Back to Journals » Clinical and Experimental Gastroenterology » Volume 10
Primary sclerosing cholangitis: diagnostic and management challenges
Received 21 February 2017
Accepted for publication 14 August 2017
Published 6 November 2017 Volume 2017:10 Pages 265—273
DOI https://doi.org/10.2147/CEG.S105872
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Andreas M. Kaiser
Sanjeev Sirpal,1 Natasha Chandok2
1Department of Medicine, Centre Hospitalier de l’Université de Montréal (CHUM), University of Montreal, Montreal, QC, 2Department of Medicine, University of Western Ontario, London, ON, Canada
Abstract: Primary sclerosing cholangitis (PSC) is a chronic immune-mediated disease affecting intra- and extrahepatic bile ducts, primarily the large biliary ducts. Clinical manifestations are broad, and the spectrum encompasses asymptomatic cholestasis, icteric cholangitis with pruritis, cirrhosis, and cholangiocarcinoma. Though rare, PSC has a propensity to affect young to middle-aged males and is strongly associated with inflammatory bowel disease. There is an unmet need for effective medical treatments for PSC, and to date, the only curative therapy is liver transplantation reserved for those with end-stage liver disease. This article addresses the diagnostic and management challenges of PSC, with a succinct analysis of existing therapies, their limitations, and a glimpse into the future of the management of this multifaceted pathologic entity.
Keywords: primary sclerosing cholangitis, management, PSC
Introduction
Primary sclerosing cholangitis (PSC) is a chronic immune-mediated disease of intra-and extrahepatic bile ducts, primarily affecting large ducts. Its insidious course related to progressive fibrostenotic stricturing of the biliary tree causes important clinical sequelae, including liver cirrhosis, portal hypertension, and end-stage liver disease. Unlike primary biliary cirrhosis (PBC), which affects small bile ducts and predominantly occurs in females, PSC has a proclivity toward males, with a median age of presentation of ~40 years.1 It is strongly associated with inflammatory bowel disease (IBD), with a unique phenotype of ulcerative colitis involving rectal sparing, right colonic disease with backwash ileitis. In such cases where IBD overlaps with PSC, there is an identified higher risk of malignancy, and therefore, such a presentation necessitates rigorous colorectal cancer surveillance per multisociety guidelines.2 There is also an independent association of PSC with cholangiocarcinoma (CCA). CCA in the PSC patient is typically diagnosed within the first 2 years of the diagnosis of PSC, and as it is often detected at an advanced stage, it often portends a guarded prognosis. Surgery for early CCA, and even liver transplant in selected cases of hilar CCA, is associated with clear survival benefit.3 Why CCA is diagnosed within the first 2 years of receiving a diagnosis of PSC is unknown, but is a well-described observation. It is likely that a patient with PSC who develops “early” CCA may well have has occult PSC for a much longer period but simply did not have symptoms. At present, there is insufficient evidence on CCA screening guidelines for PSC.4,5
There are significant diagnostic and management challenges to PSC, largely owing to its frequent subclinical presentation with normal liver tests. To date, no approved or proven therapy exists for PSC, with pharmacotherapy aimed at treating symptoms and managing complications.6 Immunosuppressants, bile salts, chelators (eg, cholestyramine for pruritus), and steroids have not shown significant benefit in clinical trials. Clearly, there is an unmet need for treatment in PSC, particularly as at least a third of patients will experience liver-related death without hepatic transplantation.7 The purpose of this review is to address the diagnostic and management challenges of PSC, with a pointed focus on new developments and avenues of clinical research.
Clinical presentation and diagnosis of PSC
PSC often has a subtle presentation, with many patients presenting with asymptomatic cholestasis on blood work. If symptoms are present, they are often nonspecific, such as fatigue and pruritus. Pruritus can range from mild to disabling, resulting in severe excoriations and a decreased quality of life. The pathophysiology of pruritus is unknown, but accumulation of bile acids in the skin and endogenous opioid production are among the hypothesized etiologies.8
Other signs and symptoms include right upper quadrant abdominal pain, weight loss, and episodes of fever and chills. Cirrhotic symptomatology and portal hypertension sequelae (ascites and variceal hemorrhage) occur in advanced stages of PSC, though such manifestations are typically not present at diagnosis which usually occurs much earlier in the disease course in the current era of readily available biochemistry and liver imaging.9
On clinical exam, patients may be unremarkable, or present with jaundice, hepatomegaly, splenomegaly, and excoriations. Of these findings, hepatomegaly and splenomegaly are the most frequent. Complications of longstanding cholestatic liver diseases like PSC include metabolic bone disease and fat malabsorption with steatorrhea. Like many autoimmune diseases, PSC is associated with a range of other autoimmune conditions, features of which may also be present on clinical exam (Table 1).1–3
 | Table 1 Common autoimmune diseases Note: Data from Lerner et al.10 |
The diagnosis of PSC is made via a showing of elevated serum markers of cholestasis (AP, γGT) in combination with imaging findings – magnetic resonance cholangiopancreatography (MRCP) or endoscopic retrograde cholangiopancreatography (ERCP) demonstrating characteristic bile duct changes with multifocal strictures and segmental dilatations, when all other possible cholestatic disorders and secondary causes are excluded.1–6
One of the main diagnostic challenges of PSC is the need for it to be differentiated from secondary causes of sclerosing cholangitis and immunoglobulin G4 (IgG4)-associated cholangitis/autoimmune pancreatitis.11 In addition, PSC overlap syndromes (ie, the concurrent presence of PSC with PBC or PSC with autoimmune hepatitis) should be considered as overlap syndromes are not uncommon though underdiagnosed in PSC patients, and lack of recognition would have adverse consequences given that both PBC and autoimmune hepatitis have proven treatments.12 Clinicians should be mindful to exclude the many secondary causes to sclerosing cholangitis, especially IgG-4 disease which is imminently treatable (unlike PSC). A list of diseases that can mimic PSC is noted in Table 2:
 | Table 2 Secondary causes of sclerosing cholangitis Note: Data from Gotthardt et al.11 Abbreviation: IgG4, immunoglobulin G4. |
Biochemical tests may reveal a fluctuating pattern, owing to a transient blockage of strictured bile ducts by biliary sludge or small stones. In contrast, serum aminotransferases are not usually elevated (typically <300 IU/L) and in early disease serum albumin is normal, whereas those with active IBD may have hypoalbuminemia.1–7 Serology in PSC may reveal other findings (Table 3).
 | Table 3 Additional serological findings possible in PSC Note: Data from Pollock et al.13 Abbreviation: PSC, primary sclerosing cholangitis. |
Several other autoantibodies, including antinuclear, anti-smooth muscle, anticardiolipin, thyroperoxidase, and rheumatoid factor, may be present, but their clinical significance has not been demonstrated. Whereas PBC is typically associated with antimitochondrial antibodies, such antibodies are notably absent in PSC. One study showed that in some instances, up to 97% of patients with PSC could be positive for one or more than one autoantibody, while 81% were positive for ≥3 autoantibodies.14 That study demonstrated a lack of correlation among autoantibodies and PSC disease severity, except for the presence of anticardiolipin antibodies, which correlated well with the Mayo risk score.14
Increased serum levels of IgG4 (typically associated with autoimmune pancreatitis) has been demonstrated in some PSC patients.15,16 One report demonstrated that in one sample, 9% of PSC patients had IgG4 levels above the upper limit of normal.15 Serum IgG4 concentration >135 mg/dL makes IgG4 cholangiopathy a more likely diagnosis for which treatment with corticosteroids and other immunomodulatory agents may be warranted.17 It is vital to not miss a diagnosis of IgG4 disease given that most patients respond to treatment.
Cholangiography – via MRCP, ERCP, or percutaneous transhepatic cholangiography – is typically used to confirm a diagnosis of PSC with a showing of characteristic multifocal stricturing and dilation of intrahepatic and/or extrahepatic bile ducts. MRCP is now the mainstay modality to diagnose PSC given its safety and noninvasiveness, with far superior sensitivity and specificity over both ultrasound and computer tomography. A meta-analysis of six studies demonstrated that the sensitivity and specificity of diagnosing PSC with MRCP were 86% and 94%, respectively.18 MRCP demonstrates the classic “beaded” appearance of the bile duct, resulting from multifocal, short, annular strictures that alternate with normal or mildly dilated segments. The presence of dominant strictures on MRCP may indicate possible underlying CCA, often necessitating ERCP. ERCP is additionally indicated in patients with cholangitis to relieve biliary obstruction, and vigilance should always be exerted to exclude CCA vs benign strictures amenable to dilatation and stent insertion.
In classic PSC, focal strictures are present in the biliary tree, with normal intervening areas. One report found a distribution of strictures in the following areas: intrahepatic and extrahepatic bile ducts – 87%; intrahepatic bile ducts alone – 11%; and extrahepatic bile ducts alone – 2%. Gallbladder and cystic duct abnormalities may also be present – one study showed gallbladder abnormalities in 41% of patients, especially the cohort with extrahepatic PSC.19 Six percent of patients had a gallbladder mass lesion, and more than half of those were gallbladder carcinoma, highlighting the importance of cholecystectomy following detection of such lesions.20
Early stage disease may not present with the typical “beaded” appearance of the biliary tree, and shallow ulcerations may indeed be the only cholangiographic finding. ERCP is indicated in such patients.
Small duct PSC (or “pericholangitis”) presents a diagnostic challenge in many instances owing to the fact that such patients present with normal cholangiography, attributable to the fact that only small-caliber bile ducts are affected, and this entity is undetected with cholangiographic techniques.21 Although small ductal PSC is comparable to classic PSC on biochemical and histologic terms, extant studies show a significantly better prognosis than classic PSC,22–24 with a potential for degeneration into classic PSC.23,25 One case series study showed that small duct PSC degenerated to classic PSC in 4 of 27 patients studied after a median of 72 months.22
Continuous bile duct destruction in PSC can cause end-stage liver disease, and other sequelae include:
- Fat-soluble vitamin deficiencies (A, D, E, and K)
- Metabolic bone disease
- Dominant biliary strictures
- Cholangitis and cholelithiasis
- Cholangiocarcinoma
- Gallbladder cancer
- Hepatocellular carcinoma (in patients with cirrhosis)
- Colon cancer (in patients with concomitant ulcerative colitis)
Diagnosis
Patients with underlying IBD presenting with a cholestatic pattern on liver biochemistry (especially an elevated alkaline phosphatase) should be investigated for possible PSC. Once a high degree of clinical suspicion is achieved, a firm diagnosis can be made via a showing of cholangiographic evidence of characteristic bile duct changes (multifocal strictures, segmental dilations) and the “notable absence of secondary explanations of sclerosing cholangitis.” Liver biopsy is not required in patients with typical cholangiographic findings and a suggestive biochemical profile, although it is when small duct PSC is suspected in the differential. As stated earlier, patients with autoimmune liver diseases not uncommonly have an overlap with another condition. As such, clinicians should actively exclude concurrent autoimmune hepatitis or PBC in patients with PSC.
Liver biopsy
Although percutaneous liver biopsy may reinforce a diagnosis of PSC, it is rarely used diagnostically26 and may not be diagnostic given the patchy nature of PSC. Liver biopsy is reserved for instances of suspected small duct disease or overlap syndrome with autoimmune hepatitis. If biopsy is indicated, prophylactic antibiotic therapy prior to biopsy minimizes the risk of subsequent cholangitis.
On biopsy, PSC presents “classically” with fibrous obliteration of small bile ducts, with concentric replacement by connective tissue in an “onion skin” pattern, although this histology is found in less than 25% of liver biopsies. More often, histologic abnormalities in PSC are nonspecific and are similar to those in PBC. PSC staging, similar to that used in PBC, is based on the degree of involvement of portal triads and/or hepatic lobules (Table 4).27
 | Table 4 Staging of PSC Abbreviation: PSC, primary sclerosing cholangitis. |
Imaging
Transient elastography, a noninvasive measure of hepatic fibrosis via the assessment of liver stiffness properties, can be used to estimate the degree of hepatic fibrosis in PSC.28 A recent study showed that magnetic resonance elastography (MRE) is also able to detect cirrhosis with high specificity. Moreover, liver stiffness obtained by MRE is predictive of hepatic decompensation in PSC.29
Etiology
The etiology of PSC disease development and progression is largely unknown. However, multiple mechanisms, imputing both the innate and adaptive immune systems, have been hypothesized.
First and foremost, the frequent coexistence of PSC and ulcerative colitis (with a known autoimmune pathophysiologic mechanism) suggests a common autoimmune pathway.4 However, temporal differences in copresentation or the fact that only a small percentage of ulcerative colitis patients have PSC suggests a multifactorial etiology.30 Portal venous bacteremia has also been proposed as a mechanistic pathway in PSC pathogenesis – an extension of the idea of an infectious trigger for autoimmunity. Specifically, chronic entry of bacteria in the portal circulation can result in an inflammatory reaction in the liver and bile ducts. Alternatively (or in addition), exposure to toxic bile acids produced by infectious mediators (colonic bacteria or viruses) can reinforce immunoactivation pathways.31–33
An alternate pathway proposed is tied to ischemic ductal injury – support for this hypothesis stems from studies demonstrating a remarkable resemblance in clinical, biochemical, and cholangiographic profiles in bile ductal ischemic injury and PSC – also confirmed via intra-arterial infusion of floxuridine.34 Hence, ischemic injury to peribiliary arterioles may be one of the pathophysiological processes that occurs in PSC.
The general scientific consensus is that PSC is a multifactorial disease with a similar clinical presentation. Although the precise pathophysiological process is yet to be determined, it is believed that immunologically mediated bile duct injury in PSC is the common downstream sequela resulting in its clinicopathological presentation. Further, this immunopathological process often occurs in the backdrop of patients with a favorable genetic predisposition upon exposure to environmental triggers.
Management and treatment: current practices and challenges
Management of PSC
There are two major treatment goals in PSC: slow or reverse disease progression and management of symptoms of disease progression.
This section will review the current medical, endoscopic, and surgical therapies aimed at managing progressive PSC and cancer screening in patients with PSC. Unfortunately, no proven treatment slows or reverses progression of the disease. However, excellent outcomes may be achieved after liver transplantation (LT) for advanced disease. Best evidence suggests fertility does not seem to be reduced in patients with PSC, who are able to deliver healthy children without an apparent increase in risk for mother or child.35 The management guidelines here are in accordance with a 2015 guideline from the American College of Gastroenterology2 and a 2010 guideline from the American Association for the Study of Liver diseases,1 along with the European Association for the Study of the Liver.
Pharmacological treatments
Several immunosuppressive and anti-inflammatory agents have been studied in PSC,5,36 a summary of which is presented in Table 5.
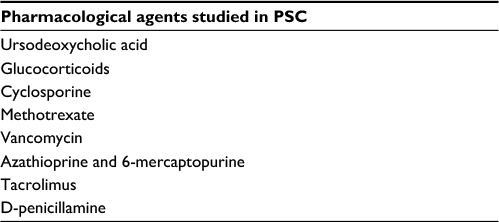 | Table 5 Pharmacological agents studied in PSC Abbreviation: PSC, primary sclerosing cholangitis. |
Inasmuch as a plethora of agents have been studied in PSC, none has been conclusively proven to alter the natural history of the disease. Some data do suggest, however, that patients with decreased alkaline phosphatase during follow-up may have improved survival.37
Ursodeoxycholic acid (UDCA) is a hydrophilic bile acid, and it has been the most extensively studied putative pharmacological treatments for PSC. Its role in PSC management is very controversial. UDCA is expected to exert protective effects on cholangiocytes against cytotoxic hydrophobic bile acids (at doses between 13 and 15 mg/kg per day) via the stimulation of hepatobiliary secretion, protection against bile acid-induced hepatocyte apoptosis, and induction of antioxidants.38
Studies have shown that high-dose UDCA may improve liver biochemistry and curb hepatic inflammation, but there is definitively no transplant-free survival benefit.39–46 Moreover, Lindor et al demonstrated that long-term high-dose UDCA is associated with higher rates of serious adverse events, including death.2 As such, high-dose UDCA should not be used in the management of PSC.
A management complication in PSC is that once prescribed, one study showed that withdrawal of UDCA can precipitate a worsening of pruritus and biochemical test results.47 In this study, 3 months post-UDCA treatment, study patients noted 76% increase in mean alkaline phosphatase, 118% increase in gamma-glutamyl transpeptidase, 50% increase in bilirubin, 64% increase in alanine aminotransferase, 45% increase in aspartate aminotransferase, and 0.5-point increase in Mayo risk score (from baseline). Relevant clinical symptomatology, including pruritus, worsened post-UDCA discontinuation. However, certain limitations of the study limit its application. For instance, the study assessed surrogate outcomes (ie, biochemical profiles in lieu of clinically relevant outcomes such as survival), had possible selection bias, was of short duration, and notably was unblinded.47,48
The American Association for the Study of Liver Diseases 2010 guideline recommends against UDCA treatment in PSC,1 whereas a 2015 guideline from the American College of Gastroenterology does not explicitly recommend against or for make a recommendation about using UDCA other than to note that it should not be used in doses >28 mg/kg/day.2 The guidelines acknowledge the paucity of evidence pertaining to UDCA usage in PSC, safety concerns, and the lack of proven effect on clinically relevant end points.
Some studies suggest that UDCA treatment at a dose of 13–15 mg/kg/day can be safely pursued in patients, provided that after 6 months of treatment levels of alkaline phosphatase normalize or decrease by at least 40%.48 An animal model study has shown promising results for a derivative of UDCA (24-norursodeoxycholic acid) in the treatment of PSC.49
Other treatments
A summary of other treatment options and evidence of their efficacy is presented in Table 6.
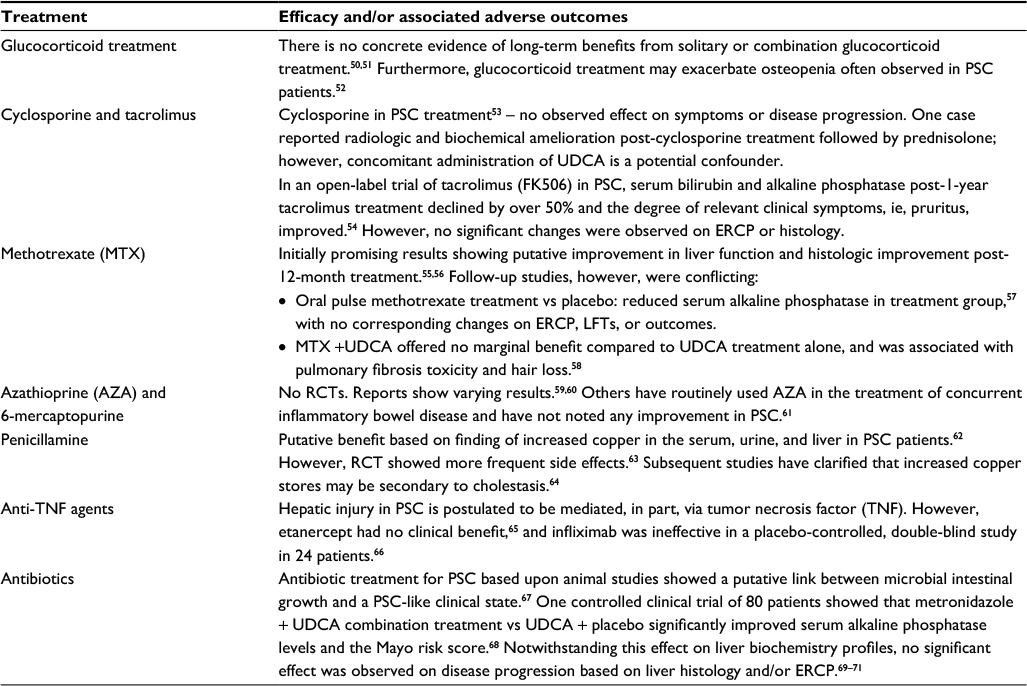 | Table 6 Other treatment options in PSC Abbreviations: PSC, primary sclerosing cholangitis; UDCA, ursodeoxycholic acid; ERCP, endoscopic retrograde cholangiopancreatography; LFT, liver function tests; RCT, randomized clinical trial. |
Endoscopic therapy
There exists an established cohort of PSC patients with a dominant extrahepatic biliary stricture that could be compatible with endoscopic therapy.72–74 One study of 125 PSC patients showed that the proportion of patients with disease amenable to endoscopic therapy could be as high as 45%.75 The proportion in such a cohort is dynamic, however, since patients who do not initially have a stricture may develop one over time, eg, 40% of patients without a stricture could develop one after 5 years of follow-up.74
However, no controlled trial has evaluated whether endoscopic treatment of a dominant stricture improves outcomes in PSC. In fact, one study showed no significant benefit in endoscopic treatment of PSC patients and warned against routine usage until clear benefit is established.75 Notwithstanding this study, others have shown clinical and radiographic improvement in PSC patients following endoscopic dilation with or without placement of a biliary stent.72–75 These findings suggest that short-term endoscopic dilation/stenting may be beneficial for patients with a dominant extrahepatic biliary stricture.2 However, the quality of evidence in this domain is weak, as most studies are observational and/or retrospective in nature, and therefore susceptible to significant selection bias.
Surgical therapy
PSC may also be managed surgically, and options include biliary reconstructive procedures, proctocolectomy (in patients with ulcerative colitis), and LT.76
Biliary reconstruction
Studies have shown that biliary reconstruction surgery with/without intraoperative stent insertion is very effective in avoiding jaundice and cholangitis, even for several years after the procedure.77–79 However, cirrhotic patients may experience higher morbidity and mortality, with an increased risk of postoperative infection and fibrosis of the porta hepatis, potentially complicating future LT.77
A retrospective study has shown that LT is superior to biliary surgical procedures.80 Hence, much attention has since shifted away from biliary reconstruction to liver transplantation.81
Proctocolectomy
Extant evidence suggests lack of a significant advantage in the usage of proctocolectomy in terms of clinical outcomes in patients with concomitant PSC and chronic ulcerative colitis.82 In a retrospective examination of 45 patients, the study reported no significant differences in new onset of hepatomegaly, splenomegaly, esophageal varices, and ascites in patients with and without proctocolectomy. Biochemically, the serial changes in bilirubin, alkaline phosphatase, aspartate aminotransferase, prothrombin time, and albumin were similar. Histologic progression on liver biopsy did not differ between groups, nor did changes on serial cholangiograms. Proctocolectomy also had no effect on survival. Hence, current best practice is reserving proctocolectomy for situations when indicated as a result of the colitis.
Liver transplantation
Patients with advanced liver disease secondary to PSC should be referred for LT provided that the Model for End-Stage Liver Disease (MELD) score is ≥15.1 Outcomes for LT in PSC are comparable to transplants for other indications, with 5-year survival rates approximating 85%.83–85 Evaluation for LT is muddled by the unpredictability of the disease course and high risk of biliary tract malignancy. The Mayo risk score is a prognostic tool useful for predicting the natural history of PSC. The indications for LT in PSC are, in large part, the same as those for other categories of end-stage liver disease.86
Special situations, notwithstanding a low MELD score, may necessitate LT on a case-by-case basis:
- Recurrent or refractory cholangitis
- Intractable pruritus
- Peripheral or hilar CCA <3 cm in diameter
Looking forward: prospective on novel treatment approaches in PSC
There is a notable gap in effective treatment strategies to improve survival or curb and/or prevent the need for LT in PSC. Existing evidence shows a lack of transplant-free survival benefit in patients receiving UDCA treatment (notwithstanding a reported improvement in liver biochemistry and histology), as described earlier. However, current research suggests that farnesoid X receptor (FXR) agonists (eg, obeticholic acid, 6α-ethyl chenodeoxycholic acid) and 24-norursodeoxycholic acid, a side-chain-modified UDCA derivate resistant to amidation which undergoes cholehepatic shunting, may be novel treatment options. An animal model of PSC demonstrated the plausible effectiveness of the 24-norursodeoxycholic acid derivative.87,88 Fibrates, with their global pleiotropic and anti-inflammatory properties, may be an alternative novel treatment strategy.89,90
Summary
As discussed, there are several challenges in the diagnosis and management of PSC. From a diagnostic standpoint, PSC patients are often asymptomatic, which can lead to difficulties in establishing a firm diagnosis. Sometimes, a PSC diagnosis is achieved in the course of abnormal laboratory biochemical findings with or without the presence of compatible clinical symptoms, ie, fatigue and pruritus. Abnormal physical examination findings include jaundice, hepatomegaly, splenomegaly, and excoriations. On biochemistry, biochemical tests demonstrate a cholestatic pattern, with elevation of the serum alkaline phosphatase predominating in most patients. Imaging, namely MRCP in the current era, confirms the diagnosis with the presence of abnormal appearing bile ducts with wall thickening, dilations, and strictures.
There are two major goals of treatment in PSC: delay or reversal of the disease process and the management of progressive disease and its complications. While several immunosuppressive and anti-inflammatory agents have been studied in patients with PSC, none has shown a consistent benefit on overall or transplant-free survival, highlighting the dire need for medical management of this rare disease. LT is currently the treatment of choice for patients with advanced liver disease secondary to PSC.
Disclosure
The authors report no conflicts of interest in this work.
References
Chapman R, Fevery J, Kalloo A et al. Diagnosis and management of primary sclerosing cholangitis. Hepatology. 2009;51(2):660–678. | ||
Lindor K, Kowdley K, Harrison M. ACG clinical guideline: primary sclerosing cholangitis. Am J Gastroenterol. 2015;110(5):646–659. | ||
Rea DJ, Heimbach JK, Rosen CB, et al. Liver transplantation with neoadjuvant chemoradiation is more effective than resection for hilar cholangiocarcinoma. Ann Surg. 2005;242(3):451–461. | ||
Lee Y, Kaplan M. Primary sclerosing cholangitis. N Engl J Med. 1995; 332(14):924–933. | ||
Angulo P, Lindor K. Primary sclerosing cholangitis. Hepatology. 1999;30(1):325–332. | ||
Lindor K. New treatment strategies for primary sclerosing cholangitis. Dig Dis. 2011;29(1):113–116. | ||
Ponsioen C. Natural history of primary sclerosing cholangitis and prognostic value of cholangiography in a Dutch population. Gut. 2002;51(4):562–566. | ||
Talwalkar J, Lindor K. Natural history and prognostic models in primary sclerosing cholangitis. Best Pract Res Clin Gastroenterol. 2001; 15(4):563–575. | ||
Housset C. Updates on primary sclerosing cholangitis. Curr Opin Gastroenterol. 2017;33(2):69–70. | ||
Lerner A, Jeremias P, Matthias T. The world incidence and prevalence of autoimmune diseases is increasing. Int J Celiac Dis. 2016;3(4):151–155. | ||
Gotthardt D, Chahoud F, Sauer P. Primary sclerosing cholangitis: diagnostic and therapeutic problems. Dig Dis. 2011;29(s1):41–45. | ||
Boberg K, Chapman R, Hirschfield G, Lohse A, Manns M, Schrumpf E. Overlap syndromes: The International Autoimmune Hepatitis Group (IAIHG) position statement on a controversial issue. J Hepatol. 2011;54(2):374–385. | ||
Pollock G, Minuk G. Diagnostic considerations for cholestatic liver disease. J Gastroenterol Hepatol. 2017;32(7):1303–1309. | ||
Angulo P, Peter J, Gershwin M et al. Serum autoantibodies in patients with primary sclerosing cholangitis. J Hepatol. 2000;32(2):182–187. | ||
Mendes F, Jorgensen R, Keach J, et al. Elevated serum IgG4 concentration in patients with primary sclerosing cholangitis. Am J Gastroenterol. 2006;101(9):2070–2075.. | ||
Hirano K, Kawabe T, Yamamoto N, et al. Serum IgG4 concentrations in pancreatic and biliary diseases. Clinica Chimica Acta. 2006;367(1–2):181–184. | ||
Wallace Z, Deshpande V, Mattoo H, et al. IgG4-related disease: clinical and laboratory features in one hundred twenty-five patients. Arthritis Rheumatol. 2015;67(9):2466–2475. | ||
Dave M, Elmunzer B, Dwamena B, Higgins P. Primary sclerosing cholangitis: meta-analysis of diagnostic performance of MR cholangiopancreatography. Radiology. 2010;256(2):387–396. | ||
Said K, Glaumann H, Bergquist A. Gallbladder disease in patients with primary sclerosing cholangitis. J Hepatol. 2008;48(4):598–605. | ||
Buckles D, Lindor K, LaRusso N, Petrovic L, Gores G. In primary sclerosing cholangitis, gallbladder polyps are frequently malignant. Am J Gastroenterol. 2002;97(5):1138–1142. | ||
Wee A, Ludwig A. Pericholangitis in chronic ulcerative colitis: primary sclerosing cholangitis of the small bile ducts? Ann Intern Med. 1985;102(5):581. | ||
Broomé U, Glaumann H, Lindstöm E, et al. Natural history and outcome in 32 Swedish patients with small duct primary sclerosing cholangitis (PSC). J Hepatol. 2002;36(5):586–589. | ||
Angulo P, Maor-Kendler Y, Lindor K. Small-duct primary sclerosing cholangitis: a long-term follow-up study. Hepatology. 2002;35(6):1494–1500. | ||
Björnsson E, Olsson R, Bergquist A, et al. The natural history of small-duct primary sclerosing cholangitis. Gastroenterology. 2008;134(4):975–980. | ||
Bjornsson E. Patients with small duct primary sclerosing cholangitis have a favourable long term prognosis. Gut. 2002;51(5):731–735. | ||
Burak K, Angulo P, Lindor K. Is there a role for liver biopsy in primary sclerosing cholangitis?. Am J Gastroenterol. 2003;98(5):1155–1158. | ||
Ludwig J, Barham S, Larusso N, Elveback L, Wiesner R, McCall J. Morphologic features of chronic hepatitis associated with primary sclerosing cholangitis and chronic ulcerative colitis. Hepatology. 1981;1(6):632–640. | ||
Corpechot C, El Naggar A, Poujol-Robert A, et al. Assessment of biliary fibrosis by transient elastography in patients with PBC and PSC. Hepatology. 2006;43(5):1118–1124. | ||
Eaton J, Dzyubak B, Venkatesh S, et al. Performance of magnetic resonance elastography in primary sclerosing cholangitis. J Gastroenterol Hepatol. 2016;31(6):1184–1190. | ||
Fausa O, Schrumpf E, Elgjo K. Relationship of inflammatory bowel disease and primary sclerosing cholangitis. Semin Liver Dis. 1991;11(01):31–39. | ||
Palmer K, Duerden B, Holdsworth C. Bacteriological and endotoxin studies in cases of ulcerative colitis submitted to surgery. Gut. 1980;21(10):851–854. | ||
Vinnik IE, Kern F Jr, Struthers JE Jr, Hill RB, Guzak S. Experimental chronic portal vein bacteremia. Proc Soc Exp Biol Med. 1964;115(2):311–314. | ||
Lichtman S, Sartor R, Keku J, Schwab J. Hepatic inflammation in rats with experimental small intestinal bacterial overgrowth. Gastroenterology. 1990;98(2):414–423. | ||
Woolf G, Vierling J. Disappearing intrahepatic bile ducts: the syndromes and their mechanisms. Semin Liver Dis. 1993;13(03):261–275. | ||
Wellge B, Sterneck M, Teufel A, et al. Pregnancy in primary sclerosing cholangitis. Gut. 2011;60(8):1117–1121. | ||
Eaton J, Talwalkar J, Lazaridis K, Gores G, Lindor K. Pathogenesis of primary sclerosing cholangitis and advances in diagnosis and management. Gastroenterology. 2013;145(3):521–536. | ||
Lindström L, Hultcrantz R, Boberg K, Friis–Liby I, Bergquist A. Association between reduced levels of alkaline phosphatase and survival times of patients with primary sclerosing cholangitis. Clin Gastroenterol Hepatol. 2013;11(7):841–846. | ||
Paumgartner G. Ursodeoxycholic acid in cholestatic liver disease: mechanisms of action and therapeutic use revisited. Hepatology. 2002;36(3):525–531. | ||
Chen W, Gluud C. Bile acids for primary sclerosing cholangitis (A Cochrane systematic review). J Hepatol. 2003;38:205. | ||
Charatcharoenwitthaya P, Angulo P, Enders F, Lindor K. Impact of inflammatory bowel disease and ursodeoxycholic acid therapy on small-duct primary sclerosing cholangitis. Hepatology. 2007;47(1):133–142. | ||
Stiehl A, Walker S, Stiehl L, Rudolph G, Hofmann W, Theilmann L. Effect of ursodeoxycholic acid on liver and bile duct disease in primary sclerosing cholangitis. A 3-year pilot study with a placebo-controlled study period. J Hepatol. 1994;20(1):57–64. | ||
Harnois D. High dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis. Year Bk Gastroenterol. 2010;2010:240–241. | ||
Chapman R. Medical treatment of primary sclerosing cholangitis with ursodeoxycholic acid. Dig Liver Dis. 2003;35(5):306–308. | ||
Van Hoogstraten H, Wolfhagen F, van de Meeberg P, et al. Ursodeoxycholic acid therapy for primary sclerosing cholangitis: results of a 2-year randomized controlled trial to evaluate single versus multiple daily doses. J Hepatol. 1998;29(3):417–423. | ||
Stiehl A, Rudolph G, Sauer P, et al. Efficacy of ursodeoxycholic acid treatment and endoscopic dilation of major duct stenoses in primary sclerosing cholangitis. J Hepatol. 1997;26(3):560–566. | ||
Shi J, Li Z, Zeng X, Lin Y, Xie W. Ursodeoxycholic acid in primary sclerosing cholangitis: meta-analysis of randomized controlled trials. Hepatol Res. 2009;39(9):865–873. | ||
Wunsch E, Trottier J, Milkiewicz M, et al. Prospective evaluation of ursodeoxycholic acid withdrawal in patients with primary sclerosing cholangitis. Hepatology. 2014;60(3):931–940. | ||
Tabibian J, Lindor K. Ursodeoxycholic acid in primary sclerosing cholangitis: if withdrawal is bad, then administration is good (right?). Hepatology. 2014;60(3):785–788. | ||
Fickert P, Wagner M, Marschall H, et al. 24-norUrsodeoxycholic acid is superior to ursodeoxycholic acid in the treatment of sclerosing cholangitis in Mdr2 (Abcb4) knockout mice. Gastroenterology. 2006;130(2):465–481. | ||
Angulo P. Oral budesonide in the treatment of primary sclerosing cholangitis. Am J Gastroenterol. 2000;95(9):2333–2337. | ||
Kochhar R, Goenka M, Das K, et al. Primary sclerosing cholangitis: an experience from India. J Gastroenterol Hepatol. 1996;11(5):429–433. | ||
Hay J. The metabolic bone disease of primary sclerosing cholangitis. Hepatology. 1991;14(2):257–261. | ||
Wiesner R, Ludwig J, Lindor K, et al. A controlled trial of cyclosporine in the treatment of primary biliary cirrhosis. N Eng J Med. 1990;322(20):1419–1424. | ||
Van Thiel DH, Carroll P, Abu-Elmagd K, et al. Tacrolimus (FK 506), a treatment for primary sclerosing cholangitis: results of an open-label preliminary trial. Am J Gastroenterol. 1995; 90:455. | ||
Kaplan MM, Arora S, Pincus SH. Primary sclerosing cholangitis and low-dose oral pulse methotrexate therapy. Clinical and histologic response. Ann Intern Med. 1987;106:231. | ||
Knox TA, Kaplan MM. Treatment of primary sclerosing cholangitis with oral methotrexate. Am J Gastroenterol. 1991;86:546. | ||
Knox TA, Kaplan MM. A double-blind controlled trial of oral-pulse methotrexate therapy in the treatment of primary sclerosing cholangitis. Gastroenterology. 1994;106:494. | ||
Lindor KD, Jorgensen RA, Anderson ML, et al. Ursodeoxycholic acid and methotrexate for primary sclerosing cholangitis: a pilot study. Am J Gastroenterol. 1996;91:511. | ||
Javett SL. Azathioprine in primary sclerosing cholangitif. Lancet. 1971;1:810. | ||
Wagner A. Azathioprine treatment in primary sclerosing cholangitis. Lancet. 1971;2:663. | ||
Kaplan MM. Medical approaches to primary sclerosing cholangitis. Semin Liver Dis. 1991;11:56. | ||
Gross JB Jr, Ludwig J, Wiesner RH, et al. Abnormalities in tests of copper metabolism in primary sclerosing cholangitis. Gastroenterology. 1985; 89:272 | ||
LaRusso NF, Wiesner RH, Ludwig J, et al. Prospective trial of penicillamine in primary sclerosing cholangitis. Gastroenterology. 1988;95:1036. | ||
Kowdley KV, Knox TA, Kaplan MM. Hepatic copper content is normal in early primary biliary cirrhosis and primary sclerosing cholangitis. Dig Dis Sci. 1994;39:2416. | ||
Epstein MP, Kaplan MM. A pilot study of etanercept in the treatment of primary sclerosing cholangitis. Dig Dis Sci. 2004;49:1. | ||
Hommes DW, Erkelens W, Ponsioen C, et al. A double-blind, placebo-controlled, randomized study of infliximab in primary sclerosing cholangitis. J Clin Gastroenterol. 2008;42:522. | ||
Lichtman SN, Sartor RB, Keku J, Schwab JH. Hepatic inflammation in rats with experimental small intestinal bacterial overgrowth. Gastroenterology. 1990;98:414. | ||
Färkkilä M, Karvonen AL, Nurmi H, et al. Metronidazole and ursodeoxycholic acid for primary sclerosing cholangitis: a randomized placebo-controlled trial. Hepatology. 2004;40:1379. | ||
Davies YK, Cox KM, Abdullah BA, et al. Long-term treatment of primary sclerosing cholangitis in children with oral vancomycin: an immunomodulating antibiotic. J Pediatr Gastroenterol Nutr. 2008;47:61. | ||
Silveira MG, Torok NJ, Gossard AA, et al. Minocycline in the treatment of patients with primary sclerosing cholangitis: results of a pilot study. Am J Gastroenterol. 2009;104:83. | ||
Tabibian JH, Weeding E, Jorgensen RA, et al. Randomised clinical trial: vancomycin or metronidazole in patients with primary sclerosing cholangitis – a pilot study. Aliment Pharmacol Ther. 2013;37:604. | ||
MacCarty RL, LaRusso NF, Wiesner RH, Ludwig J. Primary sclerosing cholangitis: findings on cholangiography and pancreatography. Radiology. 1983;149:39. | ||
May GR, Bender CE, LaRusso NF, Wiesner RH. Nonoperative dilatation of dominant strictures in primary sclerosing cholangitis. AJR Am J Roentgenol. 1985;145:1061. | ||
Stiehl A, Rudolph G, Klöters-Plachky P, et al. Development of dominant bile duct stenoses in patients with primary sclerosing cholangitis treated with ursodeoxycholic acid: outcome after endoscopic treatment. J Hepatol. 2002;36:151. | ||
Björnsson E, Lindqvist-Ottosson J, Asztely M, Olsson R. Dominant strictures in patients with primary sclerosing cholangitis. Am J Gastroenterol. 2004;99:502. | ||
Eckhauser FE, Colleti LM, Knol JA. The changing role of surgery for sclerosing cholangitis. Dig Dis. 1996;14:180. | ||
Cameron JL, Pitt HA, Zinner MJ, et al. Resection of hepatic duct bifurcation and transhepatic stenting for sclerosing cholangitis. Ann Surg. 1988;207:614. | ||
Goldenring JR, Cahow CE. Intrahepatic cholangiojejunostomy as a palliative procedure in primary sclerosing cholangitis. Arch Surg. 1989;124:565. | ||
Myburgh JA. Surgical biliary drainage in primary sclerosing cholangitis. The role of the Hepp-Couinaud approach. Arch Surg. 1994; 129:1057. | ||
Farges O, Malassagne B, Sebagh M, Bismuth H. Primary sclerosing cholangitis: liver transplantation or biliary surgery. Surgery. 1995;117:146. | ||
Pawlik TM, Olbrecht VA, Pitt HA, et al. Primary sclerosing cholangitis: role of extrahepatic biliary resection. J Am Coll Surg. 2008; 206:822. | ||
Cangemi JR, Wiesner RH, Beaver SJ, et al. Effect of proctocolectomy for chronic ulcerative colitis on the natural history of primary sclerosing cholangitis. Gastroenterology. 1989);96(3):790–794. | ||
Langnas AN, Grazi GL, Stratta RJ, et al. Primary sclerosing cholangitis: the emerging role for liver transplantation. Am J Gastroenterol. 1990;85:1136. | ||
McEntee G, Wiesner RH, Rosen C, et al. A comparative study of patients undergoing liver transplantation for primary sclerosing cholangitis and primary biliary cirrhosis. Transplant Proc. 1991;23:1563. | ||
Graziadei IW, Wiesner RH, Marotta PJ, et al. Long-term results of patients undergoing liver transplantation for primary sclerosing cholangitis. Hepatology. 1999;30:1121. | ||
Dickson ER, Murtaugh PA, Wiesner RH, et al. Primary sclerosing cholangitis: refinement and validation of survival models. Gastroenterology. 1992;103:1893. | ||
Fickert P, Wagner M, Marschall HU, et al. 24-norUrsodeoxycholic acid is superior to ursodeoxycholic acid in the treatment of sclerosing cholangitis in Mdr2 (Abcb4) knockout mice. Gastroenterology. 2006;130: 465–481. | ||
Halilbasic E, Fiorotto R, Fickert P, et al. Side chain structure determines unique physiologic and therapeutic properties of norursodeoxycholic acid in Mdr2-/- mice. Hepatology. 2009;49:1972–1981. | ||
O Chazouilleres CC, Gaouar F, Poupon R. Fenofibrate improves liver tests in primary sclerosing cholangitis with incomplete biochemical response to ursodeoxycholic acid. Hepatology. 2010;52:488A. | ||
Nakamuta M, Fujino T, Yada R, et al. Therapeutic effect of bezafibrate against biliary damage: a study of phospholipid secretion via the PPARalpha-MDR3 pathway. Int J Clin Pharmacol Ther. 2010;48:22–28. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.