Back to Journals » Clinical Ophthalmology » Volume 13
Primary vitreoretinal lymphoma: prevalence, impact, and management challenges
Authors Venkatesh R ![]() , Bavaharan B, Mahendradas P, Yadav NK
, Bavaharan B, Mahendradas P, Yadav NK
Received 16 November 2018
Accepted for publication 23 January 2019
Published 14 February 2019 Volume 2019:13 Pages 353—364
DOI https://doi.org/10.2147/OPTH.S159014
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Scott Fraser
Ramesh Venkatesh,1 Bharathi Bavaharan,1 Padmamalini Mahendradas,2 Naresh Kumar Yadav1
1Department of Retina and Vitreous, Narayana Nethralaya, Bengaluru – 560010, India; 2Department of Uvea and Intraocular Inflammation, Narayana Nethralaya, Bengaluru – 560010, India
Abstract: Primary vitreoretinal lymphoma (PVRL) is a rare and potentially fatal intraocular malignancy. More than half of PVRL cases eventually involve the central nervous system (CNS). PVRL frequently masquerades as chronic uveitis. Advanced imaging tests, such as optical coherence tomography and fundus autofluorescence, have been applied in the diagnosis of PVRL. Histology and immunohistochemistry, in combination with molecular tests and IL-10 analysis, have been demonstrated as reliable in diagnosing PVRL. Mortality is high in patients with PVRL associated with CNS involvement, and relapses are common. The use of systemic chemotherapy in addition to the local therapies has proved to extend the mean survival time of these patients. Local therapies, including intravitreal injections of methotrexate and/or rituximab and low-dose radiotherapy to the eye, have been shown to be extremely effective in controlling intraocular lymphoma.
Keywords: CNS lymphoma, malignancy, ocular tumor, treatment, investigations, prognosis
Introduction
Primary intraocular lymphoma (PIOL), previously known as ocular reticulum cell sarcoma, was first described by Cooper and Riker1 in 1951, followed by Givner2 in 1955, as a malignant lymphoma of the uveal tract. Lymphomatous proliferations seen in the eye can essentially be subdivided into two groups: 1) those which occur in the vitreous and/or retina and 2) those which occur in the uvea.3 Vitreoretinal lymphomas are lymphomas that arise primarily in the vitreous and/or retina and are considered as a part of the primary central nervous system lymphoma (PCNSL). Uveal lymphomas can be further divided into those which start as a primary disease in the uveal tract or those which occur as an ocular manifestation of systemic non-Hodgkin lymphoma.3,4 In this review, we will concentrate on primary vitreoretinal lymphoma (PVRL) only. PVRL is a subset of PCNSL, where the lymphocytic neoplastic cells primarily affect the retina with or without involving the vitreous or the optic nerve and may not have brain or cerebrospinal fluid (CSF) involvement at presentation.4–6 PVRL is a rare but potentially fatal intraocular malignancy. Ocular manifestations of PCNSL can occur or ultimately develop in approximately a quarter of patients with PCNSL.7,8 With an increase in the incidence of PCNSL in recent times, there has been a similar increase in PVRL incidence worldwide.8,9 PVRLs are usually diffuse large B-cell lymphomas with very few cases of primary T-cell VRL being described in the literature.10,11 Presently, PVRL is associated with a poor prognosis, mainly due to delays in diagnosis and lack of effective therapies.12,13 Systemic chemotherapy, along with ocular chemotherapy/radiation, forms the basis of treatment in PVRL. Many papers have been published recently, highlighting the clinical features, diagnosis, and treatment modalities available in PVRL.14–16 With increase in PVRL cases due to better understanding and diagnosis of the disease, it is important to keep the readers updated of the varying clinical features and newer treatment options available. With this review article, we intend to discuss the clinical and pathological features of PVRL followed by an overview of current diagnostic and treatment options, prognosis, and also highlight the areas where further research can be considered in the management of PVRL.
Demographic profile
PVRL, a rare intraocular malignancy, is a subset of PCNSL It is a rare disease, with an approximate incidence of 0.047 cases per 100,000 people per year.17 This represents 4%–6% of all brain tumors and less than 1% of all non-Hodgkin’s lymphomas.9,18 Fifteen percent of all PCNSL patients have intraocular disease, whereas over 50% of patients with PVRL develop CNS disease.19 PVRL usually affects the adults in the fifth–sixth decades of life.20–22 A few cases of PVRL seen during early childhood and adolescence have also been documented in the literature,23,24 particularly in those who are immunocompromised as a result of treatment or due to HIV.
There seems to be no sex or racial predilection to the disease. However, a few reports suggest women to be more commonly affected than men, by 2:1 or even greater.21,25–28
Etiopathogenesis
The etiology of PIOL/PCNSL is not very clear. Two theories have been implicated in the etiology of PVRL, namely 1) infectious theory and 2) hematological spread. Infections with Epstein–Barr virus or HIV virus, especially in immunocompromised patients, attracts the lymphoid cells while the neoplastic transformation to lymphoid malignancy takes place later, at the eye and/or CNS. This is supported by the finding that Epstein–Barr virus is invariably found in AIDS patients with PCNSL and usually runs a more aggressive clinical course.29 Toxoplasma gondii has also been found in B-cell lymphoma cells in two out of ten PIOL samples, leading to speculation on the possible role of this organism in the etiology of PVRL.30 Another theory is the hematological spread of neoplastic cells from nodal and extranodal sites to ocular and CNS structures.26 According to this theory, B-cell chemokines may selectively attract the lymphoma cells from the choroidal circulation to the retinal pigment epithelium (RPE) and/or retina. B-cell chemokine receptors CXCR4 and CXCR5 were detected in the lymphoma cells, whereas the ligands BLC and SDF-1 were detected only in the RPE, thus supporting this vascular theory.31
Clinical features
Ocular features
PVRL has often been termed as a masquerade syndrome as its clinical signs can mimic a wide variety of ocular diseases. The clinical signs of PVRL vary significantly between patients. The ocular signs are usually bilateral in 64%–83% of the cases, but are often asymmetrical at presentation due to the uneven disease distribution.3,4 Most patients with PVRL visit the ophthalmologist with symptoms of hazy vision and/or floaters. The ocular symptoms are noticed by the patient long before PVRL is suspected. Some studies have reported the incidence of blurred vision in 40%–50% of cases, decreased visual acuity in 25%–30%, and floaters in 20%–25%.3,4 Anterior segment findings are less frequently seen compared to posterior segment ophthalmoscopic findings. Anterior segment findings range from a few anterior chamber cells and keratitic precipitates32,33 to the presence of pseudohypopyon,34,35 and iris and angle infiltration36,37 in rare cases. Examination of the posterior segment reveals vitritis, ranging from mild-to-severe, in the majority of cases. Visual acuity in most cases of PVRL is better than that expected compared to the extent of vitritis on clinical examination. The lymphoma cells in the vitreous cavity may form clumps, strands, sheets, and membranes with mild-to-severe vitreous haze. Lymphoma cells are homogeneous, larger than inflammatory cells, and do not cluster with reactive cells. This gives an “aurora borealis” like appearance from cells lining along the vitreous fibrils. Creamy lesions with yellowish infiltrates located deep in the retina and/or RPE is another important clinical finding seen in PVRL patients.21,33,38,39 They can give rise to a characteristic “leopard skin” pigmentation overlying the mass.40 Other retinal findings include isolated subretinal lesions,41 exudative retinal detachment,21 RPE atrophy with subretinal fibrosis, and disciform scarring at the macula (Figures 1A and 2B). Optic nerve infiltration may also occur.42 In comparison to uveitis cases of similar cellularity, cystoid macular edema is usually absent.
 | Figure 1 Clinical and imaging features of a patient diagnosed with PVRL. (A) Left eye color fundus image of a 56-year-old male treated with oral corticosteroids for recurrent posterior uveitis with vitritis (2+). An ill-defined yellowish lesion is noted at the posterior pole of the fundus and vitreous biopsy confirmed it is a case of intraocular lymphoma (arrow with black outline). (B) Fundus autofluoroscence showing areas of hypo with hyper autofluorescence in the posterior pole of the left eye (arrow with black outline). (C) SDOCT reveals hyperreflective sub-RPE deposits characteristic of intraocular lymphoma (white arrow with black outline). |
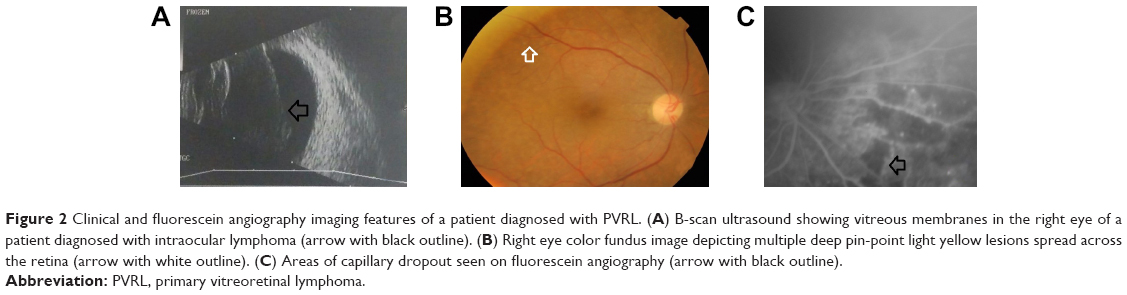 | Figure 2 Clinical and fluorescein angiography imaging features of a patient diagnosed with PVRL. (A) B-scan ultrasound showing vitreous membranes in the right eye of a patient diagnosed with intraocular lymphoma (arrow with black outline). (B) Right eye color fundus image depicting multiple deep pin-point light yellow lesions spread across the retina (arrow with white outline). (C) Areas of capillary dropout seen on fluorescein angiography (arrow with black outline). |
CNS features
At presentation, 16%–34% of PVRL cases have CNS involvement,21,43–45 whereas 13%–25% of patients with PCNSL demonstrate ocular findings.21,46,47 The interval between the onset of ocular or neurological symptoms and a positive diagnosis usually varies from 4 to 40 months, although progression may occur rapidly in some cases.21,48 Neurological symptoms may occur at any time during the disease course and can be focal and/or diffuse in nature. Symptoms like behavioral changes and alteration in cognitive function, focal neurological deficits like hemiparesis and cerebellar signs, including ataxia, are noted in patients with PCNSL. Occurrence of new seizures is a strong indicator of CNS involvement. Infiltration of the meninges by malignant lymphoma cells without intracerebral involvement can also be noted.49 It is estimated that between 42% and 92% of PVRL cases go on to exhibit intracranial lymphoma within a mean interval of <30 months.21,43,44,50,51
Differential diagnosis
Both infectious and noninfectious uveitis can masquerade as PVRL (Table 1). Systemic malignancies with ocular metastasis present with multiple focal pale retina lesions with associated exudative retinal detachment and absence of vitreous involvement. Brownish pigmentary mottling overlying the retinal pigment epithelium in these eyes can resemble the “leopard skin” pattern seen in PVRL.
 | Table 1 Differential diagnosis of primary vitreoretinal lymphoma |
Imaging findings
Angiographic imaging
A wide variety of angiographic features have been described in eyes with PVRL. Cassoux et al21 reported the fluorescein angiographic (FA) findings in 44 patients with PVRL. They observed punctate hyperfluorescent window defects in 55% cases of PVRL followed by circular hypofluorescent lesions in 34% and vasculitis in 14%. Angiographically cystoid macular edema was noted in only 2% of cases.21 Fardeau et al45 studied the angiographic and topographic features of 53 eyes with cytologically proven diagnosis of PVRL. On FA, numerous small hypofluorescent lesions corresponding to punctate whitish lesions in the fundus were associated with the diagnosis of intraocular lymphoma. Indocyanine green angiography (ICGA) also showed round clustered hypofluorescent lesions at the corresponding areas. The hypofluorescent lesions were identified more in number on FA than ICGA. In conjunction, the FA and ICGA findings had a positive predictive value of 89% and a negative predictive value of 85%45 (Figure 2C). Other angiographic features like granularity, blockage, late staining at the level of the RPE, and focal areas of capillary nonperfusion have also been described in the literature.52,53 The hypofluorescent lesions seen on FA in both the early and late phases of the angiogram are mainly due to the accumulation of the lymphoma infiltrates between the retinal pigment epithelium and Bruch membrane, causing blockage of the choroidal fluorescence. The Bruch membrane is impermeable to the lymphoma cells and prevents the malignancy from progressing toward the choroid. This pattern is seen in 45% of eyes with PIOL, compared to 2% of uveitis cases.45
Fundus autofluorescence (FAF)
On FAF images, both hypo- and hyperautofluorescent areas are seen in eyes with PVRL. Hypoautofluorescent areas are suggestive of RPE atrophy or blockage of RPE by the overlying lymphoma cells, whereas hyperautofluorescent spots indicate the overlapping of PVRL and RPE cells. The hyperautofluorescent spots change to hypoautofluorescent spots following treatment with intravitreal methotrexate (Figure 1B). Thus, in recent times, FAF has been used to detect the activity of PVRL and response to treatment.
Optical coherence tomography (OCT)
Recently, OCT has been utilized in the diagnosis and monitoring treatment in patients with PVRL.54 On SD-OCT, lymphoma cells appear as nodular elevated lesions underneath the RPE. This is due to the direct focal infiltration of the retina and pre-Bruch membrane space/sub-RPE space by the malignant lymphoma cells.54,55 Nodular or band hyperreflective spots were noted in 43% of PVRL eyes.56 Other notable features on spectral domain optical coherence tomography (SDOCT) in eyes with severe PVRL include disruption of the photoreceptor inner segment/outer segment junction, multiple hyperreflective infiltrations within the inner retina, and exudative retinal detachment with subretinal hyporeflective fluid. Cystoid macular edema is not frequently reported. Resolution of the lymphoma deposits along Bruch’s membrane was seen on the OCT after intravitreal chemotherapy.57 This noninvasive method may be convenient for further monitoring of relapse or resolution of PVRL. However, these OCT findings of PVRL needs to be differentiated from those seen in age-related macular degeneration or idiopathic polypoidal choroidal vasculopathy (Figure 1C).58
B-scan ultrasound imaging
The most common findings seen on ophthalmic ultrasound are sheets of vitreous debris, retinal detachment, elevated chorioretinal lesions, and optic nerve widening (Figure 2A). However, none of these changes are specific for PVRL.
Neuroimaging
In a mean interval of 8–29 months, 42%–92% of PVRL cases presented with intracranial lymphoma.21,43,44,50,51 Thus, it is imperative for every patient to undergo contrast-enhanced MRI on a regular basis during the disease course after the definitive diagnosis of PIOL has been made.59 MRI helps in identifying CNS involvement and in monitoring the treatment response of neurological lesions following high-dose systemic chemotherapy. On MRI, lymphoma lesions appear hypodense on T1-weighted and hyperdense on T2-weighted scans with discrete or diffuse borders.47 Positron emission tomography/computed tomography has been used to identify both CNS and ocular activity, both of which take up the 2-[(18)F] fluoro-2-deoxy-D-glucose dye.57
The characteristic features of different imaging modalities are mentioned in Table 2.
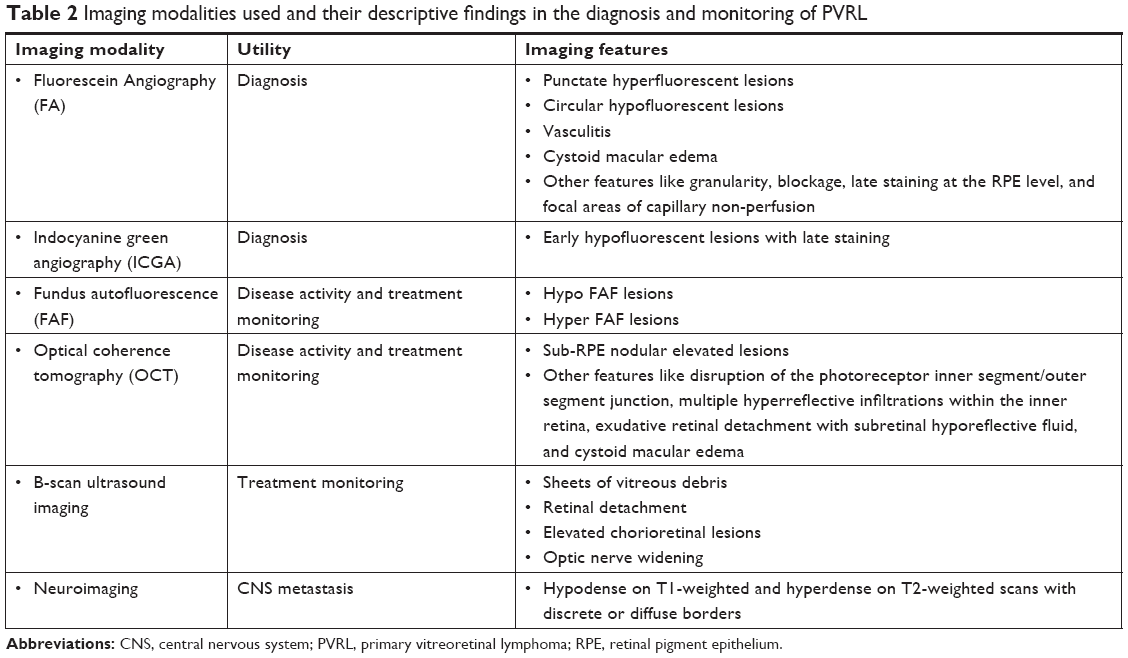 | Table 2 Imaging modalities used and their descriptive findings in the diagnosis and monitoring of PVRL |
Biopsy
Ocular tissue biopsy
Identification of the malignant lymphoid cells in ocular tissue using biopsy still remains the cornerstone of diagnosis and treatment in malignant lymphomas, including PVRL.60,61 Usually, vitreous biopsy either by vitrectomy or direct vitreous aspiration is performed, especially when CSF evaluation is negative or when intracranial lymphoma lesions are not identified on neuroimaging. Clinically, pars plana vitrectomy is better than vitreous aspirate as it provides a superior diagnostic yield.61 In addition, pars plana vitrectomy helps in improving vision by clearing the vitreous debris and maximizing the sample size,60,61 although extension of the lymphoma through the sclerotomy port to the epibulbar space following vitrectomy may occur.62 A small gage transconjunctival sutureless vitrectomy performed using the 25 G or 27 G instrumentation is a safe and effective technique for the diagnosis of PVRL.63 A low cut rate of 600 cpm is recommended in order to obtain a higher diagnostic yield and more viable lymphoma cells.64 Routinely, both the undiluted vitreous (1–2 mL) and diluted vitreous samples are obtained as much as possible after turning on the infusion. Retinal biopsy is seldom used clinically, and retinal samples are usually taken from the deeper part of the lesion, near the choriocapillaris, where viable lymphoma cells are most likely to be found.61 Aqueous aspirate is used in special conditions, such as pseudohypopyon. It is important to transfer the sample immediately to the pathologist once it has been obtained. Biopsy results have a high false-negative rate (about 70%). This is mainly due to the poor sample volume, paucicellular specimens, and necrotic lymphoma cells. Hence, more than one biopsy is often required to make a confident diagnosis in PVRL cases.5 In many cases, the paucity of the cells in the biopsy specimens may be the result of corticosteroid treatment, which must be discontinued for at least 2 weeks before the procedure.65
Cerebrospinal fluid
CSF is obtained in cases with suspected PCNSL. About 25% of patients with identifiable lesions on MRI will have positive CSF cytology.66,67 If a diagnosis of PCNSL can be made from the identification of lymphoma cells in CSF, and there is simultaneous ocular involvement, then the patient is spared from the need for invasive ocular tissue biopsy for diagnosis.
Cytology and histopathology
The diagnosis of PVRL requires a multi-disciplinary approach. The pathologist’s role is to accurately identify the malignant lymphoma cells on the basis of morphology, and then to assess the clonality using immunocytochemistry and molecular techniques like flow cytometry and PCR analysis. Morphologically, the typical lymphoma cells are large B-cell lymphoid cells with a scanty basophilic cytoplasm, an increased nucleus:cytoplasm ratio, with hypersegmented, multivariable-shaped nuclei, multiple nucleoli, and a coarse chromatin pattern.43,44,68,69 The correlation between pathological diagnosis and clinical diagnosis on long-term follow-up is as high as 96%.44 Davis et al70 studied the vitreous sample using cytology evaluation, flow cytometry, and culture techniques in 28 patients with suspected intraocular lymphoma based on their clinical features. Vitreous evaluation confirmed the diagnosis of PVRL in 14 of the 28 (50%) patients. Thirteen out of the 27 vitreous samples that underwent cytological evaluation tested positive for lymphoma. In only four samples with positive cytological evaluation, the clinical diagnosis of lymphoma was confirmed. In the remaining ten eyes with confirmed diagnosis of lymphoma, cytology was unable to identify the abnormal malignant lymphoid cells. Thus, for lymphoma, the positive predictive value of cytologic evaluation was 30.8% (95% CI =12.7–57.6) and the negative predictive value was 33.3% (95% CI=15.2–58.3).70,71 This is due to the sparse number of cells in vitreous sample and the contamination of the vitreous specimen with other cellular structures like reactive T-lymphocytes, necrotic cells, debris, and fibrin.44 Molecular testing of vitreous in 80 eyes of PVRL showed the IL-10:IL-6 >1 in 56 out of 80 eyes; 53/56 had the correct diagnosis of PVRL. In 24 eyes with IL-10:IL-6 <1, 17 eyes had the correct diagnosis of uveitis. Thus, the positive predictive value of molecular testing for lymphoma was 94.6% (95% CI =85.4–98.5) and the negative predictive value was 70.8% (95% CI =50.8–85.1).71 In cases with scanty cytology, phenotyping of cells by their surface markers are useful in identifying the lymphomatous cells. Immunocytological techniques use a cell-mounted slide with antibodies directed at specific cell markers (Figure 3).72,73 In flow cytometry, the cells are separated using a fluorescence activated cell sorter in a fluid medium. Immunocytology increases the rate of diagnosis from 30% (using cytology alone) to 70%74 but requires more cells. Flow cytometry allows for multiple monoclonal antibodies to be applied to an aliquot of suspected lymphoma cells simultaneously, which allows the use of a larger detection panel.75–77 In B-cell lymphomas, a B-cell population stains positively for B-cell markers (CD19, CD20, or CD22) with restricted expression of either κ or λ. In T-cell lymphomas, the T-cell population stains positively for T-cell markers (CD3, CD4).57 A ratio of κ:λ of >3 or <0.6 is useful as a marker for clonality.57
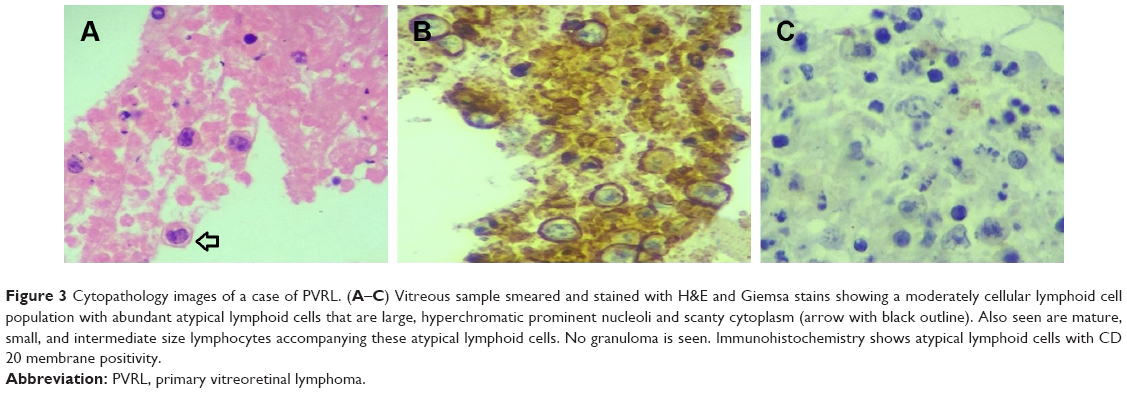 | Figure 3 Cytopathology images of a case of PVRL. (A–C) Vitreous sample smeared and stained with H&E and Giemsa stains showing a moderately cellular lymphoid cell population with abundant atypical lymphoid cells that are large, hyperchromatic prominent nucleoli and scanty cytoplasm (arrow with black outline). Also seen are mature, small, and intermediate size lymphocytes accompanying these atypical lymphoid cells. No granuloma is seen. Immunohistochemistry shows atypical lymphoid cells with CD 20 membrane positivity. |
PCR analysis
Immunoglobulin heavy chain (IgH) gene sequences, particularly in the third complementarity-determining region (CDR3) of the variable region of IgH and T-cell receptor gene rearrangements have been used in assessing the clonal expansion of B and T lymphocytes, respectively. Studies using PCR on systemic B-cell lymphoma78 and in PIOL79–81 reveal rearrangement of IgH gene and bcl-2 proto-oncogene translocations. Cellular microdissection and PCR amplification can be applied together if malignant cells are too scanty to yield a greater cell population from vitreous specimens.71,81–83 Wang et al,71 in 114 cases of B-cell PIOL using this technique, showed rearrangement of the CDR3 of IgH gene in all, but this change was found in only one of 86 cases of uveitis. PCR-based studies have suggested that the B-cells of PIOL are mature B-cells that have undergone the germinal center reaction and the identical clone can be identified in ocular and cerebral tissues in cases of CNS involvement.84
Biochemical tests
High levels of IL-10 are produced by B-cell lymphomas. IL-10 is a Th2 cytokine and linked to rapid disease progression. Normal inflammatory cells secrete high levels of IL-6.4,85 Chan et al86 were the first to report high levels of vitreous IL-10 in three B-cell PVRL patients. IL-10 is elevated in the presence of malignant B-lymphocytes in PIOL/PCNSL, whereas IL-6 or IL-12 is elevated in inflammatory states.86–89 Higher IL-10 levels have been correlated with more aggressive disease.90 Currently, a IL-10:IL-6 ratio >1 in ocular fluid has become an adjunctive and supportive biomarker for the diagnosis and prognosis of PVRL, particularly the B-cell PVRL.4,57
Blood tests
Blood investigations should be done to rule out other causes of infectious and noninfectious uveitis.91 Additionally a complete blood count and HIV virus testing is useful.18
Others
The presence of MYD88 mutations, especially L265P, are very frequent in VRL, and their detection significantly improves the diagnostic yield of vitrectomy specimens.92,93
The interpretation of any test should be done in the context of clinical findings and should involve a multi-disciplinary approach.
Treatment
PVRLs are radiosensitive and chemosensitive tumors. The overall mortality rate of PVRL is quite high, ranging between 9% and 81% in follow-up periods ranging from 12 months to 35 months.3,25,38,43 A professional team of experts in ophthalmology, oncology (particularly neuro-oncology or hematooncologist), and pathology are essential for optimizing patient management.85 The International Primary CNS Lymphoma Collaborative Group in 2011 recommended the guidelines for the treatment and follow-up of patients with PVRL with/without PCNSL.4 In 2015, the European Association of neuro-oncologists laid the guidelines for the diagnosis and treatment of PCNSL94 (Figure 4). As per the recent guidelines, the first line of treatment of PIOL is high-dose systemic chemotherapy along with intravitreal chemotherapy and/or ocular radiotherapy, even in the absence of PCNSL. In patients with contraindications for systemic chemotherapy or for elderly patients with relapsing intraocular disease, local treatment alone (intravitreal chemotherapy or ocular radiotherapy) is an acceptable and valid approach.94
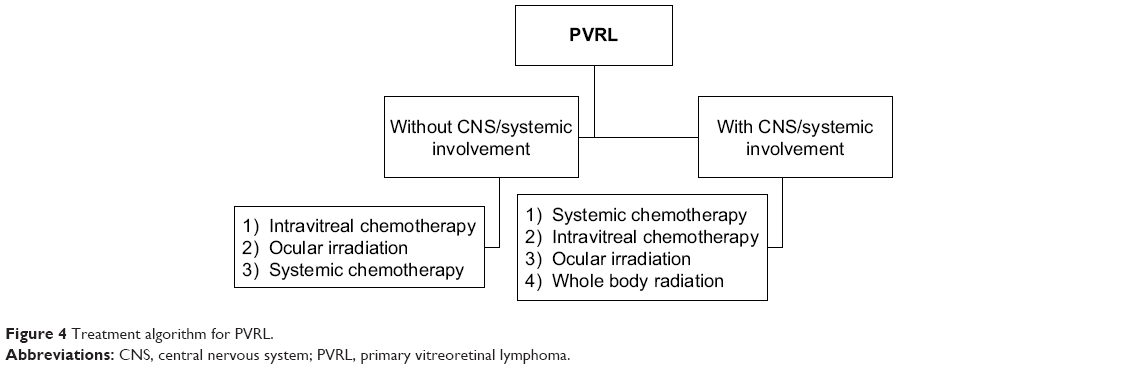 | Figure 4 Treatment algorithm for PVRL. |
Radiotherapy
Ocular irradiation using low-dose external beam radiotherapy has been recommended as local treatment for ocular only PVRL patients.4 Berenbom et al25 treated 12 patients of PVRL with radiotherapy at a dose of 30–35 Gy in 15 fractions during a mean follow-up of 19 months. No ocular relapses were reported in any of these patients. If PVRL patients having concurrent CNS involvement failed systemic chemotherapy and were also unavailable to aggressive therapy, whole brain and eye radiotherapy may be added. The complication of whole brain radiation often induces a delayed neurotoxicity with a decrease in cognitive function, ataxia, and sometimes even death, whereas ocular complications were cataract, dry eye, and radiation retinopathy.25,95 Currently, whether to use intravitreal chemotherapy or ocular radiation as first-line therapy is still controversial.
Systemic chemotherapy
High-dose chemotherapy with intravenous methotrexate is the first line agent used in the treatment of patients with PVRL with CNS or systemic involvement. The efficiency of high-dose methotrexate is demonstrated to produce a remission rate of up to 72% when used alone, and up to 94%–100% in combinations.43,78 Median survival improved from 28 to 85 months in the high-dose chemotherapy group.96 High dose chemotherapy with thiotepa, busulfan, and cyclophosphamide, followed by autologous peripheral blood stem cell transplantation, is reserved for cases demonstrating relapse or refractory response.96–99 Recurrent PIOL with PCNSL has been treated with intrathecal methotrexate and cytarabine.98,100 Oral trofosphamide or intravenous iraphosphamide may produce 6–18 months of progression-free survival.101–104
The IELSG32 trial provided a high level of evidence supporting the use of MATRix combination (methotrexate, cytarabine, thiotepa, and rituximab) as the new standard chemoimmunotherapy for patients aged up to 70 years with newly diagnosed primary CNS lymphoma.105 At median follow-up of 30 months (IQR = 22–38), patients treated with MATRix regime had a complete remission rate of 49% (95% CI =38–60), compared with 23% (14–31) of those treated with methotrexate-cytarabine alone (HR =0.46, 95% CI =0.28–0.74), and 30% (21–42) of those treated with methotrexate-cytarabine plus rituximab (0.61, 0.40–0.94).
Role of systemic chemotherapy and/or irradiation to prevent CNS involvement
In a study by Hormigo et al106 to investigate the role of prophylactic aggressive therapy for ocular only disease, 17 PVRL patients treated with chemotherapy and/or irradiation had a median survival period of 60 months after onset of ocular symptoms, compared to 35 months for 14 patients treated only after CNS disease developed. Another study also demonstrated that prophylactic systemic chemotherapy did not inhibit, but did significantly delay the onset of CNS involvement in PVRL patients.107
Ocular chemotherapy
Two local chemotherapy agents used in the treatment of PVRL are intravitreal methotrexate and intravitreal rituximab.
Intravitreal methotrexate
Local tumor control can be achieved with intravitreal chemotherapy with methotrexate at a dose of 400 μg in 0.1 mL in relapsed PIOL,108–110 in ocular relapse of PCNSL,111 and as a primary treatment in combination with systemic chemotherapy,112 intrathecal chemotherapy,113 or sub-Tenon steroid injections.114 The schedule for intravitreal methotrexate injections was bi-weekly for 4 weeks, every week for 8 weeks, and then monthly for 9 months, comprising a total of 25 injections. Remission was usually reached after a mean of 6.4 injections, with only 5% of the eyes requiring ≥13 injections. In a 10-year study using intravitreal injections of methotrexate as first line treatment in PVRL, none of the total 26 patients had an intraocular recurrence.115 The common complications were transient intraocular pressure elevations and corneal epitheliopathy.85,116
Intravitreal rituximab
Rituximab is an anti-CD20 monoclonal antibody, used recently to treat CD20-positive PVRL.96 Intravitreal injections of 1 mg/0.1 mL-rituximab weekly for 4 weeks as a one-course protocol showed encouraging results in 20 eyes with CD20-positive PVRL. However, 50% of the cases showed recurrences in the study.117 Relapses following intravitreal rituximab may require treatment with intravitreal methotrexate and radiation.118 Rituximab may be a less toxic alternative to methotrexate.
Newer emerging therapies
In recent times, research groups have been testing newer innovative methods for treating PIOL. This includes the use of membrane FasL vesicles, the membrane-only form of Fas ligand, to activate innate immunity and terminate the immune privilege of the eye.119 Recombinant immunotoxin therapy is an emerging immunotherapeutic approach, where a single intravitreal injection of immunotoxin HA22 into eyes of a mouse model of intraocular lymphoma resulted in complete regression of the tumor, demonstrating B-cell-specific immunotoxin therapy.120,121 Other monoclonal antibodies, such as daclizumab, efalizumab, and alemtuzumab, showed positive results in animal models, and had the potential for useful adjuvant therapy for ocular lymphoma.122 Venetoclax, a BCL-2 inhibitor, has been used for the treatment of patients with chronic lymphocytic leukemia (CLL), small lymphocytic lymphoma (SLL), or diffuse large B-cell lymphoma.123 The use of stem cell transplantation for PVRL has been described in the literature. However, limited data is available on the use of high-dose chemotherapy and autologous stem cell transplantation in the treatment of PVRL, with most trials either including only PCNSL or combining PCNSL with PVRL. The results of autologous stem cell transplantation in the treatment of PVRL seems to be promising, and can be considered mainly for refractory and recurrent cases of PVRL.96–99,124 Newer drugs like pomalidomide,125 ibrutinib,126 and a combination of lenalidomide and rituximab127 have also been tried in the treatment of PCNSL and PVRL.
Prognosis
The diagnosis of PVRL is often delayed. The delay in the diagnosis is due to the late presentation to the ophthalmologist. Most patients of PVRL present persistent floaters affecting their visual quality with relatively better visual acuity. The reporting of mortality in the literature is inconsistent due to variations in patient populations, variation in treatment regimens, and small case series. Mortality rates range between 9% and 81% in follow-up periods, ranging from 12 to 35 months.3,25,38,43 In PCNSL, median survival of patients treated with radiotherapy with/without chemotherapy ranged from 10 to 16 months, which can be extended to greater than 30 months with methotrexate-based chemotherapy128 or by ifosfamide or trofosfamide.105 Grimm et al67 reported a median progression-free interval of 18 months and overall survival period of 31 months in 221 patients with PCNSL with intraocular involvement collected from 16 centers in seven countries receiving a mix of therapies. They concluded that ocular therapy improved local tumor control but had no effect on survival. In contrast, another series found a survival advantage if PIOL was diagnosed before CNS involvement 60 months survival vs 35 months, although the group with initial ocular diagnosis was younger.95
Conclusion
PVRL is a masquerading ocular disease, making it difficult to diagnose and treat. With recent advances in diagnostic imaging and molecular techniques, the detection of this highly aggressive disease has improved. High relapse rates and CNS involvement are common, which result in poor prognosis of PVRL, despite aggressive treatment with chemotherapy and/or radiotherapy. Further, with the advent of new targeted therapies for both PVRL and PCNSL, there is hope that, if PVRL is detected sufficiently early, the prognosis will improve. Collaborative multicenter efforts are essential to improve data collection to have a better understanding of the pathogenesis and biology of this disease. In PVRL, despite the multiple encouraging diagnostic and therapeutic advances that have been achieved, the optimal standard clinical diagnosis and treatment strategies still merit further research.
Disclosure
The authors report no conflicts of interest in this work.
References
Cooper EL, Riker JL. Malignant lymphoma of the uveal tract. Am J Ophthalmol. 1951;34(8):1153–1158. | ||
Givner I. Malignant lymphoma with ocular involvement: a clinico-pathologic report. Am J Ophthalmol. 1955;39(1):29–32. | ||
Coupland SE, Damato B. Understanding intraocular lymphomas. Clin Exp Ophthalmol. 2008;36(6):564–578. | ||
Chan CC, Rubenstein JL, Coupland SE, et al. Primary vitreoretinal lymphoma: a report from an International Primary Central Nervous System Lymphoma Collaborative Group Symposium. Oncologist. 2011;16(11):1589–1599. | ||
Coupland SE, Chan CC, Smith J. Pathophysiology of retinal lymphoma. Ocul Immunol Inflamm. 2009;17(4):227–237. | ||
Char DH, Ljung BM, Deschênes J, Miller TR. Intraocular lymphoma: immunological and cytological analysis. Br J Ophthalmol. 1988;72(12):905–911. | ||
Mochizuki M, Singh AD. Epidemiology and clinical features of intraocular lymphoma. Ocul Immunol Inflamm. 2009;17(2):69–72. | ||
Schabet M. Epidemiology of primary CNS lymphoma. J Neurooncol. 1999;43(3):199–201. | ||
Sagoo MS, Mehta H, Swampillai AJ, et al. Primary intraocular lymphoma. Surv Ophthalmol. 2014;59(5):503–516. | ||
Kluin PM, Deckert M, Ferry JA. Primary diffuse large B-cell lymphoma of the CNS. In: Swerdlow SH, Campo E, Harris NL, editors. WHO Classification of Tumours of the Haematopoietic and Lymphoid Tissues. Lyon, France: IARC Press; 2008:240–241. | ||
Coupland SE, Anastassiou G, Bornfeld N, Hummel M, Stein H. Primary intraocular lymphoma of T-cell type: report of a case and review of the literature. Graefes Arch Clin Exp Ophthalmol. 2005;243(3):189–197. | ||
Fend F, Ferreri AJ, Coupland SE. How we diagnose and treat vitreoretinal lymphoma. Br J Haematol. 2016;173(5):680–692. | ||
Grimm SA, Pulido JS, Jahnke K, et al. Primary intraocular lymphoma: an International Primary Central Nervous System Lymphoma Collaborative Group Report. Ann Oncol. 2007;18(11):1851–1855. | ||
Pulido JS, Johnston PB, Nowakowski GS, Castellino A, Raja H. The diagnosis and treatment of primary vitreoretinal lymphoma: a review. Int J Retina Vitreous. 2018;4(1):18. | ||
Araujo I, Coupland SE. Primary vitreoretinal lymphoma: a review. Asia Pac J Ophthalmol. 2017;6(3):283–289. | ||
Reichstein D. Primary vitreoretinal lymphoma: an update on pathogenesis, diagnosis and treatment. Curr Opin Ophthalmol. 2016;27(3):177–184. | ||
Levasseur SD, Wittenberg LA, White VA. Vitreoretinal lymphoma: a 20-year review of incidence, clinical and cytologic features, treatment, and outcomes. JAMA Ophthalmol. 2013;131(1):50–55. | ||
Meunier J, Lumbroso-Le Rouic L, Vincent-Salomon A, et al. Ophthalmologic and intraocular non-Hodgkin’s lymphoma: a large single centre study of initial characteristics, natural history, and prognostic factors. Hematol Oncol. 2004;22(4):143–158. | ||
Pe’er J, Hochberg FH, Foster CS. Clinical review: treatment of vitreoretinal lymphoma. Ocul Immunol Inflamm. 2009;17(5):299–306. | ||
Brown SM, Jampol LM, Cantrill HL. Intraocular lymphoma presenting as retinal vasculitis. Surv Ophthalmol. 1994;39(2):133–140. | ||
Cassoux N, Merle-Beral H, Leblond V, et al. Ocular and central nervous system lymphoma: clinical features and diagnosis. Ocul Immunol Inflamm. 2000;8(4):243–250. | ||
Hoffman PM, Mckelvie P, Hall AJ, Stawell RJ, Santamaria JD. Intraocular lymphoma: a series of 14 patients with clinicopathological features and treatment outcomes. Eye. 2003;17(4):513–521. | ||
Wender A, Adar A, Maor E, Yassur Y. Primary B-cell lymphoma of the eyes and brain in A 3-year-old boy. Arch Ophthalmol. 1994;112(4):450–451. | ||
Sobrin L, Dubovy SR, Davis JL, Murray TG, Isolated MTG. Isolated, bilateral intraocular lymphoma in a 15-year-old girl. Retina. 2005;25(3):370–373. | ||
Berenbom A, Davila RM, Lin H-S, Harbour JW. Treatment outcomes for primary intraocular lymphoma: implications for external beam radiotherapy. Eye. 2007;21(9):1198–1201. | ||
Buettner H, Bolling JP. Intravitreal large-cell lymphoma. Mayo Clin Proc. 1993;68(10):1011–1015. | ||
Peterson K, Gordon KB, Heinemann MH, Deangelis LM. The clinical spectrum of ocular lymphoma. Cancer. 1993;72(3):843–849. | ||
Qualman SJ, Mendelsohn G, Mann RB, Green WR. Intraocular lymphomas. Natural history based on a clinicopathologic study of eight cases and review of the literature. Cancer. 1983;52(5):878–886. | ||
Margolis L, Fraser R, Lichter A, Char DH. The role of radiation therapy in the management of ocular reticulum cell sarcoma. Cancer. 1980;45(4):688–692. | ||
Shen DF, Herbort CP, Tuaillon N, Buggage RR, Egwuagu CE, Chan CC. Detection of Toxoplasma gondii DNA in primary intraocular B-cell lymphoma. Mod Pathol. 2001;14(10):995–999. | ||
Chan CC, Shen D, Hackett JJ, Buggage RR, Tuaillon N. Expression of chemokine receptors, CXCR4 and CXCR5, and chemokines, BLC and SDF-1, in the eyes of patients with primary intraocular lymphoma. Ophthalmology. 2003;110(2):421–426. | ||
Coupland SE, Heimann H, Bechrakis NE. Primary intraocular lymphoma: a review of the clinical, histopathological and molecular biological features. Graefes Arch Clin Exp Ophthalmol. 2004;242(11):901–913. | ||
Buggage RR, Chan CC, Nussenblatt RB. Ocular manifestations of central nervous system lymphoma. Curr Opin Oncol. 2001;13(3):137–142. | ||
Corriveau C, Easterbrook M, Payne D. Lymphoma simulating uveitis masquerade syndrome. Can J Ophthalmol. 1986;21(4):144–149. | ||
Lobo A, Larkin G, Clark BJ, Towler HM, Lightman S. Pseudo-hypopyon as the presenting feature in B-cell and T-cell intraocular lymphoma. Clin Exp Ophthalmol. 2003;31(2):155–158. | ||
Velez G, de Smet MD, Whitcup SM, Robinson M, Nussenblatt RB, Chan CC. Iris involvement in primary intraocular lymphoma: report of two cases and review of the literature. Surv Ophthalmol. 2000;44(6):518–526. | ||
Chan SM, Hutnik CM, Heathcote JG, Orton RB, Banerjee D. Iris lymphoma in a pediatric cardiac transplant recipient: clinicopathologic findings. Ophthalmology. 2000;107(8):1479–1482. | ||
Char DH, Ljung BM, Miller T, Phillips T. Primary intraocular lymphoma (ocular reticulum cell sarcoma) diagnosis and management. Ophthalmology. 1988;95(5):625–630. | ||
Hormigo A, Deangelis LM. Primary ocular lymphoma: clinical features, diagnosis, and treatment. Clin Lymphoma. 2003;4(1):22–29. | ||
Gass JD, Sever RJ, Grizzard WS, et al. Multifocal pigment epithelial detachments by reticulum cell sarcoma: a characteristic Funduscopic picture. Retina. 1984;4(3):135–143. | ||
Levy-Clarke GA, Byrnes GA, Buggage RR, et al. Primary intraocular lymphoma diagnosed by fine needle aspiration biopsy of a subretinal lesion. Retina. 2001;21(3):281–284. | ||
Hedayatfar A, Phaik Chee S. Presumptive primary intraocular lymphoma presented as an intraocular mass involving the optic nerve head. J Ophthalmic Inflamm Infect. 2012;2(1):49–51. | ||
Akpek EK, Ahmed I, Hochberg FH, et al. Intraocular-central nervous system lymphoma: clinical features, diagnosis, and outcomes. Ophthalmology. 1999;106(9):1805–1810. | ||
Coupland SE, Bechrakis NE, Anastassiou G, et al. Evaluation of vitrectomy specimens and chorioretinal biopsies in the diagnosis of primary intraocular lymphoma in patients with masquerade syndrome. Graefes Arch Clin Exp Ophthalmol. 2003;241(10):860–870. | ||
Fardeau C, Lee CP, Merle-Béral H, et al. Retinal fluorescein, indocyanine green angiography, and optic coherence tomography in non-Hodgkin primary intraocular lymphoma. Am J Ophthalmol. 2009;147(5):886–894. | ||
Ferreri AJM, Blay JY, Reni M, et al. Relevance of intraocular involvement in the management of primary central nervous system lymphomas. Ann Oncol. 2002;13(4):531–538. | ||
Herrlinger U. Primary CNS lymphoma: findings outside the brain. J Neurooncol. 1999;43(3):227–230. | ||
Hoffman PM, Mckelvie P, Hall AJ, Stawell RJ, Santamaria JD. Intraocular lymphoma: a series of 14 patients with clinicopathological features and treatment outcomes. Eye. 2003;17(4):513–521. | ||
Hochberg FH, Miller DC. Primary central nervous system lymphoma. J Neurosurg. 1988;91:835–853. | ||
Freeman LN, Schachat AP, Knox DL, Michels RG, Green WR. Clinical features, laboratory investigations, and survival in ocular reticulum cell sarcoma. Ophthalmology. 1987;94(12):1631–1639. | ||
Peterson K, Gordon KB, Heinemann MH, Deangelis LM. The clinical spectrum of ocular lymphoma. Cancer. 1993;72(3):843–849. | ||
Venkatesh R, Gurav P, Abhishek Dave P, Gandhi A. Capillary dropout: a novel fluorescein angiography finding in primary vitreoretinal lymphoma. Ocul Oncol Pathol. 2017;3(4):324–327. | ||
Velez G, Chan CC, Csaky KG. Fluorescein angiographic findings in primary intraocular lymphoma. Retina. 2002;22(1):37–43. | ||
Egawa M, Mitamura Y, Hayashi Y, Naito T. Spectral-domain optical coherence tomographic and fundus autofluorescence findings in eyes with primary intraocular lymphoma. Clin Ophthalmol. 2014;8:335–341. | ||
Jang HS, Sepah YJ, Sophie R, et al. Longitudinal spectral domain optical coherence tomography changes in eyes with intraocular lymphoma. J Ophthalmic Inflamm Infect. 2013;3(1):59. | ||
Casady M, Faia L, Nazemzadeh M, Nussenblatt R, Chan CC, Sen HN. Fundus autofluorescence patterns in primary intraocular lymphoma. Retina. 2014;34(2):366–372. | ||
Davis JL. Intraocular lymphoma: a clinical perspective. Eye. 2013;27(2):153–162. | ||
Mulay K, Narula R, Honavar SG. Primary vitreoretinal lymphoma. Indian J Ophthalmol. 2015;63(3):180–186. | ||
Sen HN, Bodaghi B, Hoang PL, Nussenblatt R. Primary intraocular lymphoma: diagnosis and differential diagnosis. Ocul Immunol Inflamm. 2009;17(3):133–141. | ||
Char DH, Kemlitz AE, Miller T. Intraocular biopsy. Ophthalmol Clin North Am. 2005;18(1):177–185. | ||
Gonzales JA, Chan CC. Biopsy techniques and yields in diagnosing primary intraocular lymphoma. Int Ophthalmol. 2007;27(4):241–250. | ||
Cursiefen C, Holbach LM, Lafaut B, Heimann K, Kirchner T, Naumann GO. Oculocerebral non-Hodgkin’s lymphoma with uveal involvement: development of an epibulbar tumor after vitrectomy. Arch Ophthalmol. 2000;118(10):1437–1440. | ||
Yeh S, Weichel ED, Faia LJ, et al. 25-gauge transconjunctival sutureless vitrectomy for the diagnosis of intraocular lymphoma. Br J Ophthalmol. 2010;94(5):633–638. | ||
Jiang T, Zhao Z, Chang Q. Evaluation of cytologic specimens obtained during experimental vitreous biopsy using B-cell lymphoma line. Eur J Ophthalmol. 2014;24(6):911–917. | ||
Sm W, Chan CC, Rr B, et al. Improving the diagnostic yield of vitrectomy for intraocular lymphoma. Arch Ophthalmol. 2000;118:446. | ||
Deangelis LM. Primary central nervous system lymphomas. Curr Treat Options Oncol. 2001;2(4):309–318. | ||
Grimm SA, Mccannel CA, Omuro AM, et al. Primary CNS lymphoma with intraocular involvement: International PCNSL Collaborative Group Report. Neurology. 2008;71(17):1355–1360. | ||
Karma A, von Willebrand EO, Tommila PV, Paetau AE, Oskala PS, Immonen IJ. Primary intraocular lymphoma: improving the diagnostic procedure. Ophthalmology. 2007;114(7):1372–1377. | ||
Ljung BM, Char D, Miller TR, Deschenes J. Intraocular lymphoma: cytologic diagnosis and the role of immunologic markers. Acta Cytol. 1988;32(6):840–847. | ||
Davis JL, Miller DM, Ruiz P. Diagnostic testing of vitrectomy specimens. Am J Ophthalmol. 2005;140(5):822–829. | ||
Wang Y, Shen D, Wang VM, Sen HN, Chan CC. Molecular biomarkers for the diagnosis of primary vitreoretinal lymphoma. Int J Mol Sci. 2011;12(9):5684–5697. | ||
Davis JL, Solomon D, Nussenblatt RB, Palestine AG, Chan CC. Immunocytochemical staining of vitreous cells indications, techniques, and results. Ophthalmology. 1992;99(2):250–256. | ||
Intzedy L, Teoh SC, Hogan A, et al. Cytopathological analysis of vitreous in intraocular lymphoma. Eye. 2008;22(2):289–293. | ||
Davis JL, Viciana AL, Ruiz P. Diagnosis of intraocular lymphoma by flow cytometry. Am J Ophthalmol. 1997;124(3):362–372. | ||
Raparia K, Chang CC, Chévez-Barrios P. Intraocular lymphoma: diagnostic approach and immunophenotypic findings in vitrectomy specimens. Arch Pathol Lab Med. 2009;133(8):1233–1237. | ||
Zaldivar RA, Martin DF, Holden JT, Grossniklaus HE. Primary intraocular lymphoma: clinical, cytologic, and flow cytometric analysis. Ophthalmology. 2004;111(9):1762–1767. | ||
Missotten T, Tielemans D, Bromberg JE, et al. Multicolor flowcytometric immunophenotyping is a valuable tool for detection of intraocular lymphoma. Ophthalmology. 2013;120(5):991–996. | ||
Abdel-Reheim FA, Edwards E, Arber DA. Utility of a rapid polymerase chain reaction panel for the detection of molecular changes in B-cell lymphoma. Arch Pathol Lab Med. 1996;120(4):357–363. | ||
Higashide T, Takahira M, Okumura H, et al. Bilaterally identical monoclonality in a case of primary intraocular lymphoma. Am J Ophthalmol. 2004;138(2):306–308. | ||
Katai N, Kuroiwa S, Fujimori K, Yoshimura N. Diagnosis of intraocular lymphoma by polymerase chain reaction. Graefes Arch Clin Exp Ophthalmol. 1997;235(7):431–436. | ||
Shen DF, Zhuang Z, Lehoang P, et al. Utility of microdissection and polymerase chain reaction for the detection of immunoglobulin gene rearrangement and translocation in primary intraocular lymphoma. Ophthalmology. 1998;105(9):1664–1669. | ||
Chan CC, Shen D, Nussenblatt RB, Böni R, Zhuang Z. Detection of molecular changes in primary intraocular lymphoma by microdissection and polymerase chain reaction. Diagn Mol Pathol. 1998;7(1):63–64. | ||
Tuaillon N, Chan CC. Molecular analysis of primary central nervous system and primary intraocular lymphomas. Curr Mol Med. 2001;1(2):259–272. | ||
Coupland SE, Loddenkemper C, Smith JR, et al. Expression of immunoglobulin transcription factors in primary intraocular lymphoma and primary central nervous system lymphoma. Invest Ophthalmol Vis Sci. 2005;46(11):3957–3964. | ||
Chan CC, Sen HN. Current concepts in diagnosing and managing primary vitreoretinal (intraocular) lymphoma. Discov Med. 2013;15(81):93–100. | ||
Chan CC, Whitcup SM, Solomon D, Nussenblatt RB. Interleukin-10 in the vitreous of patients with primary intraocular lymphoma. Am J Ophthalmol. 1995;120(5):671–673. | ||
Buggage RR, Whitcup SM, Nussenblatt RB, Chan CC. Using interleukin 10 to interleukin 6 ratio to distinguish primary intraocular lymphoma and uveitis. Invest Ophthalmol Vis Sci. 1999;40(10):2462–2463. | ||
El-Shabrawi Y, Livir-Rallatos C, Christen W, Baltatzis S, Foster CS. High levels of interleukin-12 in the aqueous humor and vitreous of patients with uveitis. Ophthalmology. 1998;105(9):1659–1663. | ||
Ohta K, Sano K, Imai H, Kikuchi T. Cytokine and molecular analyses of intraocular lymphoma. Ocul Immunol Inflamm. 2009;17(3):142–147. | ||
Ramkumar HL, Shen DF, Tuo J, et al. IL-10-1082 SNP and IL-10 in primary CNS and vitreoretinal lymphomas. Graefes Arch Clin Exp Ophthalmol. 2012;250(10):1541–1548. | ||
Chan CC, Wallace DJ. Intraocular lymphoma: update on diagnosis and management. Cancer Control. 2004;11(5):285–295. | ||
Bonzheim I, Giese S, Deuter C, et al. High frequency of MyD88 mutations in vitreoretinal B-cell lymphoma: a valuable tool to improve diagnostic yield of vitreous aspirates. Blood. 2015;126(1):76–79. | ||
Pulido JS, Salomao DR, Frederick LA, Viswanatha DS. MyD-88 L265P mutations are present in some cases of vitreoretinal lymphoma. Retina. 2015;35(4):624–627. | ||
Hoang-Xuan K, Bessell E, Bromberg J, et al. European association for neuro-oncology Task Force on primary CNS lymphoma. Diagnosis and treatment of primary CNS lymphoma in immunocompetent patients: guidelines from the European association for neuro-oncology. Lancet Oncol. 2015;16(7):e322–e332. | ||
Rosenfeld MR, Pruitt AA. Management of malignant gliomas and primary CNS lymphoma: standard of care and future directions. Continuum. 2012;18(2):406–415. | ||
Soussain C, Choquet S, Fourme E, et al. Intensive chemotherapy with thiotepa, busulfan and cyclophosphamide and hematopoietic stem cell rescue in relapsed or refractory primary central nervous system lymphoma and intraocular lymphoma: a retrospective study of 79 cases. Haematologica. 2012;97(11):1751–1756. | ||
Soussain C, Hoang-Xuan K, Taillandier L, et al. Société Française de Greffe de Moëlle Osseuse-Thérapie Cellulaire. Intensive chemotherapy followed by hematopoietic stem-cell rescue for refractory and recurrent primary CNS and intraocular lymphoma: Société Française de Greffe de Moëlle Osseuse-Thérapie Cellulaire. J Clin Oncol. 2008;26(15):2512–2518. | ||
Soussain C, Merle BH, Reux I, et al. A single-center study of 11 patients with intraocular lymphoma treated with conventional chemotherapy followed by high-dose chemotherapy and autologous bone marrow transplantation in 5 cases. Leuk Lymphoma. 1996;23(3–4):339–345. | ||
Soussain C, Suzan F, Hoang-Xuan K, et al. Results of intensive chemotherapy followed by hematopoietic stem-cell rescue in 22 patients with refractory or recurrent primary CNS lymphoma or intraocular lymphoma. J Clin Oncol. 2001;19(3):742–749. | ||
Mason JO, Fischer DH. Intrathecal chemotherapy for recurrent central nervous system intraocular lymphoma. Ophthalmology. 2003;110(6):1241–1244. | ||
Jahnke K, Bechrakis NE, Coupland SE, et al. Treatment of primary intraocular lymphoma with oral trofosfamide: report of two cases and review of the literature. Graefes Arch Clin Exp Ophthalmol. 2004;242(9):771–776. | ||
Jahnke K, Korfel A, Komm J, et al. Intraocular lymphoma 2000–2005: results of a retrospective multicentre trial. Graefes Arch Clin Exp Ophthalmol. 2006;244(6):663–669. | ||
Jahnke K, Thiel E, Bechrakis NE, et al. Ifosfamide or trofosfamide in patients with intraocular lymphoma. J Neurooncol. 2009;93(2):213–217. | ||
Jahnke K, Wagner T, Bechrakis NE, et al. Pharmacokinetics and efficacy of ifosfamide or trofosfamide in patients with intraocular lymphoma. Ann Oncol. 2005;16(12):1974–1978. | ||
Ferreri AJ, Cwynarski K, Pulczynski E, et al. International Extranodal Lymphoma Study Group (IELSG). Chemoimmunotherapy with methotrexate, cytarabine, thiotepa, and rituximab (MATRix regimen) in patients with primary CNS lymphoma: results of the first randomisation of the International Extranodal Lymphoma Study Group-32 (IELSG32) phase 2 trial. Lancet Haematol. 2016;3(5):e217–e227. | ||
Hormigo A, Abrey L, Heinemann MH, Deangelis LM. Ocular presentation of primary central nervous system lymphoma: diagnosis and treatment. Br J Haematol. 2004;126(2):202–208. | ||
Hashida N, Ohguro N, Nishida K. Efficacy and complications of intravitreal rituximab injection for treating primary vitreoretinal lymphoma. Transl Vis Scie Technol. 2012;1:1. | ||
de Smet MD, Stark-Vancs V, Kohler DR, Smith J, Wittes R, Nussenblatt RB. Intraocular levels of methotrexate after intravenous administration. Am J Ophthalmol. 1996;121(4):442–444. | ||
de Smet MD, Vancs VS, Kohler D, Solomon D, Chan CC. Intravitreal chemotherapy for the treatment of recurrent intraocular lymphoma. Br J Ophthalmol. 1999;83(4):448–451. | ||
Wang JK, Yang CM, Lin CP, Shan YD, Lo AY, Tien HF. An Asian patient with intraocular lymphoma treated by intravitreal methotrexate. Jpn J Ophthalmol. 2006;50(5):474–478. | ||
Kim E, Kim C, Lee J, Cho Y. A case of primary intraocular lymphoma treated by intravitreal methotrexate. Korean J Ophthalmol. 2009;23(3):210–214. | ||
Fishburne BC, Wilson DJ, Rosenbaum JT, Neuwelt EA. Intravitreal methotrexate as an adjunctive treatment of intraocular lymphoma. Arch Ophthalmol. 1997;115(9):1152–1156. | ||
Sou R, Ohguro N, Maeda T, Saishin Y, Tano Y. Treatment of primary intraocular lymphoma with intravitreal methotrexate. Jpn J Ophthalmol. 2008;52(3):167–174. | ||
Velez G, Boldt HC, Whitcup SM, Nussenblatt RB, Robinson MR. Local methotrexate and dexamethasone phosphate for the treatment of recurrent primary intraocular lymphoma. Ophthalmic Surg Lasers. 2002;33(4):329–333. | ||
Frenkel S, Hendler K, Siegal T, Shalom E, Pe’er J. Intravitreal methotrexate for treating vitreoretinal lymphoma: 10 years of experience. Br J Ophthalmol. 2008;92(3):383–388. | ||
Riemens A, Bromberg J, Touitou V, et al. Treatment strategies in primary vitreoretinal lymphoma: a 17-center European Collaborative study. JAMA Ophthalmol. 2015;133(2):191–197. | ||
Hashida N, Nakai K, Saitoh N, Nishida K. Association between ocular findings and preventive therapy with onset of central nervous system involvement in patients with primary vitreoretinal lymphoma. Graefes Arch Clin Exp Ophthalmol. 2014;252(4):687–693. | ||
Itty S, Olson JH, O’Connell DJ, Pulido JS. Treatment of primary intraocular lymphoma (PIOL) has involved systemic, intravitreal or intrathecal chemotherapy and/or radiotherapy. Retina. 2009;29:415–416. | ||
Gregory MS, Koh S, Huang E, et al. A novel treatment for ocular tumors using membrane FasL vesicles to activate innate immunity and terminate immune privilege. Invest Ophthalmol Vis Sci. 2005;46(7):2495–2502. | ||
Li Z, Mahesh SP, Shen DF, et al. Eradication of tumor colonization and invasion by a B cell-specific immunotoxin in a murine model for human primary intraocular lymphoma. Cancer Res. 2006;66(21):10586–10593. | ||
Pastan I, Beers R, Bera TK. Recombinant immunotoxins in the treatment of cancer. Methods Mol Biol. 2004;248:503–518. | ||
Rodrigues EB, Farah ME, Maia M, et al. Therapeutic monoclonal antibodies in ophthalmology. Prog Retin Eye Res. 2009;28(2):117–144. | ||
Cabanillas F, Shah B. Advances in diagnosis and management of diffuse large B-cell lymphoma. Clin Lymphoma Myeloma Leuk. 2017;17(12):783–796. | ||
Illerhaus G, Marks R, Ihorst G, et al. High-dose chemotherapy with autologous stem-cell transplantation and hyperfractionated radiotherapy as first-line treatment of primary CNS lymphoma. J Clin Oncol. 2006;24(24):3865–3870. | ||
Li Z, Qiu Y, Personett D, et al. Pomalidomide shows significant therapeutic activity against CNS lymphoma with a major impact on the tumor microenvironment in murine models. PLoS One. 2013;8(8):e71754. | ||
Wilson WH, Young RM, Schmitz R, et al. Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat Med. 2015;21(8):922–926. | ||
Ghesquieres H, Houillier C, Chinot O, et al. Rituximab-Lenalidomide (REVRI) in relapse or refractory primary central nervous system (PCNSL) or vitreo retinal lymphoma (PVRL): results of a “proof of concept” phase II study of the French LOC network (Abstract 785). Blood. 2016;128:785. | ||
Plotkin SR, Batchelor TT. Primary nervous-system lymphoma. Lancet Oncol. 2001;2(6):354–365. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.