%), alongside progressive right heart enlargement, deteriorating tricuspid regurgitation, and a gradual decline in right ventricular systolic function (TAPSE measuring 11.5 mm, TDI s’ 6 cm/sec) (refer to Table 2). Surgical intervention to address severe tricuspid valve pathology was recommended to counteract further deterioration in right heart function; however, the patient opted against this course of action once more.
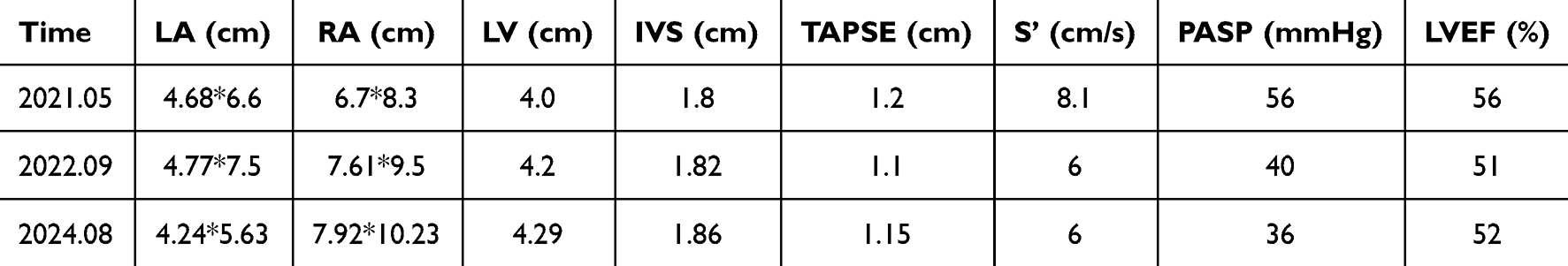 |
Table 2 A Summary Table Showing the Key Findings from Cardiac Ultrasound Over Time |
Discussion
We present a case of a patient with non-obstructive HCM with progressive deterioration of right heart function. The presence of the c.470C>T (p.Ala157Val) mutation within exon 7 of the TNNI3 gene was confirmed. Mutations in the TNNI3 gene have been documented in about 2% of individuals with HCM, exhibiting a distinctive concentration within exons 7 and 8.11 The clinical manifestations associated with TNNI3 mutations display considerable heterogeneity, spanning from asymptomatic carriers to individuals who experience sudden cardiac death.12 Notably, the most severe prognostic outcomes have been observed in carriers of the Ala157Val mutation located in exon 7.13 Roughly 30% of individuals with HCM harboring TNNI3 mutations manifest supraventricular tachycardia (SVT), encompassing conditions such as atrial fibrillation14 and atrial flutter. When juxtaposed with individuals possessing alternative genetic variants, those with TNNI3 mutations face an elevated risk for heart failure composite endpoints as well as combined heart failure and arrhythmia composite endpoints.15,16
HCM is associated with complex pathophysiological mechanisms, including left ventricular outflow tract obstruction (LVOTO), mitral regurgitation and diastolic dysfunction.2 In cases of non-obstructive HCM, diastolic dysfunction emerges as the predominant factor contributing to diminished exercise capacity and the onset of heart failure symptoms.17 As a result of impaired ventricular relaxation, the process of ventricular filling becomes increasingly dependent on atrial contraction. Therefore, when atrial arrhythmias, such as atrial fibrillation, manifest, patients with HCM demonstrate diminished tolerance to these alterations.9
The HCM patient exhibited progressive right ventricular dysfunction, likely attributable to multiple factors. Firstly, the mechanism by which HCM leads to right heart failure is related to ventricular interdependence18. The right and left ventricles are connected by the interventricular septum. Structural and functional changes to the left ventricle in patients with HCM affect the right ventricle through movement of the septum, leading to right ventricular dysfunction and right heart failure. Secondly, patients with HCM have impaired diastolic function in the left ventricle, resulting in elevated end-diastolic pressure. This, in turn, increases pulmonary artery pressure. In order to pump blood into the pulmonary arteries, the right ventricle needs to overcome higher pressures, and the increased afterload can lead to dilatation and impaired function of the right ventricle, and ultimately right heart failure in the long term.19 Finally, right ventricular hypertrophy leads to increased right ventricular diastolic pressure, resulting in right ventricular dysfunction.20 In our patient, right ventricular involvement in HCM is correlated with an increased incidence of supraventricular arrhythmias, advancing heart failure, and a heightened risk of sudden cardiac death.21 Our patient presented with a combination of atrial fibrillation occurring concurrently with HCM, which further diminished left ventricular filling, thereby increasing right ventricular afterload and contributing to progressive right ventricular enlargement. This ongoing enlargement leads to increasingly severe tricuspid regurgitation, establishing a vicious cycle in which each condition exacerbates the other. In light of these findings, we contemplated whether interventions such as atrial fibrillation ablation or surgical correction of the tricuspid valve could potentially mitigate the progressive decline in right heart function. Atrial fibrillation ablation may offer significant benefits to heart failure patients22,23 by enhancing electrical-mechanical synchronization of the heart, restoring atrial systolic function, and optimizing ventricular filling. Additionally, rhythm control can reverse ventricular remodeling and restore sinus rhythm, which in turn reduces neuroendocrine activation and slows ventricular dilatation. In patients with atrial fibrillation and heart failure, the combination of catheter ablation and guideline-directed medical therapy was associated with a reduced risk of all-cause mortality, left ventricular assist device implantation, or urgent cardiac transplantation compared with drug therapy alone.24
Consequently, it is advisable to incorporate routine evaluation of the right ventricle into the assessment protocols for patients with HCM. Furthermore, there are currently no targeted treatment modalities specifically available for HCM patients exhibiting right ventricular dysfunction. In the present case, the patient experienced a progressive decline in right ventricular systolic function, as evidenced by the worsening of peripheral edema and ascites. Treatment strategies focused on volume management, necessitating adjustments to the diuretic regimen by incorporating torasemide alongside tolvaptan therapy due to observed diuretic resistance. Lastly, traditional echocardiography lacks sensitivity in detecting early-stage right ventricular dysfunction; thus, advanced diagnostic techniques such as cardiac magnetic resonance (CMR) imaging and right heart catheterization are recommended for a more precise assessment.25
Historically, HCM combined with right ventricular dysfunction has received little attention due to the limitations of previous imaging techniques. With the advent of advanced imaging modalities, including CMR, an increasing amount of right heart data is now available.2
This case report has several limitations. First, patient compliance was relatively low, and they did not fully adopt more advanced diagnostic techniques, such as CMR imaging or right heart catheterization. Second, we did not conduct further analysis of the gene mutation to determine its potential association with a progressive decline in the patient’s right heart function.
Conclusion
Hypertrophic cardiomyopathy with right heart failure as the main manifestation is rare in clinical practice and has relatively few therapeutic options. The specific mechanisms involved are unclear and may include the following: elevated pressure in the pulmonary circulation, activation of the neuroendocrine system, and abnormal interventricular interactions. Consequently, clinicians should consider the possibility of left ventricular hypertrophy coexisting with right ventricular insufficiency in their practice and continue to explore the underlying pathophysiological mechanisms. This case provides ideas for the diagnosis and treatment of right heart failure and needs more research attention in the future.
Abbreviations
HCM, Hypertrophic cardiomyopathy; NGS, Next generation sequencing; HFpEF, Heart failure with preserved ejection fraction; LV:, Left ventricular; PASP, pulmonary artery systolic pressure; ICD, Implantable cardioverter defibrillator; CMR, Cardiac magnetic resonance.
Ethics and Consent
Informed Consent: Written informed consent was obtained from the patient for the publication of this case report, including authorization to use any accompanying images. At the Central Hospital of Enshi Tujia and Miao Autonomous Prefecture, ethical approval from the Institutional Review Board was not required for the publication of case reports.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the Natural Science Foundation of China (Grant number: 82400566,82160072), the Youth Project of the Natural Science Foundation of Hubei Province (Grant number:2023AFB127), Enshi Prefecture Science and Technology Bureau, Selenium citation special (Grant number: D20230072).
Disclosure
The authors have declared no conflicts of interest in this work.
References
1. Maron BJ. Clinical course and management of hypertrophic cardiomyopathy. N Engl J Med. 2018;379(7):1977. doi:10.1056/NEJMc1812159
2. Ommen SR, Mital S, Burke MA, et al. AHA/ACC guideline for the diagnosis and treatment of patients with hypertrophic cardiomyopathy: executive summary: a report of the American college of cardiology/American heart association joint committee on clinical practice guidelines. J Am Coll Cardiol. 2020;76:3022–3055. doi:10.1016/j.jacc.2020.08.044
3. Lopes LR, Ho CY, Elliott PM. Genetics of hypertrophic cardiomyopathy: established and emerging implications for clinical practice. Eur Heart J. 2024;45(30):2727–2734. doi:10.1093/eurheartj/ehae421
4. Maron BJ, Maron MS, Maron BA, Loscalzo J. Moving beyond the sarcomere to explain heterogeneity in hypertrophic cardiomyopathy: JACC review topic of the week. J Am Coll Cardiol. 2019;73(15):1978–1986. doi:10.1016/j.jacc.2019.01.061
5. Rowin EJ, Maron BJ, Carrick RT, et al. Outcomes in patients with hypertrophic cardiomyopathy and left ventricular systolic dysfunction. J Am Coll Cardiol. 2020;75(24):3033–3043. doi:10.1016/j.jacc.2020.04.045
6. Maron MS, Rowin EJ, Olivotto I, et al. Contemporary natural history and management of nonobstructive hypertrophic cardiomyopathy. J Am Coll Cardiol. 2016;67(12):1399–1409. doi:10.1016/j.jacc.2016.01.023
7. Polovina M, Tschope C, Rosano G, et al. Incidence, risk assessment and prevention of sudden cardiac death in cardiomyopathies. Eur J Heart Fail. 2023;25(12):2144–2163. doi:10.1002/ejhf.3076
8. Writing Committee M, Ommen SR, Ho CY, et al. AHA/ACC/AMSSM/HRS/PACES/SCMR guideline for the management of hypertrophic cardiomyopathy: a report of the American heart association/American college of cardiology joint committee on clinical practice guidelines. J Am Coll Cardiol. 2024;83:2324–2405. doi:10.1016/j.jacc.2024.02.014
9. Rowin EJ, Hausvater A, Link MS, et al. Clinical profile and consequences of atrial fibrillation in hypertrophic cardiomyopathy. Circulation. 2017;136(25):2420–2436. doi:10.1161/CIRCULATIONAHA.117.029267
10. Maron BJ, Desai MY, Nishimura RA, et al. Management of hypertrophic cardiomyopathy: JACC state-of-the-art review. J Am Coll Cardiol. 2022;79(4):390–414. doi:10.1016/j.jacc.2021.11.021
11. Chiswell K, Zaininger L, Semsarian C. Evolution of genetic testing and gene therapy in hypertrophic cardiomyopathy. Prog Cardiovasc Dis. 2023;80:38–45. doi:10.1016/j.pcad.2023.04.009
12. Maron BJ. Clinical course and management of hypertrophic cardiomyopathy. N Engl J Med. 2018;379(7):655–668. doi:10.1056/NEJMra1710575
13. Curila K, Benesova L, Penicka M, et al. Low prevalence and variable clinical presentation of troponin I and troponin T gene mutations in hypertrophic cardiomyopathy. Genet Test Mol Biomarkers. 2009;13(5):647–650. doi:10.1089/gtmb.2009.0041
14. Carrick RT, Maron MS, Adler A, et al. Development and validation of a clinical predictive model for identifying hypertrophic cardiomyopathy patients at risk for atrial fibrillation: the HCM-AF score. Circ Arrhythm Electrophysiol. 2021;14(6):e009796. doi:10.1161/CIRCEP.120.009796
15. Ho CY, Day SM, Ashley EA, et al. Genotype and lifetime burden of disease in hypertrophic cardiomyopathy: insights from the sarcomeric human cardiomyopathy registry (SHaRe). Circulation. 2018;138(14):1387–1398. doi:10.1161/CIRCULATIONAHA.117.033200
16. Coppini R, Ho CY, Ashley E, et al. Clinical phenotype and outcome of hypertrophic cardiomyopathy associated with thin-filament gene mutations. J Am Coll Cardiol. 2014;64(24):2589–2600. doi:10.1016/j.jacc.2014.09.059
17. Kitaoka H, Tsutsui H, Kubo T, et al. JCS/JHFS 2018 guideline on the diagnosis and treatment of cardiomyopathies. Circ J. 2021;85:1590–1689. doi:10.1253/circj.CJ-20-0910
18. Harjola VP, Mebazaa A, Celutkiene J, et al. Contemporary management of acute right ventricular failure: a statement from the heart failure association and the working group on pulmonary circulation and right ventricular function of the European society of cardiology. Eur J Heart Fail. 2016;18(3):226–241. doi:10.1002/ejhf.478
19. Konstam MA, Kiernan MS, Bernstein D, et al. Evaluation and management of right-sided heart failure: a scientific statement from the American heart association. Circulation. 2018;137(20):e578–e622. doi:10.1161/CIR.0000000000000560
20. Reddy S, Bernstein D. Molecular mechanisms of right ventricular failure. Circulation. 2015;132(18):1734–1742. doi:10.1161/CIRCULATIONAHA.114.012975
21. Nugent AW, Daubeney PE, Chondros P, et al. National Australian childhood cardiomyopathy S. Clinical features and outcomes of childhood hypertrophic cardiomyopathy: results from a national population-based study. Circulation. 2005;112(9):1332–1338. doi:10.1161/CIRCULATIONAHA.104.530303
22. Richter S, Di Biase L, Hindricks G. Atrial fibrillation ablation in heart failure. Eur Heart J. 2019;40(8):663–671. doi:10.1093/eurheartj/ehy778
23. Reddy YNV, Borlaug BA, Gersh BJ. Management of atrial fibrillation across the spectrum of heart failure with preserved and reduced ejection fraction. Circulation. 2022;146(4):339–357. doi:10.1161/CIRCULATIONAHA.122.057444
24. Sohns C, Fox H, Marrouche NF, et al. Catheter ablation in end-stage heart failure with atrial fibrillation. N Engl J Med. 2023;389(15):1380–1389. doi:10.1056/NEJMoa2306037
25. Rowin EJ, Maron BJ, Maron MS. The hypertrophic cardiomyopathy phenotype viewed through the prism of multimodality imaging: clinical and etiologic implications. JACC Cardiovasc Imaging. 2020;13(9):2002–2016. doi:10.1016/j.jcmg.2019.09.020
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.