Back to Journals » OncoTargets and Therapy » Volume 8
Primary paraesophageal Ewing’s sarcoma: an uncommon case report and literature review
Authors Tarazona N, Navarro L, Cejalvo JM, Gambardella V, Pérez-Fidalgo J, Sempere A, Navarro S, Cervantes A
Received 14 January 2015
Accepted for publication 12 March 2015
Published 14 May 2015 Volume 2015:8 Pages 1053—1059
DOI https://doi.org/10.2147/OTT.S80879
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Daniele Santini
Noelia Tarazona,1,* Lara Navarro,2,* Juan Miguel Cejalvo,3,* Valentina Gambardella,3,* J Alejandro Pérez-Fidalgo,3 Alejo Sempere,2 Samuel Navarro,2 Andrés Cervantes3
1Department of Medicine, GI and Lymphoma Unit, The Royal Marsden NHS Foundation Trust, London and Surrey, UK; 2Department of Pathology, 3Department of Hematology and Medical Oncology, Biomedical Research Institute, INCLIVA, University of Valencia, Valencia, Spain
*These authors contributed equally to this work
Abstract: Ewing’s sarcoma is a rare and highly aggressive cancer most frequently arising in people under 20 years of age. We report an uncommon case of primary paraesophageal Ewing’s sarcoma in a 25-year-old male harboring the infrequent EWSR1/ERG fusion transcript with multiple splice variants coexisting in the same tumor. The patient was totally refractory to chemotherapy and died 17 months after diagnosis. We underscore the need for better understanding of the molecular pathogenesis of the disease and improved systemic therapy options.
Keywords: Ewing’s sarcoma, recurrence, immunohistochemistry, fusion genes
Introduction
Ewing’s sarcoma is the second most frequent primary malignant bone cancer, after osteosarcoma.
It was first described by James Ewing in 1921, as an undifferentiated tumor developing in the diaphysis of the ulna of a young female patient.1
Ewing’s sarcoma is a rare disease, with approximately three cases per million per year. It is slightly more common in males than in females (55/45 ratio) and its incidence is nine times greater in Caucasians than in African-Americans. Over 50% of all patients are adolescents, although 20%–30% of the cases are diagnosed in the first decade of life.2,3
The tumor typically develops in the diaphyseal portion of flat bones of the axial skeleton and in long bones. At time of diagnosis, metastases are detectable in approximately 25% of the patients. The lungs, bone, or bone marrow are usually involved, while metastatic spread to the lymph nodes, liver, or central nervous system is infrequent.
Extraskeletal Ewing’s sarcoma is a very rare disease, accounting for 6%–47% of all cases of Ewing’s sarcoma. It is mainly diagnosed in the trunk, extremities, retroperitoneum, and head and neck region. Patients with extraosseous Ewing’s sarcoma are more likely to be older, female, and not of Caucasian origin. An extraskeletal origin of the disease is correlated to poor prognosis.4–6 We present an uncommon case of extraskeletal Ewing’s sarcoma, and discuss its rare presentation and evolution. To our knowledge, this is the first reported case of paraesophageal primary Ewing’s sarcoma and primitive neuroectodermal tumor.
Case report
A 25-year-old man with a history of herpes esophagitis 6 years prior to admission presented with progressive dysphagia and a palpable and painless right cervical mass for the past approximately 5 months.
Computed tomography (CT) and F-18 fluorodeoxyglucose-positron emission tomography revealed an eccentric hypermetabolic mass (4.1×6.8 cm; SUV >12) in the right esophageal wall, causing an important mass effect with tracheal and esophageal deviation. Neither lymph nodes nor other organs were involved (Figure 1).
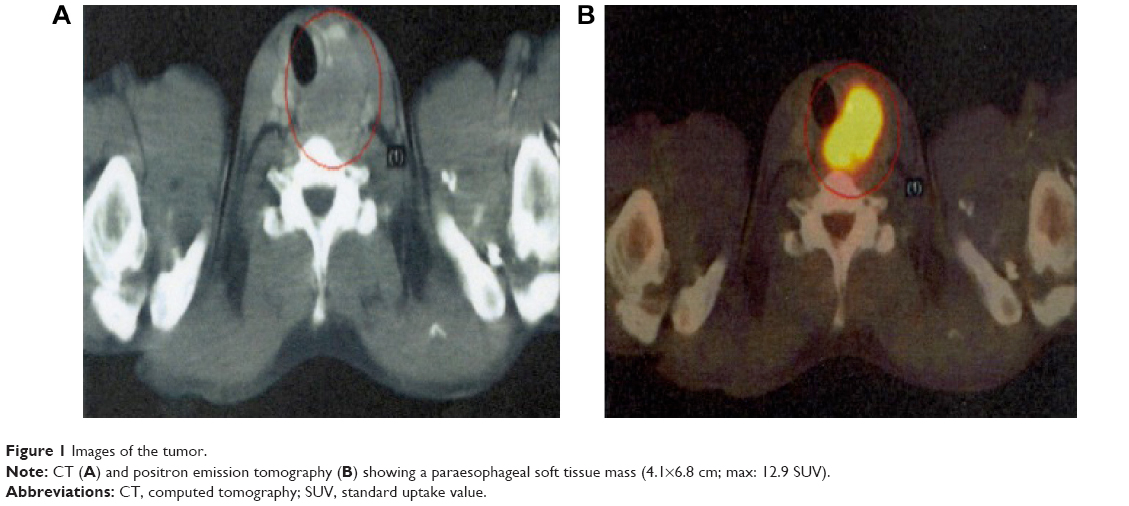 | Figure 1 Images of the tumor. |
The pathology report on the neck biopsy informed of an immature neoplasm with round and basophil cell proliferation forming a diffuse infiltrate compatible with round cell malignancy (Figure 2). The immunohistochemical study revealed intense expression of CD99/MIC2 +, c-kit +, FLI-1 +, and Ki67 + in 60% of the neoplastic cells (Figure 3). The rest of the tumor markers were negative. Reverse-transcription polymerase chain reaction (RT-PCR) identified a chimeric EWSR1/ERG fusion transcript distinctive of the t(21;22)(q22;q21) translocation. To our surprise, RT-PCR revealed two splice variants coexisting within the same tumor (EWSR1(ex7)-ERG(ex6) and EWSR1(ex7)-ERG(ex7)) (Figure 4). The diagnosis of extraskeletal Ewing’s sarcoma with high proliferation was confirmed.
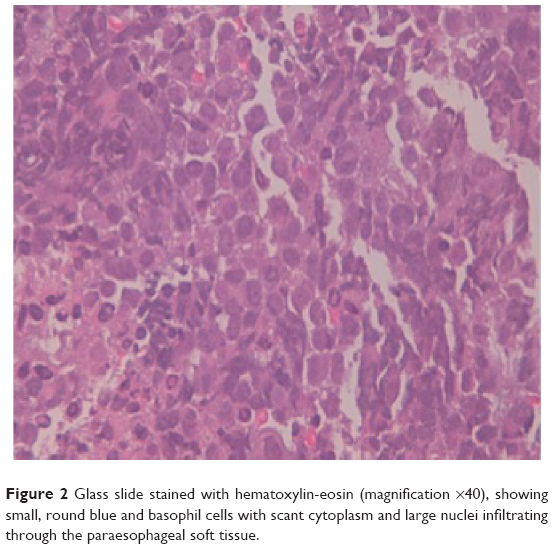 | Figure 2 Glass slide stained with hematoxylin-eosin (magnification ×40), showing small, round blue and basophil cells with scant cytoplasm and large nuclei infiltrating through the paraesophageal soft tissue. |
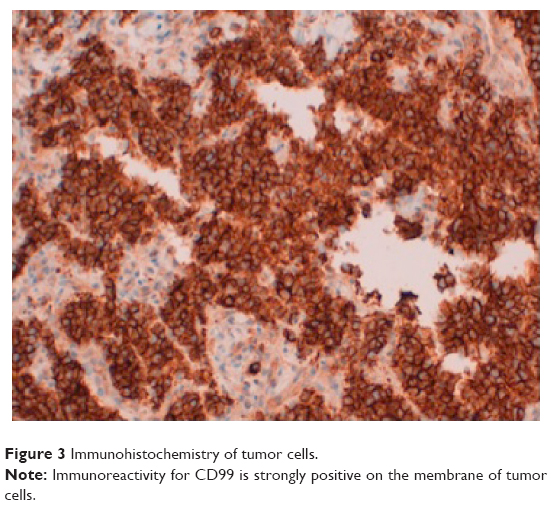 | Figure 3 Immunohistochemistry of tumor cells. |
 | Figure 4 Detection of a chimeric fusion transcript by RT-PCR analysis. |
Chemotherapy based on the therapeutic schedule vincristine, doxorubicin, and cyclophosphamide alternated with ifosfamide and etoposide every 3 weeks was administered for a total of 17 cycles. Locoregional treatment consisted of radiation therapy with a total dose of 54 Gy performed from the fourth month, while receiving concomitant chemotherapy.
Complete response after eight cycles was achieved, and the patient remained asymptomatic for 1 month after the last chemotherapy cycle. Subsequently, progressive dysphonia was detected, and laryngoscopy revealed right vocal cord paralysis, while the CT scan showed a well-delimited paraesophageal mass (4.5 cm) suggesting local recurrence. The latter was confirmed by the histological study (Figure 4).
After relapse, the patient was treated with a combination of irinotecan and temozolomide. Following the first cycle, he experienced rapid clinical progression. High-dose ifosfamide was therefore administered, with no clinical benefit after one cycle. Considering the aggressiveness of the disease, a gemcitabine-docetaxel regimen was started. The tumor remained totally refractory to chemotherapy after three cycles, with both locoregional and lung progression.
Regrettably, the patient died suddenly secondary to airway obstruction caused by progression of the disease.
Discussion
Paraesophageal Ewing’s sarcoma is an extremely rare entity. To our knowledge, only three cases of esophageal Ewing’s sarcoma have been reported in the literature to date.7–9 All cases occurred in people over 20 years of age. Skeletal and extraskeletal Ewing’s sarcoma are both characterized by the presence of monomorphic round cells with small hyperchromatic nuclei, inconspicuous nucleoli, scant cytoplasm, and extensive necrotic areas. The diagnosis of such infrequent presentations is a challenge. The histological and immunophenotypic features of Ewing’s sarcoma overlap with those of other small round cell tumors of childhood. For this reason, an expanded panel of immunohistochemical studies, fluorescent in situ hybridization and RT-PCR are strictly necessary to exclude other entities such as neuroblastoma, lymphoblastic lymphoma, poorly differentiated synovial sarcoma, etc.10
Typically, Ewing’s sarcoma cells express glycoprotein CD99/MIC2 and may be immunoreactive for VIM, NSE, S-100 protein, Leu-7, and/or PgP 9.5.11,12
The cytogenetic aberrations leading to activation of chimeric transcription factors are the most relevant features of these tumors. Among recurrent cytogenetic alterations, the t(11;22)(q24;q12) translocation has been detected in approximately 90% of all cases, leading to fusion between the EWSR1 on chromosome 22 and the FLI1 on chromosome 11, but t(21;22)(q22,q12)(EWSR1/ERG) and others may also occur.13,14
The EWSR1 gene is one of the genes most sensitive to translocation in soft tissue tumors and encodes the EWS protein, which is a member of a growing family of highly conserved RNA-binding proteins mediating interaction with RNA or single-stranded DNA. The codified protein takes part in transcriptional regulation for specific genes and in mRNA splicing. Specifically, EWRS1 is involved in transcription initiation. Concerning EWSR1 breakpoints, the main areas susceptible to breakage are EWSR1 exons 7, 8, 9, or 10.15,16 The proteins encoded by FLI1 and ERG genes are members of the ETS family of transcription factors, which represent the main gene family translocated with EWSR1, and are implicated in the control of cellular proliferation, development, and tumorigenesis.17 The chimeric protein resulting from fusion interferes with different molecular pathways crucial for cell growth, differentiation and proliferation, and which are frequently involved in the pathogenesis of soft tissue tumors.18
The transcription factor Erg is necessary in definitive hematopoiesis, adult hematopoietic stem cell function, and for the upkeep of peripheral blood platelet numbers.19 Chromosomal rearrangements involving ERG are found in myeloid leukemia, acute lymphoblastic leukemia, Ewing’s sarcoma, and most prostate cancers. However, the normal physiological function of Erg is unknown. Approximately, 10% of all Ewing’s sarcomas carry the t(21;22)(q22;q12)(EWSR1-ERG) translocation. ERG shares 68% overall amino acid identity with FLI1 and 98% identity within their ETS DNA-binding domains. Considering the structural similarities of EWSR1/FLI1 and EWSR1/ERG fusions, it is likely that the two proteins deregulate similar target genes in Ewing’s sarcoma. A retrospective study comparing EWSR1/FLI1 to EWSR1/ERG fusions in Ewing’s sarcoma cases demonstrated no significant differences in pathological and clinical features, in addition to overall survival. The most common breakpoint described for EWSR1 and ERG is EWSR1 exon 7, which translocates to ERG exon 6, 7, or 9.20
Interestingly, in our patient two different fusion transcripts within the same tumor, EWSR1(ex7)-ERG(ex6) and EWSR1(ex7)-ERG(ex7), were detected. However, this circumstance is a well-known phenomenon in human tumors, such as desmoplastic small round cell tumors, synovial sarcomas, clear cell sarcomas, and ETV6-ABL1 or PML-RARA-positive leukemia. Likewise, the coexistence of multiple fusion transcripts in the same patient seems to be distinctive but atypical for Ewing’s sarcoma. Of the 17 cases with different fusion transcripts within the same tumor reported in the literature, just one case exhibited splice variants derived from an ERG gene fragment, (erg-3), and the rest showed alternative splicing of exons from EWSR1 or FLI1 gene. 21 Thus, we present the first case reported in the literature in which two fusion transcripts in the same patient arise from splicing out of exons from the ERG gene. Two feasible explanations have been suggested to account for this phenomenon. One may be the occurrence of two distinct chromosomal rearrangements in the same patient, leading to either polyclonality of the same tumor or the unlikely development of two different Ewing’s sarcomas. The second and most probable explanation would be mRNA splicing based on most of the reported cases to date.
The etiology of Ewing’s sarcoma is unknown. Our patient had a history of herpes esophagitis 6 years before diagnosis of the tumor. However, the association between viral infections and Ewing’s sarcoma remains unclear. There is no evidence that herpes simplex virus is related to the origin of Ewing’s sarcoma. In the past, it was believed that cytomegalovirus and Epstein Barr virus could play a role in the pathogenesis of Ewing’s sarcoma though no sound supporting evidence has been obtained to date.22,23
Ewing’s sarcoma and primitive neuroectodermal tumor requires multimodal therapeutic strategies. The standard treatment for localized Ewing’s sarcoma is similar to that used in metastatic disease, and is based on the combination of doxorubicin, cyclophosphamide, ifosfamide, vincristine, dactinomycin and etoposide, along with local therapy for 10–12 months.24 The choice of local therapy includes surgery, radiotherapy, or both. The INT-0091 trial was the first randomized trial to demonstrate that there is no difference among the different local control modalities in terms of local recurrence and event-free survival.25 With surgery or radiotherapy alone, 5-year survival was <10% but on including chemotherapy, the survival rate increased by 60%–70% in localized and 20%–40% in metastatic disease. Although relevant progress has been made in the treatment of Ewing’s sarcoma in recent years, patients with localized tumor disease at the time of diagnosis show a 4-year relapse rate of 20%.
Ewing’s sarcoma patients presenting with recurrence or progressive disease less than 2 years from initial diagnosis present a 5-year survival of only 30%. At the same time, patients with metastatic disease at diagnosis, females, those with elevated LDH at diagnosis, and patients whose sites of first recurrence include both local and distant metastatic disease have a significantly poorer post-recurrence survival rate.26
There is no a standard treatment for refractory Ewing’s sarcoma. Treatment options including irinotecan, temozolomide, ifosfamide, gemcitabine, docetaxel, etoposide, platinum or vincristine have demonstrated response rate between 29% and 66%.27–31 However, many patients do not respond to these agents, and further chemotherapeutic options are limited in this setting. In the clinical setting, target cancer therapies under investigation in Ewing’s sarcoma include inhibitors of the IGF-1 receptor plus an inhibitor of mTOR, which has resulted in tumor regression in approximately 25%–30% of patients with refractory metastatic disease. In turn, bevacizumab, a VEGF inhibitor, administered as monotherapy, resulted in stable disease at the most during at least 4 months in three out of five patients enrolled in the COG Phase I trial. In preclinical models of Ewing’s sarcoma, a dual PI3K/AKT inhibitor and a combination of PARP inhibitors and temozolomide enhanced sensitivity to actinomycin D and synergistic effects in Ewing’s sarcoma, respectively.32–35 Unfortunately, the prognosis of patients with refractory or recurrent Ewing’s sarcoma remains dismal. This underlines the need for further research in this field.
Acknowledgment
Noelia Tarazona and Valentina Gambardella hold a grant from the European Society for Medical Oncology.
Disclosure
The authors state that they have no potential conflicts of interest.
References
Ewing J. The classic: Diffuse endotelioma of bone. Proc N Y Pathol Soc. 1921;21:17–24. | ||
Hense HW, Ahrens S, Paulussen M, Lehnert M, Jürgens H. [Descriptive epidemiology of Ewing’s tumor – analysis of German patients from EICESS 1980–1997]. Klin Padiatr. 1999;211(4):271–275. German. | ||
Jawad MU, Cheung MC, Min ES, Schneiderbauer MM, Koniaris LG, Scully SP. Ewing sarcoma demonstrates racial disparities in incidence-related and sex-related differences in outcome: an analysis of 1631 cases from the SEER database, 1973–2005. Cancer. 2009;115(15):3526–3536. | ||
Siegel RD, Ryan LM, Antman KH. Adults with Ewing’s sarcoma: an analysis of 16 patients at the Dana-Farber Cancer Institute. Am J Clin Oncol.1988;11(6):614–617. | ||
Fizazi K, Dohollou N, Blay JY, et al. Ewing’s family of tumors in adults: multivariate analysis of survival and long-term results of multimodality therapy in 182 patients. J Clin Oncol. 1983;16(12):3736–3743. | ||
Granowetter L, West DC. The Ewing’s sarcoma family tumors: Ewing’s sarcoma and peripheral neuroectodermal tumor of bone and soft tissue. Cancer Treat Res. 1997;92:253–308. | ||
Terrier-Lacombe MJ, Guillou L, Chibon F, et al. Superficial primitive Ewing’s sarcoma: a clinicophalogic and molecular cytogenetic analysis of 14 cases. Mod Pathol. 2009;22(1):87–94. | ||
Cabarcos Ortiz Barrón A, Cinza Sanjurjo S, Rivero Velasco C, Mariño Rozados A, Antúnez López J. [Esophageal Ewing’s sarcoma with tracheal invasion with presentation as asthmatic episode]. Rev Clin Esp. 2007;207(6):310–311. Spanish. | ||
Johnson AD, Pambuccian SE, Andrade RS, Dolan MM, Asian DL. Ewing sarcoma and primitive neuroectodermal tumor of the esophagus: report of a case and review of literature. Int J Surg Pathol. 2010;18(5):388–393. | ||
Machado I, Noguera R, Pellin A, et al. Molecular diagnosis of Ewing sarcoma family tumors: a comparative analyses of 560 cases with FISH and RT-PCR. Diagn Mol Pathol. 2009;18(14):189–199. | ||
Navarro S, Cavazzana AO, Llombart-Bosh A, Triche TJ. Comparison of Ewing’s sarcoma of the bone and peripheral neuropithelioma, an immunocytochemical and ultrastructural analysis of two primitive neuroectodermal neoplasms. Arch Pathol Lab Med. 1994;118(6):608–615. | ||
Dierick AM, Roeles H, Langlois M. The immunophenotype of Ewing’s Sarcoma. An immunohistochemical analysis. Pathol Res Pract. 1993;189(1):26–32. | ||
Delattre O, Zucman J, Plougastel B, et al. Gene fusion with an ETS DNA binding domain caused by chromosome traslocation in human tumours. Nature. 1992;359(6391):162–165. | ||
Sorensen PH, Lessnick SL, Lopez-Terrada D, Liu XF, Triche TJ, Denny CT. A second Ewing’s sarcoma translocation, t(21;22), fuses the EWS gene to another ETS-family transcription factor, ERG. Nat Genet. 1994;6(2):146–151. | ||
Stolow DT, Haynes SR. Cabeza, a Drosophila gene encoding a novel RNA binding protein, shares homology with EWS and TLS, two genes involved in human sarcoma formation. Nucleic Acids Res. 1995;23(5):835–843. | ||
Sankar S, Lessnick SL. Promiscuous partnerships in Ewins’s sarcoma. Cancer Genet. 2011;204(7):351–365. | ||
Hromas R, Klemsz M. The ETS oncogene family in development, proliferation and neoplasia. Int J Hematol. 1994;59(4):257–265. | ||
Cantile M, Marra L, Franco R, et al. Molecular detection and targeting of EWSR1 fusion transcripts in soft tissue tumors. Med Oncol. 2013;30(1):412. | ||
Loughran SJ, Kruse EA, Hacking DF, et al. The transcription factor Erg is essential for definitive haematopoiesis and the function of adult hematopoietic stem cells. Nat Immunol. 2008;9(7):810–819. | ||
Zoubek A, Dockhorn-Dworniczak B, Delattre O, et al. Does expression of different EWS chimeric transcripts define clinically distinct risk groups of Ewing tumor patients? J Clin Oncol. 1996;14(4):1245–1251. | ||
Patócs B, Németh K, Garami M, et al. Multiple splice variants of EWSR1-ETS fusion transcripts co-existing in the Ewing sarcoma family of tumors. Cell Oncol (Dordr). 2013;36(3):191–200. | ||
Cope JU. A viral etiology for Ewing’s sarcoma. Med Hypotheses. 2000;55(5):369–372. | ||
Kebudi R, Bilgic B, Görgun O, Ayan I, Demiryont M. Is the Epstein Barr virus implicated in Ewing’s sarcoma? Med Pediatr Oncol. 2003;40(4):256–257. | ||
Grier H, Krailo M, Tarbell NJ, et al. Addition of ifosfamide and etoposide to standard chemotherapy for Ewing’s sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med. 2003;348(8):694–701. | ||
Yock TI, Krailo M, Fryer CJ, et al. Local control in pelvic Ewing sarcoma: analysis from INT-0091-a report from the children’s Oncology Group. J Clin Oncol. 2006;24(24):3838–3843. | ||
Leavey PJ, Mascarenhas L, Marina N, et al. Prognostic factors for patients with Ewing sarcoma (EWS) at first recurrence following multi-modality therapy: A report from the Children’s Oncology Group. Pediatr Blood Cancer. 2008;51(3):334–338. | ||
Casey DA, Wexler LH, Merchant MS, et al. Irinotecan and temozolomide for Ewing sarcoma: the Memorial Sloan-Kettering experience. Pediatr Blood Cancer. 2009;53(6):1029–1034. | ||
Ferrari S, del Prever AB, Palmerini E, et al. Response to high-dose ifosfamide in patients with advanced/recurrent Ewing sarcoma. Pediatr Blood Cancer. 2009;52(5):581–584. | ||
Mora J, de Torres C, Parareda A, et al. Treatment of Ewing Sarcoma Family of Tumors With a Modified P6 Protocol in Children and Adolescents. Pediatr Blood Cancer. 2011;57(1):69–75. | ||
EI Weshi A, Memon M, Raja M, et al. VIP (etoposide, ifosfamide, cisplatin) in adult patients with recurrent or refractory Ewing sarcoma family of tumors. Am J Clin Oncol. 2004;27(5):529–534. | ||
Raciborska A, Bilska K, Drabko K, et al. Vincristine, irinotecan, and temozolomide in patients with relapsed and refractory Ewing sarcoma. Pediatr Blood Cancer. 2013;60(10):1621–1625. | ||
Naing A, Kurzrock R, Burger A, et al. Phase I trial of cixutumumab combined with temsirolimus in patients with advanced cancer. Clin Cancer Res. 2011;17(18):6052–6060. | ||
Glade Bender JL, Adamson PC, Reid JM, et al. Phase I trial and pharmacokinetic study of bevacizumab in pediatric patients with refractory solid tumors: a Children’s Oncology Group Study. J Clin Oncol. 2008;26(3):399–405. | ||
Yamamoto T, Ohno T, Wakahara K, et al. Simultaneous inhibition of mitogen-activated protein kinase and phosphatidylinositol 3-kinase pathways augment the sensitivity to actinomycin D in Ewing sarcoma. J Cancer Res Clin Oncol. 2009;135(8):1125–1136. | ||
Brenner JC, Feng FY, Han S, et al. PARP-1 inhibition as a targeted strategy to treat Ewing’s sarcoma. Cancer Res. 2012;72(7):1608–1613. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.