Back to Journals » Journal of Inflammation Research » Volume 12
Pro-inflammatory agents released by pathogens, dying host cells, and neutrophils act synergistically to destroy host tissues: a working hypothesis
Authors Ginsburg I ![]() , Korem M, Koren E
, Korem M, Koren E ![]() , Varani J
, Varani J
Received 7 October 2018
Accepted for publication 5 December 2018
Published 23 January 2019 Volume 2019:12 Pages 35—47
DOI https://doi.org/10.2147/JIR.S190007
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Isaac Ginsburg,1 Maya Korem,2 Erez Koren,3 James Varani4
1Institute of Dental Sciences, Faculty of Dental Medicine, The Hebrew University, Jerusalem, Israel; 2Department of Clinical Microbiology and Infectious Diseases, Hadassah-Hebrew University Medical Center, Jerusalem, Israel; 3Research and Development Department, Clexio Biosciences Ltd, Petah Tikva, Israel; 4Department of Pathology, The University of Michigan Medical School, Ann Arbor, MI, USA
Abstract: We postulate that the extensive cell and tissue damage inflicted by many infectious, inflammatory and post-inflammatory episodes is an enled result of a synergism among the invading microbial agents, host neutrophils and dead and dying cells in the nidus. Microbial toxins and other metabolites along with the plethora of pro-inflammatory agents released from activated neutrophils massively recruited to the infectious sites and high levels of cationic histones, other cationic peptides, proteinases and Th1 cytokines released from activated polymorphonuclear neutrophils (PMNs) and from necrotized tissues may act in concert (synergism) to bring about cell killing and tissue destruction. Multiple, diverse interactions among the many potential pro-inflammatory moieties have been described in these complex lesions. Such infections are often seen in the skin and aerodigestive tract where the tissue is exposed to the environment, but can occur in any tissue. Commonly, the tissue-destructive infections are caused by group A streptococci, pneumococci, Staphylococcus aureus, meningococci, Escherichia coli and Shigella, although many other microbial species are seen on occasion. All these microbial agents are characterized by their ability to recruit large numbers of PMNs. Given the complex nature of the disease process, it is proposed that, to treat these multifactorial disorders, a “cocktail” of anti-inflammatory agents combined with non-bacteriolytic antibiotics and measures to counteract the critical toxic role of cationic moieties might prove more effective than a strategy based on attacking the bacteria alone.
Keywords: synergism, tissue damage, bacterial toxin, bacteriolysis, cationic proteins, sepsis
Plain language summary
The present article offers a working hypothesis suggesting that cell and tissue damage induced in many infectious and inflammatory processes is an end result of a synergy among microbial toxins, metabolites and the rich arsenal of pro-inflammatory agents released by activated neutrophils accumulating in the tissues. Clinical disorders caused by group A streptococci (GAS), staphylococci, pneumococci, toxigenic Escherichia coli, Shigella and inflammatory bowel disease are characterized by recruiting huge numbers of neutrophils (polymorphonuclear neutrophils, PMNs). The major agents delivered by activated PMNs include the following: ROS and nitrogen species, myeloperoxidase, hypochlorous acid, cationic elastase, the cationic peptides such as LL37, cathepsin G, nuclear histone and cathelicidin, gelatinase, several acid hydrolases and T-helper 1 (TH1) cytokines. However, tissue destruction also delivers large amounts of cytotoxic histones. It is proposed that synergistic tissue damage might be controlled by combinations of antibiotics and cocktails of pro-inflammatory agents.
Introduction
We are all exposed to life-threatening microbial, fungal and viral infections. The most notorious microbial postinfectious sequelae is septic shock, which is among the least understood human disorders often develop rapidly but mainly in hospitals with considerable mortality.1,2 Other severe tissue-destructive inflammatory disorders that include acute respiratory distress syndrome, acute lung inflammation (ALI), bacterial pneumonia and meningitis, the intestinal bowel disorders such as ulcerative colitis (UC), Crohn’s disease (CD) and necrotizing enterocolitis (NE) as well as certain types of urinary tract infections, staphylococcal skin abscesses and periodontal disease can be defined as multifactorial events. Our understanding of the pathogenesis and our ability to offer effective treatments are still controversial. In many cases, no effective therapy is available. It is of note that, in contrast, human disorders such as tetanus, diphtheria, botulism, cholera and many of the viral infections, which are prototypic typical monofactorial disorders, can be contained to a large extent by immunizations.
Among the acute tissue-destructive inflammatory diseases, there is a distinct common denominator, ie, the accumulation of large numbers of neutrophils (polymorphonuclear neutrophils, PMNs) at the infectious site.3 Neutrophils, attracted to infectious sites by chemotactic agents generated by bacteria of the complement cascade, are armed with numerous pro-inflammatory agents. Many of the factors in PMNs are stored within granules and released upon their activation and/or demise. Other mediators (primarily oxidants) are enzymatically generated during the activation of PMNs,4,5 and histones are released by netosis.6 Ideally, when recruited to the site of inflammation, the well-armed neutrophils are “programmed” to use their vast armamentarium to, engulf, destroy and, ultimately, remove the eliciting agent. However, during the heat of battle, significant damage to PMNs and to host tissue may also occur leading to sepsis-induced immunosuppression.7 In the cases of the abovementioned processes, host tissue damage is often so severe as to, itself, be life-threatening; one example is necrotizing fasciitis (NF). It is not clear what are the factors that dictate whether a well-orchestrated inflammatory response effectively eliminates the eliciting agent without significant host tissue damage, or whether the same players (PMNs) quickly lose control of the battle and, in desperation, proceed to rampage through the tissue destroying host tissue and eliciting agent indiscriminately. We suggest that when the inflammatory site becomes an indiscriminate battlefield, it is not the neutrophil, per se, that is responsible for the widespread tissue damage but a synergistic interaction involving the eliciting agent, dead and dying host tissue cells, neutrophils and microbial agents released following bacteriolysis.8,9 With mediators of destruction coming from many sources, it can be understood why therapies focused on one source of pro-inflammatory mediators (antibacterial agents) or the other (neutrophils) – never mind the dead and dying host cells – often fail.
Attacked from all sides: the synergism concept and cell damage
The idea that tissue destruction in acute inflammation is the result of mediators from multiple sources working together is not novel. Many in vitro studies and preclinical animal models support this view. As far back as 1950s, the combination of streptococcal membrane-damaging hemolysins and proteolytic enzymes was shown to destroy tumor cells in culture under conditions in which neither agent was lethal alone.10 In a parallel study, combining antibodies and streptokinase (SK)-activated plasmin on tumor cells, a similar synergistic cell-killing synergism was also seen.11 In both cases, the increase in membrane permeability inflicted on the target cells by the membrane-active agents facilitated the intracellular penetration of proteolytic enzymes that ultimately induced massive cell damage.12,13 The voluminous literature subsequent to these early publications has shown numerous in vitro studies that support the synergism concept of cell damage in inflammation and infections.14–24 Is this important? In fact, one could argue, whether subsequent work has served primarily to identify an ever-increasing number of players and identify multiple ways in which synergistic actions may take place.
The synergism concept may therefore help to explain why neither antibiotics alone nor single antagonists tested in clinical trials of sepsis have failed to save most septic patients. This led Opal et al15 to question: “What is the next generation of sepsis trials after the demise of the promising singly administered recombinant human activated protein C (APC)?” The involvement of multiple agents in tissue damage supports the proposition that multidrug strategies may better cope with the multifactorial toxic episodes.16 This, too, is not novel. After all, cocktails of antagonists are currently used successfully to treat AIDS, tuberculosis and many malignancies.
Major participants in cell and tissue destruction: group A streptococci (GAS)
Catalase-negative, penicillin-sensitive GAS are causative agents linked to pharyngitis, tonsillitis, rheumatic fever, rheumatic heart disease, scarlet fever, arthritis, St Vitus’s dance, NF and toxic shock syndrome.17,18 Fortunately, as of today, GAS have never developed resistance to beta-lactam antibiotics which are the drugs of choice to treat GAS infections and to prevent rheumatic fever. However, treatment by such antibiotics may also aggravate the pathological changes, since they can induce bacteriolysis and the release of the pro-inflammatory agents such as lipoteichoic acid (LTA) and peptidoglycan (PPG).9,10
During growth, GAS can release into the surrounding media a plethora of extracellular pro-inflammatory toxic agonists. These include the following: SK that activates plasminogen to plasmin and hyaluronidase which both facilitate the spread of GAS in tissues, a cysteine proteinase, cationic elastase and cathepsin G, the extracellular membrane perforator sulfhydryl-dependent streptolysin O (SLO) and non-immunogenic streptolysin S (SLS) released by plasma albumin, the cell-bound hemolysin (CBH) which can inflict an irreversible membrane damage,19 DNases, four RNAses, a complement inhibitor, superoxide dismutase (SOD) and immunoglobulin-degrading enzymes.17,18
Role of GAS LTA in cell damage
In inflammatory sites caused by Gram positives, one may also expect to locate LTA.20,21 This membrane-associated agent was shown to avidly bind to surfaces of PMNs which may then interact with anti-LTA antibodies present in nearly every patient’s serum. This led to the production of large amounts of superoxide anion and hydrogen peroxide (H2O2) and also to the release of lysosomal enzymes.22 Although tested in vitro, GAS are relatively resistant to degradation either by leukocyte extracts or by a cocktail containing membrane-active lysozyme,21 the cells become susceptible to lysis by leukocyte extracts if grown in the presence of subinhibitory amounts of penicillin and bacteriolysis becomes even more pronounced when the reaction mixtures are incubated at 41°C.
The role of GAS cell wall components in tissue damage
Another major contributor to tissue damage in chronic inflammatory disorders induced by GAS may be the long persistence in macrophages of nonbiodegradable microbial cell wall components that can be released following bacteriolysis.9,10,23 These studies assessed the interaction of GAS-derived PPG and polysaccharide polymers in arthritis models.24 Arthritis was induced in rats by a single intraperitoneal injection of an aqueous suspension of peptidoglycan-polysaccharide fragments derived by sonication. This led to a chronic, remittent, erosive arthritis lasting several months.24,25 The cell wall materials also accumulated rapidly in the liver, spleen and lymph nodes, where they caused little injury. The in vivo localization of GAS cell wall components was detected by measuring muramic acid which has a unique microbial structure.26
The role of GAS cell wall components in the pathogenesis of GAS infections: the Rockefeller Institute Symposium 2000
In 2000, an important symposium was held at the Rockefeller Institute8 in which the discussants were requested to evaluate the role played by the major microbial cell wall components, endotoxin (lipopolysaccharide, LPS), LTA and PPG released by bacteriolysis induced either by antibiotics or by neutrophil-derived cationic peptides9,27 and a synergism among them as major agents implicated in the pathogenesis of sepsis, a typical multifactorial synergistic disorder. The participants in the symposium were also requested to comment and discuss the question: is sepsis a one-hit or a multiple-hit (synergistic) episodes among microbial components and host-derived agonists as major events in the pathogenesis of sepsis?8 It was also questioned whether antibiotics can induce the release of pro-inflammatory constituents from both Gram negatives and Gram positives as major events in sepsis and to offer a potential opportunity to limit the extent of inflammation by the selection of appropriate antibiotics. This is because microbes may be killed or also inhibited from growth by certain antibiotics. Experimental evidences supported the concept that different antibiotics that manifest similar efficacy in their ability to kill Gram-negative microbes may show significant differences in their capacities to cause the release of soluble endotoxin from Gram negatives and cell wall components from Gram positives.8 In this respect, the old Jarisch–Herxheimer phenomenon which had implicated bacteriolysis raises a very important dilemma whether highly bacteriolytic antibiotics could seriously aggravate septic patients’ conditions and, therefore, it should be avoided.9,27
However, there are conflicting approaches to this issue as clinicians at intensive care units (ICUs) argue that the mortality rates among septic patients administered bacteriolytic agents were not significantly greater than those administered non-bacteriolytic antibiotics. However, patients who are exposed in ICUs to prolonged intravenous infusions of antibiotics may also develop unanticipated injury to internal organs. Indeed, one common complication in Gram-negative sepsis is tubular necrosis. This is when circulating microbial cell wall agents, mainly PPG released by antibiotics, generate the release from PMNs, the toxic cytokine TNF-α.
After publishing the minutes of the Rockefeller Symposium in 2000,8 it was anticipated that new studies either supporting or refuting the synergism concept of cell damage and role of microbial cell wall components would be published. Surprisingly, no new publications on the subject have appeared either in the basic science or in the clinical literature. In addition, the term bacteriolysis is not included in clinical papers.
Gram-positive catalase-positive Staphylococcus aureus
S. aureus is a normal skin resident that can turn highly pathogenic by developing high resistance to methicillin (methicillin-resistant S. aureus, MRSA). Staphylococci can not only induce the formation of boils and carbuncles but also NF, pneumonia, endocarditis, septic shock along with additional manifestations.28,29 However, MRSA can also hide in skin keratinocytes and in PMNs, awaiting an attack when host resistance is lowered. In skin boils and carbuncles, MRSA also hides under a heavy mantle of fibrin induced by coagulase which prevents phagocytosis and killing by PMNs. In addition, hypoxia inside the lesion may prevent killing of the microbial cells by oxidants generated by activated PMNs. However, staphylococci may also release several agents that can retard PMN chemotaxis, phagocytosis and bactericidal activity as given in the following sections. In addition, during growth, staphylococci can elaborate and secrete hemolysins, toxic shock syndrome toxin, Panton–Valentine leukocidin, a phospholipase, coagulase deoxyribonuclease, staphylokinase, protease and hyaluronate lyase.
Human neutrophils (PMNs)
Mature human PMNs are found in large numbers in the bone marrow from which they migrate and marginate in the lung vasculature. Upon an alarm by chemotactic agents such as Formyl-Met-Leu-Phe (f-MLP) or C5a, from complement, they enter the bloodstream and migrate to infectious and inflammatory sites. There they intercept the invaders and engulf and kill them by oxidant- and non-oxidant-dependent systems.45
There are four types of granules in neutrophils, such as azurophilic (or primary) granules, specific (or secondary) granules, gelatinase (or tertiary) granules, and secretory vesicles. Granular contents except for secretary granules are formed during neutrophil maturation; primary, secondary and tertiary granules are formed in this order. Secretory granules, on the other hand, are formed through endocytosis at the end stage of neutrophil maturation, and therefore they contain plasma-derived proteins such as albumin. Among these granules, azurophilic granules are the most difficult to mobilize, followed by specific granules, gelatinase granules and, finally, secretory vesicles.
Activated PMNs may elaborate and secrete the following: superoxide (generated via NADPH oxidase), SOD (SOD to H2O2), hydroxyl radical and myeloperoxidase (MPO) which interacts with H2O2 and with a halide to generate toxic hypochlorous acid. In addition, nitric oxide synthase generates nitric oxide which then forms a highly toxic peroxynitrite. Other active agents include bactericidal histone,6 the polycations such as LL37, permeability-inducing agents, cathelicidin, cationic elastase, cathepsins C and G, gelatinase, several acid hydrolases, PLA2 and TH1 cytokines. However, many of these pro-inflammatory agents might also be released from non-phagocytizing PMNs necrotized by microbial leukocidins such as staphylococcal hemolysins, the streptococcal hemolysis SLO, SLS by the cell-bound streptolysin and also by Escherichia coli hemolysins (as given in the following sections). The released PMN agents may now also be available to interact synergistically/cooperatively with the microbial exo and cell wall agents and also with histones released from PMN neutrophil extracellular traps (NETs) and from necrotized tissues. These complicated combinations can amplify cell damage.
Experimental models conducted in vitro employing tissue cultures of human umbilical cord endothelial cells were studied many years ago testing combinations among oxidants, membrane proteinase, hemolysins cationic peptides and additional agents.13,30–38
Human macrophages
Macrophages have major functions including antigen presentation, phagocytosis, microbial killing and immunomodulation through the production of various cytokines and growth factors. They also play a critical role in the initiation, maintenance and resolution of inflammation.39 Macrophages can also store nonbiodegradable microbial cell wall components and are also involved in granulomatous inflammation such as seen in streptococcal arthritis.24,25,25–40 Classically, activated macrophages release oxidants, neutral proteases, cationic elastase, gelatinase, matrix metalloproteinases, collagenase, plasminogen activator, acid hydrolases, lipases, cathepsins, ribonucleases, phosphatases and glycosidases as well as lysozyme. Therefore, even dead macrophages may act in synergy with microbial exo products and cell wall components (LPS, LTA, PPG) to provide nondigestible materials suitable for macrophage engulfment. The role of cationic peptides in pathology is important and necessitates adequate neutralization by anionic agents such as heparin.
Overall, scores of toxic pro-inflammatory agonists generated by bacteria and by phagocytes may act in cooperation/synergy to induce, to enhance and also to perpetuate in acute and chronic inflammation and, ultimately, organ destruction. One of the characteristic features of NF and of other chronic inflammatory conditions is the presence in the infectious tissues of combinations among microbes, intact and necrotic PMNs, host macrophages and necrotic host tissue – all are rich in toxic histone, oxidants and proteinases.
The major contributors to cell damage in inflammation infection and in postinfectious sequelae such as sepsis and septic shock are synergistic interactions among PMNs – and macrophages – derived pro-inflammatory agents, pattern recognition receptors recognizing the toxic microbial cell wall components such as LPS, LTA and PPG. These toxic agents are mainly released from bacteria following the release of cationic peptides from activated PMNs or by certain beta-lactam antibiotics. This is why it is recommended that bacteriolytic antibiotics should be avoided.9,41
The role of cationic polyelectrolytes in cell damage
Neutrophils are equipped with several highly cationic proteins that have been shown to injure cell membranes. Already, in 1952, Katchalski et al42 and Hirsch43 showed the microbicidal effects of histone and of the synthetic poly-α basic amino acids–poly-l-lysine and poly-l-arginine (actually histone mimics).
Studies on polycations acting as cell-damaging agents for endothelial cells were already published previously but only seldom cited today.31,32,35,36,44,45,47,48 In 2009, two “provocative” publications in Nature Medicine49,50 have suggested for the first time that in human beings, the major cause of death in sepsis may be the result of the release from neutrophil NETs6 of highly toxic cationic histone. PMNs, which adhered to endothelial cells and became activated, caused endothelial cell dysregulation and enhanced their permeability leading to organ failure and patient demise. It was also shown49 that, being highly cationic, histone action could be targeted pharmacologically either by antibodies, heparin or APC; the latter cleaves histones. Antibodies to histone were also shown to reduce the mortality of mice in LPS, TNF-α or cecal ligation and puncture models of sepsis. The authors of these studies showed that extracellular histones were cytotoxic toward endothelium in vitro and were also lethal to mice. In vivo, histone administration resulted in neutrophil margination, vacuolated endothelium, intra-alveolar hemorrhage and macro- and microvascular thrombosis.
However, the study by Xu et al49 failed to consider one pivotal phenomenon which had suggested that the exclusive “unique” release of histones from NETs could not really explain the main cause of cell death in sepsis. This is because this highly toxic polycation released from PMN granules most probably actually acted in synergy with oxidants, highly basic elastase, cathepsin C and G, the protein LL37, phospholipases and TH1 cytokines simultaneously released from activated PMNs. Therefore, histone may be considered only as an additional marker released from damaged cells.51
This assumption may also be strengthened by many reports since 2009 suggesting that high levels of circulating cationic histone are also found in cardiovascular, renal, hepatic, pancreatic and in pulmonary disorders, all totally unrelated to sepsis. Histones and other polycations released from necrotic tissues and from necrotized PMNs such as seen in NF and in inflammatory bowel disease (IBD; given in the following sections) might also act synergistically with oxidants, proteinases and with additional PMN pro-inflammatory agents to enhance cell damage. In a rabbit model, decomposition products from necrotic tissues were found to contain toxic substances that possess a different toxicity profile from endotoxin, and their toxicity could be even stronger.52 In the serum, the levels of TNF-α, IL-1 and IL-6 in the rabbits increased from 2 to 4 hours after injection of the necrotic tissue homogenate and reached the peak level at 12 hours, followed by a gradual reduction since 36 hours. Compared with endotoxin, necrotic tissue homogenate resulted in an early increment (2–4 vs 5–6 hours) and significantly higher peak levels (at 30 hours).
Overall, one may suggest that in all the infectious disorders which mobilize huge numbers of PMNs to the infectious sites, synergistic cross talk among pro-inflammatory agents may expect to occur among microbial toxins and metabolites, PMNs and histone from necrotic tissues act in concert to produce the tissue damage. Since many of the potent mediators are cationic, it may further be suggested that treatment with anionic substances such as an anticoagulant heparin53 which might also prevent the formation of coagulation might prove effective by neutralizing polycation toxic effects.
Sepsis, septic shock and coagulation
Sepsis and disseminated intravascular coagulopathy are multifactorial synergistic episodes which often develop due to microbial infections which in many cases may turn worst by developing shock, organ failure and death.54 In severe disease, the coagulation system becomes diffusely activated, with the consumption of multiple clotting factors resulting in disseminated intravascular coagulation (DIC). DIC portends a higher mortality rate. A better understanding of the mechanisms that tie infection and inflammation and diffuse thrombosis will allow therapeutic interventions to be developed. Whole blood testing of coagulation may provide more clinically useful information than classical tests, and natural anticoagulants that regulate thrombosis may be downregulated in sepsis. Neutrophils (PMNs) possessing a large arsenal of toxic pro-inflammatory agents such as ROS and nitrogen species, a large assortment of highly cationic peptide histones, LL37, elastase and pro-inflammatory cytokines may act synergistically and are instrumental in the initiation of cell and tissue damage. Being typical multifactorial episodes, we are not surprised why most of the clinical trials of sepsis which had tried one single antagonist at a time had invariably failed.55
In 1999, we proposed that only multidrug strategies including a highly anionic non-anticoagulant heparin and proper non-bacteriolytic antibiotics might be ideal to cope with sepsis provided that patients will receive treatments as early as possible.16,23,27
Inflammatory bowel diseases
IBDs include a group of intestinal disorders where no unique microbial damage-associated molecular patterns–alarmins have been identified that if successfully inhibited, may prevent tissue damage characteristic of these disorders. The three major clinical conditions include the following: UC, CD and NE.56,57 These disorders are associated with various nonspecific enteric bacteria including bacteroides, Clostridium, enterobacteriaceae and a substantial presence of Pseudomonas. Histologically, all the three disorders show a massive accumulation of PMNs attacking the mucosal layers. PMNs are the first cells to be recruited to the inflammatory sites having an essential role in proper resolution of inflammation. However, if these processes are not effectively regulated, they may trigger positive amplification loops that may lead to neutrophil activation, causing significant tissue damage and chronic disease. But it may also be suggested that upon activation, and especially at the heat of the battle, PMNs may also succumb in large numbers and undergo necrosis due to microbial hemolysins and also by lysophosphatidate released from phospholipase A2 activation.45 It may therefore be suggested that in all the IBDs, the major toxic events probably involve a massive migration and accumulation of PMNs in the intestinal mucosa releasing ROS, cationic peptides, cationic proteinases and TH1 cytokines. These can now also act in synergy/cooperatively with the microbial agents such as LPS, LTA and PPG released following bacteriolysis induced either by antibiotics9,23,27 or also by cationic peptides.38 Therefore, we may assume that a use of multidrug strategies and lowering PMN activities if administered in collaboration with appropriate antibiotics may serve to mitigate tissue damage.15
Ulcerative colitis
UC is an IBD that causes long-lasting inflammation and ulcers (sores) in the digestive tract. UC affects the innermost lining of the large intestine (colon) and rectum. In UC, circulating and intestinal neutrophil activity increases substantially in driving tissue damage and extraintestinal manifestations. Calprotectin is a robust neutrophil and disease biomarker, and a few neutrophil-related targets are being clinically explored as therapeutic targets.
Targeting neutrophils and their inflammatory mediators per se is an opportunity that should be explored to identify new effective medical therapies. The overall clinical goal for neutrophil-targeted therapy will be to modulate, but not completely silence, neutrophil activity, thereby abolishing the destructive inflammation with associated acute and chronic tissue damage without compromising host defense.56
Crohn’s disease
CD is an IBD that often leads to abdominal pain, severe diarrhea, fatigue, weight loss and malnutrition.58 Inflammation in CD often spreads deep into the layers of the intestines and is characterized by immunological responses leading to chronic inflammation without tissue regeneration. Neutrophils are recruited to the site of inflammation and are also crucial to limit invasion. However, if these processes are not tightly regulated, they can trigger the release from the PMNs of oxidants via NADPH oxidase, proteinases, cationic peptides, lysophosphatides and cytokines.36–38 The PMN agents may now act in synergy with microbial metabolites and toxins to amplify cell damage. Pharmacological approaches aimed at blocking the recruitment of neutrophils to the intestinal epithelium, and the use of multidrug strategies16 might prove effective.
Necrotizing enterocolitis
NE develops mainly in premature infants, often within 2 weeks of birth.57,59 The nature of the microbial flora which are involved in pathogenicity is complex, and the identity of specific pathogens still remains elusive. NE can progress very quickly, and patients should get treatment right away. NF develops when the tissue in the inner lining of the small or large intestine becomes inflamed, damaged and begins to die. The condition usually affects only the inner lining of the intestine but the entire thickness of the intestine may become impacted eventually. In severe cases, bacteria can leak into the abdomen and cause widespread infection. A curcumin analog ameliorated intestinal necrosis.60 Activation of toll-like receptor 4 (TLR4) is also essential for the development of NE, and strategies to inhibit TLR activation or inflammatory cytokines have been proposed to help ameliorate injury in models of necrotizing enterocolitis.61 Finally, anti-PMN strategies may perhaps be found effective to protect epithelial injury and function that are associated with NE.
Meningococci
Gram-negative meningococci cause meningitis, which is a widespread blood infection that can also result in sepsis. Bacteria that enter the bloodstream and multiply can damage the walls of the blood vessels causing bleeding into the skin and organs.62 There are 12 serogroups of Neisseria meningitidis which have been identified. Appropriate antibiotic treatment started as soon as possible is ideal after the lumbar puncture has been carried. The dominant pro-inflammatory molecules in PMNs are the dominant pro-inflammatory molecules and in the cell wall of the meningococci is endotoxin which stimulates monocytes, neutrophils and endothelial cells and promotes the release of pro-inflammatory cytokines (TNF, IL-1, IL-8, interferon [IFN]-g). The release of local inflammatory cytokines and nitric oxide from activated PMNs causes breakdown of blood–brain barrier, edema, induction of cellular apoptosis, coagulation, blockage of vessels and ischemia. Meningococci are also notorious for undergoing rapid spontaneous autolysis releasing LPS and cell wall component. However, a range of non-bacteriolytic antibiotics including ampicillin, chloramphenicol and ceftriaxone may be protective.
Pneumococcal diseases
Pneumococcal diseases are caused by Streptococcus pneumoniae. These Gram positives can cause pneumonia, ear infections, sinus infections, meningitis and bacteremia.63,64 To combat this microorganism, the lung has developed a multifaceted system of defense by a vigorous recruitment of neutrophils which might, however, mediate severe inflammation. Treatment with antibiotics leads to clearance of the bacteria from the infected tissues, but the bacteriolytic nature of some antibiotics leads to an acute release of bacterial toxins and thus after antibiotic therapy the patients can be left with organ-specific deficits. Therefore, highly bacteriolytic agents should be avoided during treatment.
Toxigenic E. coli
E. coli is a bacteria commonly found in the gastrointestinal tract of people and animals.65,66 Many types of E. coli are harmless but some types of E. coli can produce toxins (Shiga toxins). Various strains of, eg, E. coli O111 and E. coli O157 infections cause a diarrheal illness. Sometimes, infections can also result in hemolytic uremic syndrome. In addition, E. coli can also release its endotoxin (LPS) mainly following antibiotic treatments which is a major phenomenon in many Gram-negative infections. Defined virulence phenotypes of uropathogens permit evasion of PMN activity. Uropathogenic (UPEC) strains, resist phagocytic killing and may dampen the production of antimicrobial ROS by PMNs. UPEC attenuated transepithelial neutrophil recruitment in an in vitro model of acute infection and in a murine model of bacterial cystitis was performed. Analysis of the transcriptional responses of PMNs to E. coli strains revealed that UPEC exposure downregulates the expression of PMN genes that direct pro-inflammatory signaling and PMN chemotaxis, adhesion and migration. UPEC strategies are important in the establishment of epithelial infection and that the findings are germane to bacterial infections at other epithelial surfaces. Histological changes evaluated were the degree of edema, mixed inflammatory infiltration, urothelial epithelial invasion by neutrophils and reactive atypia. Recently, in vivo effects of intravesical instillation of green tea extracts rich in polyphenols on bacterial cystitis were analyzed using a rat model of bacterial cystitis.67 Intravesical instillation attenuated the inflammatory response to UPEC-SR71-induced bacterial cystitis and may be a novel approach to the treatment of bacterial cystitis.
Shigella infections
Species of the genus Shigella are among the bacterial pathogens most frequently isolated from patients with diarrhea.68 Shigellosis is an acute intestinal infection; the symptoms of which can range from mild watery diarrhea to severe inflammatory bacillary dysentery characterized by strong abdominal cramps, fever and stools containing blood and mucus. After passage through the stomach and small intestine, the bacteria reach the large intestine, where they establish an infection.
Shigella flexneri ensures its survival in macrophages by rapidly inducing apoptosis. Macrophage cell death is accompanied by the release of the pro-inflammatory cytokines such as IL-1β and IL-18. Both cytokines are critical mediators of the acute and massive inflammatory response elicited by S. flexneri. Ultimately, PMNs phagocytose and kill Shigella, thus contributing to the resolution of the infection.
Many pathogenic features of Shigella intestinal infection are due to the production of potent cytotoxins. The host response to infection is characterized by the induction of an acute inflammation, which is accompanied by massive polymorphonuclear cell (PMN) infiltration, resulting in massive destruction of the colonic mucosa.
Role of red blood cells (RBCs) in infection and inflammation
RBCs oozing out from damaged capillaries may participate actively in inflammation and cell damage. ROS such as superoxide and H2O2 may oxidize proteins to make them more susceptible to digestion by proteinases.69,70 However, paradoxically, catalase-rich RBC may also altruistically protect catalase-negative bacteria against oxidants.71
Targeting PMN activities
Targeting neutrophils and their inflammatory mediators should be explored to identify new effective medical therapies.72 The overall clinical goal for neutrophil-targeted therapy should be to modulate, but not completely lower neutrophil activities. Which clinical therapeutic measures might help to control the aftermath of multifactorial synergistic microbial destructive effects? The demise of APC, the “magic” promised antisepsis agent, and the failure of single antagonists to treat sepsis and septic shock suggest that only multidrug strategies (cocktails of antagonists) might perhaps be successful.16 It suggested that in many inflammatory episodes characterized by a massive recruitment of PMNs and especially in IBD and in streptococcal NF, a direct infusion into the inflamed areas of cocktails of anti-inflammatory agents may neutralize many of the agonists which act in synergy to injure cells. Such cocktails are inexpensive and may readily be concocted in a hospital pharmacy. The cocktails suggested for clinical use may include the following: combinations and permutations among the antibiotics, chlorhexidine, antioxidants such as N-acetyl cysteine, glutathione and zinc, antioxidant polyphenols from green tea, heparin to neutralize histones and additional toxic cationic agents such a LL37, elastase and cathepsin G, aprotinin as a proteinase inhibitor and lecithin to neutralize hemolysins generated by GAS, staphylococci and by other toxigenic bacteria.16
Overall, it is stressed that after all, today, cocktails of antagonists are used with much success to treat AIDS, tuberculosis and many malignancies. Finally, if one accepts that PMN-derived pro-inflammatory agents including polycations function as double-edged swords,73 then, one may make use of antibodies to polycations neutralized by heparin in combination with cocktails of anti-inflammatory agent to ameliorate the severe tissue damage seen in conditions mediated by inflammation.
Are there any clinical conditions where PMNs lose some of their normal functions?
As stated earlier, we have postulated that many clinical disorders are characterized by the massive recruitment to infectious sites of PMNs rich in pro-inflammatory agonists. Upon arrival, PMNs encounter microorganisms, their metabolites and toxins and may engage in a synergistic manner to injure tissues. However, many strategies had been evolved in microbial infections which may retard the functions of PMNs.74,75
In addition, one important clinical status of neutrophil weakness and disabilities may be seen when intensive chemotherapy for hematological malignancies induces a breakdown of mucosal barrier leading to high neutropenic fever. It is estimated that 13%–37% of neutropenic patients develop bacteremia.
The depth and duration of neutropenia are directly related to the incidence of serious bacterial infections that can progress rapidly, leading to hypotension and life-threatening complications with a mortality rate of 50%.76 Because neutropenic patients are unable to mount robust inflammatory responses, serious infection can occur with minimal symptoms and signs. In such patients, fever is often the only sign of infection, because the endogenous pyrogen (IL-1) is produced by mononuclear cells, not by granulocytes, and these mononuclear cells include also fixed tissue macrophages that persist after chemotherapy.
The pathophysiology of sepsis in neutropenic patients cannot be attributed solely either to an initial bacterial stimulus provoking an early systemic inflammatory response syndrome or a “cytokine storm” characterized by a systemic release of inflammatory cytokines such as IL-1, IL-6, TNF-α and IFN-γ77,78 or to the release of toxic histones. During immunosuppression, the systemic release of inflammatory cytokines is either markedly lowered79 or shows the absence of selective cytokines.80 Sepsis in neutropenic patients, therefore, is probably the consequence of an imbalance between pro- and anti-inflammatory cytokines as suggested by the multimodal hypothesis of sepsis.81
Extracellular agents from GAS which modulate leukocyte functions
“SpyCEP” is a Streptococcus pyogenes protease that cleaves CXCL8/IL-8, and its activity is associated with human invasive disease severity.82 SpyCEP cleaved human CXCL1, CXCL2, CXCL6 and CXCL8 plus murine CXCL1. This protease is necessary and sufficient for systemic bacterial dissemination from a soft tissue focus in this model and also underlies dissemination in the respiratory tract protease SpyCEP (also called ScpC), which also cleaves IL-8. SpyCEP expression is strongly upregulated in vivo in the M1T1 GAS strains associated with life-threatening.
“Heparan sulfate” can modulate neutrophil and endothelial function in antibacterial innate immunity.83 Normal bactericidal activity of neutrophils is influenced by the sulfation pattern of heparan sulfate by affecting trap formation. Still, this did not affect either phagocytosis or the formation of oxidants.
“Platelets” have been reported to form heterotypic aggregates with leukocytes and may modulate their function.84 The endogenous platelet activator thrombin gave rise to platelet-dependent neutrophil activation, resulting in enhanced phagocytosis and bacterial killing. M1 protein from S. pyogenes also mediated platelet–neutrophil complex formation. However, these neutrophils were dysfunctional and exhibited diminished chemotactic ability and bacterial killing.
“SLO in PMN paralysis”:85 the secreted GAS pore-forming toxin SLO, which induces eukaryotic cell lysis in a cholesterol-dependent manner, is highly upregulated in the GAS M1T1 clone during bloodstream dissemination. SLO promotes GAS resistance to phagocytic clearance by neutrophils, a critical first element of host defense against invasive bacterial infection. SLO at sub-cytotoxic concentrations suppressed neutrophil oxidative burst, in a manner reversed by free cholesterol and anti-SLO-blocking antibodies. SLO blocked neutrophil degranulation, IL-8 secretion and responsiveness, and elaboration of DNA-based NETs, cumulatively supporting a key role for SLO as a modulator of PMNs’ functions.
“SLS inhibits neutrophil recruitment”:86 in contrast to wild-type S. pyogenes, an SLS-mutant was associated with the robust recruitment of neutrophils and significantly reduced lethal myositis in adult zebrafish. Analysis of transepithelial migration in vitro suggested that SLS inhibited the host cells’ production of signals chemotactic for neutrophils, which contrasted with the pro-inflammatory effect of an unrelated cytolytic toxin, SLO.
Although bacterial production of SLO resulted in lysis of both human keratinocytes and polymorphonuclear leukocytes, GAS expression of SLS was associated only with keratinocyte injury. Expression of SLO but not SLS impaired polymorphonuclear leukocyte killing of GAS in vitro, but this effect could only be demonstrated in the background of capsular organisms.
To facilitate invasion, S. pyogenes secretes SK, a potent plasminogen activator.17 SK–plasminogen interactions are an important determinant of GAS invasiveness in vivo, and both SK and host plasminogen activators appear to promote virulence of GAS by catalyzing plasmin formation and produce high rates of morbidity and mortality despite the implementation of aggressive treatment plans.
“DNase activity” made by GAS contributes to disease progression, degradation of NETs. These innate immune structures are composed of chromatin and granule proteins.87
“Incompetence of neutrophils to invasive GAS”:88 a panel of serotype-matched GAS, which were clinically isolated from severe invasive but not from noninvasive infections could abrogate functions of human PMNs in at least two independent ways, due to inducing necrosis to PMN by enhanced production of a pore-forming toxin SLO and due to impairment of PMN migration via digesting IL-8, a PMN-attracting chemokine, by increasing the production of a serine protease ScpC.
“Chemotactic factor”:89 human polymorphonuclear leukocyte (PMN) chemotaxis was tested during exposure to leukocyte and platelet extracts, a variety of polyelectrolytes, inflammatory exudates and bacterial products. The chemoattractants employed were either zymosan-activated serum or supernatant from autolyzed S. aureus. Chemotaxis to both chemoattractants was markedly inhibited by leukocyte and platelet extracts, inflammatory exudates, anionic polyelectrolytes, DNA, hyaluronic acid, liquid, cationic polyelectrolytes, histone, protamine base, protamine sulfate, MPO, elastase, collagenase, pepstatin, and e-aminocaproic acid. Bacterial products, such as LTA and LPSs, and extracts of human dental plaque also inhibited chemotaxis.
Conclusion
The purpose of the present review was to suggest that cell and tissue damage in infections caused by GAS, S. aureus, pneumococci, meningococci, Shigella and E. coli and patients with IBD all involve a massive recruitment of neutrophils to the site of infection. A synergistic tissue damage is observed among bacterial hemolysins, the numerous pro-inflammatory products generated by the neutrophils and products such as toxic histone released from dead and dying cells at the infectious site. It follows that effective intervention will likely need to include an equally complex cocktails of antagonists. Unfortunately, identifying the agents to be included, much less establishing their safety and efficacy, will be exceptionally difficult. Controlled trials involving combination therapy are inherently difficult to carry out with healthy subjects, much less with individuals who are already critically ill. Beyond this, it must be acknowledged that any reagent or combination of reagents that interfere with neutrophil function during a response to acute bacterial infection is also likely to interfere with neutrophil functions. Further, it should be noted that neutrophils at the inflammatory site may also have important additional functions in the final resolution of the inflammation and restoration of normal tissues lost by the destructive effects of the synergism shown in inflammatory process.3–5,12,14,13,90,38,7316 It is of interest that several GAS-derived exo agents (and also staphylococcal agents not covered in the present article) may also have inhibitory effects on PMN functions.82–89 However, the massive prolonged chemotaxis brings in fresh neutrophils which may compensate for some of the loss of the antimicrobial activities. Figure 1 shows the mediators orchestrating tissue damage in inflammation and sepsis as well as possible treatment cocktails against this process.
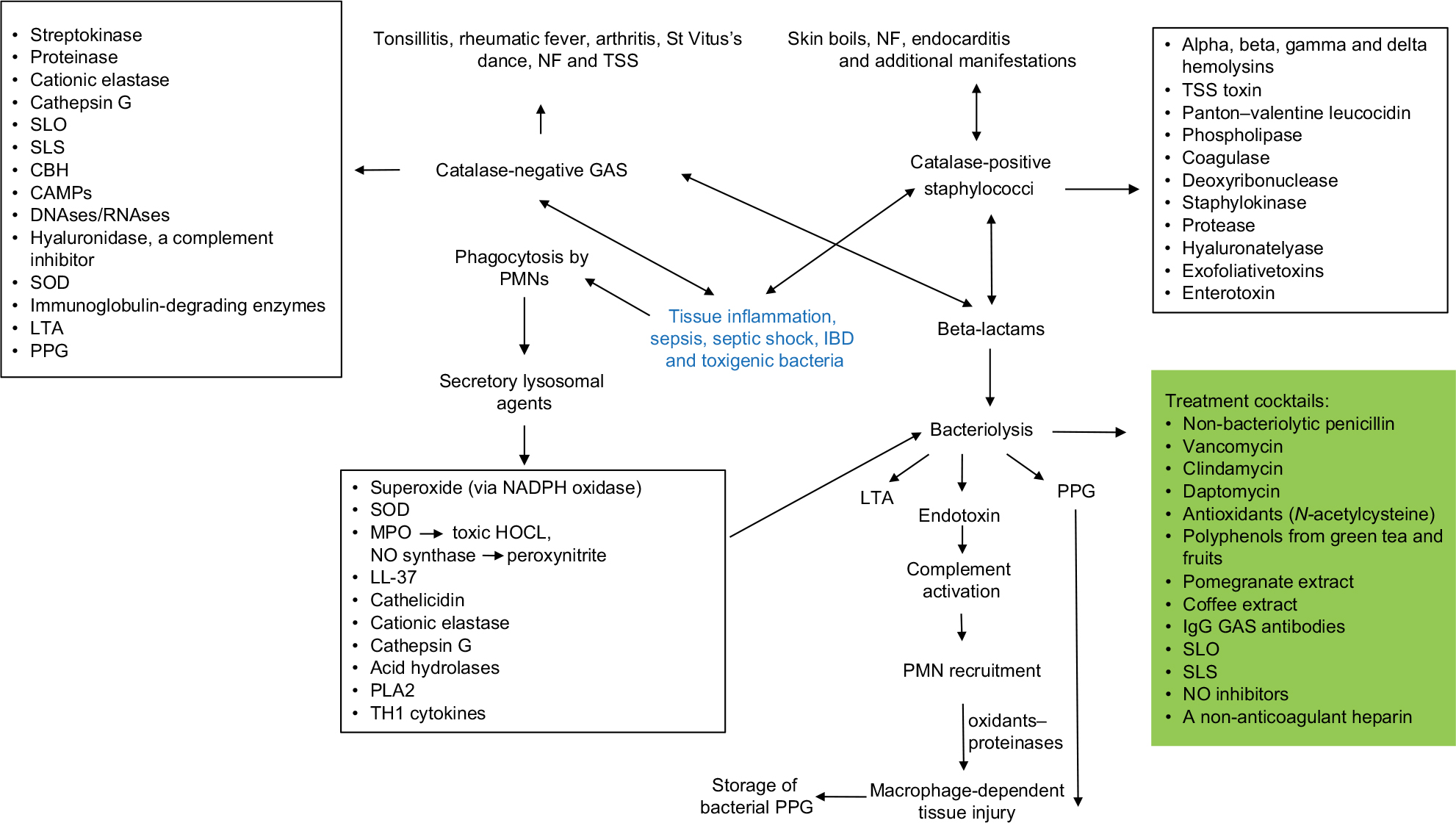 | Figure 1 Pathogenesis of tissue inflammation. Abbreviations: CAMPs, cationic antimicrobial peptides; CBH, cell-bound hemolysin; GAS, group A hemolytic streptococci; IBD, inflammatory bowel disorder; LTA, lipoteichoic acid; MPO, myeloperoxidase; NF, necrotizing fasciitis; NO, nitric oxide; PMN, polymorphonuclear neutrophil; PPG, peptidoglycan; SLO, streptolysin O; SLS, streptolysin S; SOD, superoxide dismutase; TSS, toxic shock syndrome. |
Finally, we must acknowledge that the milieu of the typical bacterial infection/inflammatory site is not simply a reflection of the mediators contributed by the invading bacteria, host neutrophils and resident tissue cells in various stages of morbidity. Rather, such lesions contain macrophages, mast cells, lymphocytes, platelets and all of the other cell types found in the tissue itself and in the circulation. Beyond this, circulating hormone, growth factors, clotting factors and fibrinolytic agents, other protease cascade proteins, circulating cytokines, etc will all be found in the lesion. One cannot fully appreciate the acute lesion without taking these additional factors into account.
Since in infectious sites, microbial PMN agonists including toxic histones and additional cationic peptides are generated, it may be recommended that, as early as possible, the morbid tissues should be either removed surgically or infused by a cocktail composed of combinations among several anti-inflammatory agents91 including anionic heparin. Such cocktails may be found effective to neutralize the synergies among the toxic agents generated by the combinations of bacterial and PMN pro-inflammatory agents. However, it should not be underestimated that arrival of PMNs and macrophages at the inflammatory sites may also have important additional functions in the final resolution of inflammation and restoration of normal tissues lost by the destructive effects of the inflammatory process.43
To better appreciate the synergistic phenomenon and how tissues may be destroyed and organs fail in postinfectious and inflammatory sequelae, we may also benefit from two older pioneering studies on the pathogenetic strategies of GAS, a typical multifactorial highly toxigenic and invasive agents.18,92 Unfortunately, a self-healing of the tissue in all necrotizing disorders may not happen too often. Therefore, the physician has to decide quickly how to proceed with treatment since the gravest danger to patients’ lives suffering from GAS NF, and necrotizing colitis is their high invasiveness and destructiveness leading to high morality. Therefore, to save patients’ lives necessitates submitting heparin to neutralize toxic histone activity and also considers a quick radical surgery and debridement of necrotic tissues. In addition, since many additional microbial infections may also be defined as multifactorial and synergistic episodes, the use of non-bacteriolysis-inducing antibiotics8,9,23,27 combined with cocktails of antagonists may perhaps be helpful to contain the infections and their aftermath.26–33,33,34,34–73
In summary, the paradigm offered in this review may explain why most of the clinical disorders characterized by a massive accumulation of PMNs may have a common pathway of pathogenicity. This may open new vistas into the treatment of several clinical disorders that cannot be effectively treated by antibiotics alone but necessitates a significant slowdown of the deleterious combinations induced when microbial and PMN toxic agents act in concert.
Finally, our older studies also suggested that we may learn a lesson taught by the pathophysiology of GAS, a typical multifactorial agent, how tissues are destroyed in infectious and inflammatory sites.18,92
Acknowledgment
This review was supported by an endowment from the late Dr SM Robbins of Cleveland, OH, USA.
Disclosure
The authors report no conflicts of interest in this work.
References
Baue AE. The horror autotoxicus and multiple-organ failure. Arch Surg. 1992;127(12):1451–1462. | ||
Opal SM. The current understanding of sepsis and research priorities for the future. Virulence. 2014;5(1):1–3. | ||
de Oliveira S, Rosowski EE, Huttenlocher A. Neutrophil migration in infection and wound repair: going forward in reverse. Nat Rev Immunol. 2016;16(6):378–391. | ||
Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013;13(3):159–175. | ||
Hampton MB, Kettle AJ, Winterbourn CC. Inside the neutrophil phagosome: oxidants, myeloperoxidase, and bacterial killing. Blood. 1998;92(9):3007–3017. | ||
Neutrophils LGM. NETs, NETosis – old or new factors in sepsis and septic shock? Anaesthesiol Intensive Ther. 2017;49(3):235–240. | ||
Boomer JS, Green JM, Hotchkiss RS. The changing immune system in sepsis is individualized immuno-modulatory therapy the answer? Virulence. 2014;5(1):45–46. | ||
Horn DL, Morrison DC, Opal SM, Silverstein R, Visvanathan K, Zabriskie JB. What are the microbial components implicated in the pathogenesis of sepsis? Report on a symposium. Clin Infect Dis. 2000;31(4):851–858. | ||
Ginsburg I. The role of bacteriolysis in the pathophysiology of inflammation, infection and post-infectious sequelae. APMIS. 2002;110(11):753–770. | ||
Ginsburg I. Action of streptococcal haemolysins and proteolytic enzymes on Ehrlich ascites tumour cells. Br J Exp Pathol. 1959;40(5):417–423. | ||
Ginsburg I, Ram M. Action of antibodies and plasmin on ehrlich ascites tumour cells. Nature. 1960;185(4709):328–330. | ||
Dallegri F, Ottonello L. Tissue injury in neutrophilic inflammation. Inflamm Res. 1997;46(10):382–391. | ||
Henson PM, Johnston RB. Tissue injury in inflammation. Oxidants, proteinases, and cationic proteins. J Clin Invest. 1987;79(3):669–674. | ||
Baird BR, Cheronis JC, Sandhaus RA, Berger EM, White CW, Repine JE. O2 metabolites and neutrophil elastase synergistically cause edematous injury in isolated rat lungs. J Appl Physiol (1985). 1986;61(6):2224–2229. | ||
Opal SM, Dellinger RP, Vincent JL, Masur H, Angus DC. The next generation of sepsis clinical trial designs: what is next after the demise of recombinant human activated protein C?*. Crit Care Med. 2014;42(7):1714–1721. | ||
Ginsburg I. Multi-drug strategies are necessary to inhibit the synergistic mechanism causing tissue damage and organ failure in post infectious sequelae. Inflammopharmacology. 1999;7(3):207–217. | ||
Ginsburg I. Mechanisms of cell and tissue injury induced by group a streptococci: relation to poststreptococcal sequelae. J Infect Dis. 1972;126(3):294–340. | ||
Ginsburg I. Can hemolytic streptococci be considered “forefathers” of modern phagocytes? Both cell types freely migrate in tissues and destroy host cells by a “synergistic cross-talk” among their secreted agonists. Comp Biochem Physiol C Pharmacol Toxicol Endocrinol. 1994;109(2):147–158. | ||
Ginsburg I. Is streptolysin S of group A streptococci a virulence factor? APMIS. 1999;107(12):1051–1059. | ||
Ginsburg I. Role of lipoteichoic acid in infection and inflammation. Lancet Infect Dis. 2002;2(3):171–179. | ||
Sela MN, Lahav M, Ginsburg I. Effect of leukocyte hydrolases on bacteria – IX. The release of lipoteichoic acid from group A streptococci and from Strep, mutans by leukocyte extracts and by lysozyme: relation to tissue damage in inflammatory sites. Inflammation. 1977;2(2):151–164. | ||
Ginsburg I, Fligiel SEG, Ward PA, Varani J. Lipoteichoic acid-antilipoteichoic acid complexes induce superoxide generation by human neutrophils. Inflammation. 1988;12(6):525–548. | ||
Ginsburg I, Koren E. Bacteriolysis – a mere laboratory curiosity? Crit Rev Microbiol. 2018;44(5):609–618. | ||
Schwab JH, Cromartie WJ, Ohanian SH, Craddock JG. Association of experimental chronic arthritis with the persistence of group A streptococcal cell walls in the articular tissue. J Bacteriol. 1967;94(5):1728–1735. | ||
Ginsburg I. Can chronic and self-perpetuating arthritis in the human be caused by arthrotropic undegraded microbial cell wall constituents? A working hypothesis. Rheumatology. 1977;16(3):141–149. | ||
Fox A, Schwab JH, Cochran T. Muramic acid detection in mammalian tissues by gas-liquid chromatography-mass spectrometry. Infect Immun. 1980;29(2):526–531. | ||
Ginsburg I, Koren E, Feuerstein O. Is bacteriolysis in vivo a friend or a foe? relation to sepsis, chronic granulomatous inflammation and to oral disorders: an overview hypothesis. SOJ Microbiol Infect Dis. 2015;3(1):1–8. | ||
Grumann D, Nübel U, Bröker BM. Staphylococcus aureus toxins – their functions and genetics. Infect Genet Evol. 2014;21:583–592. | ||
Gordon RJ, Lowy FD. Pathogenesis of methicillin-resistant Staphylococcus aureus infection. Clin Infect Dis. 2008;46(Suppl 5):S350–S359. | ||
Rodell TC, Cheronis JC, Repine JE. Endothelial cell xanthine oxidase-derived toxic oxygen metabolites contribute to acute lung injury from neutrophil elastase. Chest. 1988;93(3 Suppl):146S. | ||
Varani J, Ginsburg I, Schuger L, et al. Endothelial cell killing by neutrophils. Synergistic interaction of oxygen products and proteases. Am J Pathol. 1989;135(3):435–438. | ||
Ginsburg I, Gibbs DF, Schuger L, et al. Vascular endothelial cell killing by combinations of membrane-active agents and hydrogen peroxide. Free Radic Biol Med. 1989;7(4):369–376. | ||
Ginsburg I, Misgav R, Pinson A, Varani J, Ward PA, Kohen R. Synergism among oxidants, proteinases, phospholipases, microbial hemolysins, cationic proteins, and cytokines. Inflammation. 1992;16(5):519–538. | ||
Ginsburg I, Mitra RS, Gibbs DF, Varani J, Kohen R. Killing of endothelial cells and release of arachidonic acid – synergistic effects among hydrogen peroxide, membrane-damaging agents, cationic substances, and proteinases and their modulation by inhibitors. Inflammation. 1993;17(3):295–319. | ||
Ginsburg I, Kohen R. Synergistic effects among oxidants, membrane-damaging agents, fatty acids, proteinases, and xenobiotics: killing of epithelial cells and release of arachidonic acid. Inflammation. 1995;19(1):101–118. | ||
Ginsburg I, Kohen R. Invited review: cell damage in inflammatory and infectious sites might involve a coordinated “cross-talk” among oxidants, microbial haemolysins and ampiphiles, cationic proteins, phospholipases, fatty acids, proteinases and cytokines (an overview). Free Radic Res. 1995;22(6):489–517. | ||
Ginsburg I. Could synergistic interactions among reactive oxygen species, proteinases, membrane-perforating enzymes, hydrolases, microbial hemolysins and cytokines be the main cause of tissue damage in infectious and inflammatory conditions? Med Hypotheses. 1998;51(4):337–346. | ||
Koren E, Ginsburg I. Synergistic aspects to explain the pathophysiology of sepsis and septic shock–an opinion. J Infect Dis Ther. 2015;3:6. | ||
Ginhoux F, Jung S. Monocytes and macrophages: developmental pathways and tissue homeostasis. Nat Rev Immunol. 2014;14(6):392–404. | ||
Esser RE, Anderle SK, Chetty C, Stimpson SA, Cromartie WJ, Schwab JH. Comparison of inflammatory reactions induced by intraarticular injection of bacterial cell wall polymers. Am J Pathol. 1986;122(2):323–334. | ||
Ginsburg I, Koren E. Bacteriolysis – a mere laboratory curiosity? Crit Rev Microbiol. 2018;44(5):609–618. | ||
Katchalski E, Bichovski-Slomnitzki L, Volcani BE. Action of some water-soluble poly-α-amino-acids on bacteria. Nature. 1952;169(4313):1095–1096. | ||
Hirsch JG. Bactericidal action of histone. J Exp Med. 1958;108(6):925–944. | ||
Ginsburg I, Sadovnic M. Gamma globulin, Evan’s blue, aprotinin a PLA2 inhibitor, tetracycline and antioxidants protect epithelial cells against damage induced by synergism among streptococcal hemolysins, oxidants and proteinases: relation to the prevention of post-streptococcal sequelae and septic shock. FEMS Immunol Med Microbiol. 1998;22(3):247–256. | ||
Ginsburg I, Ward PA, Varani J. Lysophosphatides enhance superoxide responses of stimulated human neutrophils. Inflammation. 1989;13(2):163–174. | ||
Anderson BO, Brown JM, Harken AH. Mechanisms of neutrophil-mediated tissue injury. J Surg Res. 1991;51(2):170–179. | ||
Ginsburg I. Cationic polyelectrolytes: a new look at their possible roles as opsonins, as stimulators of respiratory burst in leukocytes, in bacteriolysis, and as modulators of immune-complex diseases (a review hypothesis). Inflammation. 1987;11(4):489–515. | ||
Ginsburg I, van Heerden P, Koren E. From amino acids polymers, antimicrobial peptides, and histones, to their possible role in the pathogenesis of septic shock: a historical perspective. J Inflamm Res. 2017; 10:7–15. | ||
Xu J, Zhang X, Pelayo R, et al. Extracellular histones are major mediators of death in sepsis. Nat Med. 2009;15(11):1318–1321. | ||
Chaput C, Zychlinsky A. Sepsis: the dark side of histones. Nat Med. 2009;15(11):1245–1246. | ||
Ginsburg I, Koren E, Varani J, Kohen R. Nuclear histones: major virulence factors or just additional early sepsis markers? A comment. Inflammopharmacology. 2016;24(5):287–289. | ||
Fan G, Rong X, Wang X, et al. [Effect of necrotic wound tissue decomposition products on serum inflammation factors in rabbits]. Nan Fang Yi Ke Da Xue Xue Bao. 2012;32(7):1052–1055. Chinese. | ||
Wildhagen KC, García de Frutos P, Reutelingsperger CP, et al. Nonanticoagulant heparin prevents histone-mediated cytotoxicity in vitro and improves survival in sepsis. Blood. 2014;123(7):1098–1101. | ||
Simmons J, Pittet JF. The coagulopathy of acute sepsis. Curr Opin Anaesthesiol. 2015;28(2):227–236. | ||
Korem M, Koren E, Ginsburg I. The pathogenesis of sepsis: “If we cannot beat them alone join them?”. Int J Microbiol Infect Dis. 2018;2(3):1–5. | ||
Muthas D, Reznichenko A, Balendran CA, et al. Neutrophils in ulcerative colitis: a review of selected biomarkers and their potential therapeutic implications. Scand J Gastroenterol. 2017;52(2):125–135. | ||
Grishin A, Papillon S, Bell B, Wang J, Ford HR. The role of the intestinal microbiota in the pathogenesis of necrotizing enterocolitis. Semin Pediatr Surg. 2013;22(2):69–75. | ||
Ha F, Khalil H. Crohn’s disease: a clinical update. Therap Adv Gastroenterol. 2015;8(6):352–359. | ||
Thompson AM, Bizzarro MJ. Necrotizing enterocolitis in newborns: pathogenesis, prevention and management. Drugs. 2008;68(9):1227–1238. | ||
Eckert J, Scott B, Lawrence S, Ihnat M, Chaaban H. FLLL32, a curcumin analog, ameliorates intestinal injury in necrotizing enterocolitis. J Inflamm Res. 2017;10:75–81. | ||
Sodhi CP, Neal MD, Siggers R, et al. Intestinal epithelial toll-like receptor 4 regulates goblet cell development and is required for necrotizing enterocolitis in mice. Gastroenterology. 2012;143(3):708–718. | ||
Hoffman O, Weber JR. Review: pathophysiology and treatment of bacterial meningitis. Ther Adv Neurol Disord. 2009;2(6):401–412. | ||
Mook-Kanamori BB, Geldhoff M, van der Poll T, van de Beek D. Pathogenesis and pathophysiology of pneumococcal meningitis pathogenesis and pathophysiology of pneumococcal meningitis. Clin Microbiol Rev. 2011;24(3):557–591. | ||
van Aalst M, Lötsch F, Spijker R, et al. Incidence of invasive pneumococcal disease in immunocompromised patients: a systematic review and meta-analysis. Travel Med Infect Dis. 2018;24:89–100. | ||
Loughman JA, Hunstad DA. Attenuation of human neutrophil migration and function by uropathogenic bacteria. Microbes Infect. 2011;13(6):555–565. | ||
Clements A, Young JC, Constantinou N, Frankel G. Infection strategies of enteric pathogenic Escherichia coli. Gut Microbes. 2012;3(2):71–87. | ||
Rosenberg S, Horowitz R, Coppenhagen-Glazer S, et al. Intravesical administration of green tea extract attenuates the inflammatory response of bacterial cystitis – a rat model. BJU Int. 2014;114(4):601–607. | ||
François M, Le Cabec V, Dupont MA, Sansonetti PJ, Maridonneau-Parini I. Induction of necrosis in human neutrophils by Shigella flexneri requires type III secretion, IpaB and IpaC invasins, and actin polymerization. Infect Immun. 2000;68(3):1289–1296. | ||
Stadtman ER, Levine RL. Protein oxidation. Ann N Y Acad Sci. 2000; 899(1):191–208. | ||
Halliwell B, Gutteridge JMC. Free Radicals in Biology and Medicine. 2nd ed. Oxford University Press, Oxford. 1991. | ||
Richards R. The role of erythrocytes in the inactivation of free radicals. Aust J Med Sci. 1996;50:363–367. | ||
Morgan MD, Harper L, Lu X, Nash G, Williams J, Savage CO. Can neutrophils be manipulated in vivo? Rheumatology. 2005;44(5):597–601. | ||
Ginsburg I, Koren E. Are cationic antimicrobial peptides also ‘double-edged swords’? Expert Rev Anti Infect Ther. 2008;6(4):453–462. | ||
Urban CF, Lourido S, Zychlinsky A. How do microbes evade neutrophil killing? Cell Microbiol. 2006;8(11):1687–1696. | ||
do Vale A, Cabanes D, Sousa S. Bacterial toxins as pathogen weapons against phagocytes. Front Microbiol. 2016;7:42. | ||
Madani TA. Clinical infections and bloodstream isolates associated with fever in patients undergoing chemotherapy for acute myeloid leukemia. Infection. 2000;28(6):367–374. | ||
Calandra T, Baumgartner JD, Grau GE, et al. Prognostic values of tumor necrosis factor/cachectin, interleukin-1, interferon-alpha, and interferon-gamma in the serum of patients with septic shock. Swiss-Dutch J5 Immunoglobulin Study Group. J Infect Dis. 1990;161(5):982–987. | ||
Groeneveld AB, Tacx AN, Bossink AW, van Mierlo GJ, Hack CE. Circulating inflammatory mediators predict shock and mortality in febrile patients with microbial infection. Clin Immunol. 2003;106(2):106–115. | ||
Brunialti MK, Martins PS, Barbosa de Carvalho H, Machado FR, Barbosa LM, Salomao R. TLR2, TLR4, CD14, CD11B, and CD11C expressions on monocytes surface and cytokine production in patients with sepsis, severe sepsis, and septic shock. Shock. 2006;25(4):351–357. | ||
Pruitt JH, Welborn MB, Edwards PD, et al. Increased soluble interleukin-1 type II receptor concentrations in postoperative patients and in patients with sepsis syndrome. Blood. 1996;87(8):3282–3288. | ||
Iskander KN, Osuchowski MF, Stearns-Kurosawa DJ, et al. Sepsis: multiple abnormalities, heterogeneous responses, and evolving understanding. Physiol Rev. 2013;93(3):1247–1288. | ||
Zingaretti C, Falugi F, Nardi-Dei V, et al. Streptococcus pyogenes SpyCEP: a chemokine-inactivating protease with unique structural and biochemical features. FASEB J. 2010;24(8):2839–2848. | ||
Xu D, Olson J, Cole JN, et al. Heparan sulfate modulates neutrophil and endothelial function in antibacterial innate immunity. Infect Immun. 2015;83(9):3648–3656. | ||
Hurley SM, Kahn F, Nordenfelt P, Mörgelin M, Sørensen OE, Shannon O. Platelet-dependent neutrophil function is dysregulated by M protein from Streptococcus pyogenes. Infect Immun. 2015;83(9):3515–3525. | ||
Uchiyama S, Döhrmann S, Timmer AM, et al. Streptolysin O rapidly impairs neutrophil oxidative burst and antibacterial responses to Group A Streptococcus. Front Immunol. 2015;6:581. | ||
Sierig G, Cywes C, Wessels MR, Ashbaugh CD. Cytotoxic effects of streptolysin O and streptolysin S enhance the virulence of poorly encapsulated group A streptococci. Infect Immun. 2003;71(1):446–455. | ||
Buchanan JT, Simpson AJ, Aziz RK, et al. DNase expression allows the pathogen group A Streptococcus to escape killing in neutrophil extracellular traps. Curr Biol. 2006;16(4):396–400. | ||
Ato M, Ikebe T, Kawabata H, Takemori T, Watanabe H. Incompetence of neutrophils to invasive group A Streptococcus is attributed to induction of plural virulence factors by dysfunction of a regulator. PLoS One. 2008;3(10):e3455. | ||
Ginsburg I, Quie PG. Modulation of human polymorphonuclear leukocyte chemotaxis by leukocyte extracts, bacterial products, inflammatory exudates, and polyelectrolytes. Inflammation. 1980;4(3):301–311. | ||
Lehr HA, Arfors KE. Mechanisms of tissue damage by leukocytes. Curr Opin Hematol. 1994;1(1):92–99. | ||
Pfaller MA, Messer SA, Moet GJ, Jones RN, Castanheira M. Candida bloodstream infections: comparison of species distribution and resistance to echinocandin and azole antifungal agents in Intensive Care Unit (ICU) and non-ICU settings in the SENTRY Antimicrobial Surveillance Program (2008–2009). Int J Antimicrob Agents. 2011;55(2):561–566. | ||
Ginsburg I, Ward PA, Varani J. Can we learn from the pathogenetic strategies of group A hemolytic streptococci how tissues are injured and organs fail in post-infectious and inflammatory sequelae? FEMS Immunol Med Microbiol. 1999;25(4):325–338. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.