Back to Journals » International Journal of Women's Health » Volume 6
Primary ovarian insufficiency: an update
Received 5 November 2013
Accepted for publication 25 December 2013
Published 20 February 2014 Volume 2014:6 Pages 235—243
DOI https://doi.org/10.2147/IJWH.S37636
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Leticia Cox, James H Liu
UH Case Medical Center, MacDonald Women's Hospital, Department of Obstetrics and Gynecology, Case Western Reserve University School of Medicine, Department of Reproductive Biology, Cleveland, OH, USA
Abstract: Primary ovarian insufficiency is a condition that represents impaired ovarian function on a continuum with intermittent ovulation. This condition commonly leads to premature menopause, defined as cessation of ovulation prior to the age of 40 years. Because there are potential immediate and long-term consequences of hypoestrogenism, a timely diagnosis is invaluable. This comprehensive review will discuss identifiable causes for primary ovarian insufficiency, including genetic disorders and metabolic abnormalities, as well as review current strategies for diagnosis, evaluation, and management of women with this condition.
Keywords: premature ovarian failure, premature menopause, ovarian dysfunction
Introduction
It is estimated that approximately 90% of women undergoing menopause will do so between the age of 45 and 55 years, with the average age of menopause occurring at 51 years.1,2 Unlike the age of menarche, which has decreased over the past decade, there has been no apparent drift in the age of natural menopause. These observations suggest that the age of menopause is probably an evolutionarily conserved trait. Premature menopause describes menopause that occurs prior to the age of 40 years. This condition affects approximately 1% of the population in the US.3 An earlier onset of menopause can be spontaneous or the result of medical interventions such as surgical removal of the ovaries or be due to chemotherapy or radiation treatment with subsequent ovarian damage. When menopause is surgically induced, it is associated with a rapid decline in ovarian hormone levels and consequently more severe menopausal symptoms. These symptoms include hot flashes, sleep disturbances, mood liability, and decreased energy.
Primary ovarian insufficiency
Primary ovarian insufficiency (POI) is a term that is increasingly used and has been adopted to encompass diagnostically similar conditions, including hypergonadotropic hypogonadism, premature ovarian failure, and ovarian dysgenesis. This terminology is semantically more accurate because it is a term used to describe impaired ovarian function on a continuum4 rather than a specific endpoint. This condition can be transient or progressive, and usually results in eventual premature menopause.4 The condition affects one in 10,000 women by the age of 20 years and one in 100 by the age of 40 years.5 Conceptually, POI is characterized by one of two processes, ie, ovarian follicular dysfunction or depletion of functional primordial follicles before the age of 40 years. The diagnosis should be confirmed by obtaining two follicle-stimulating hormone (FSH) levels in the menopausal range (>30 U/L) at least 1 month apart in the setting of 4–6 months of amenorrhea.
Patients typically present with oligomenorrhea or amenorrhea and may exhibit increasing symptoms of estrogen deficiency. Additionally, patients can experience long-term consequences of hypoestrogenism, including osteoporosis, accelerated cardiovascular aging, and neurocognitive disorders. Infertility is another consequence of POI. Although most patients will present with amenorrhea, about 50% will have varying degrees of residual ovarian function. It is estimated that approximately 5%–10% are able to conceive spontaneously.1
Three potential mechanisms can be associated with POI, ie, a congenital decrease in primordial follicles, accelerated follicular atresia, and an inability to recruit primordial follicles.6 Unfortunately, for most patients presenting with POI, the cause is largely unexplained. Potential etiologies for POI can be divided into genetic, autoimmune, metabolic dysfunction, infectious, and iatrogenic categories. Each are separately discussed below6 and are listed in Table 1
 | Table 1 Causes of primary ovarian insufficiency |
Genetic disorders associated with POI
X chromosome disorders
Disorders that involve the X chromosome and loci that regulate germ cell development and viability are linked to POI. Turner syndrome (45,X) is associated with streak gonads and other stigmata, including short stature, a broad and webbed neck, coarctation of the aorta, a shortened fourth metacarpal, pigmented nevi, an ogival palate, cognitive deficits, vertebral abnormalities, and renal anomalies. The prevalence is about one in 2,500 female births, with 80% of cases being maternal in origin.7 Because animal models with monosomy X have normal ovarian development, it is assumed that crucial genes on the normal X chromosome are inactivated.8
Levels of circulating adrenal androgens and their conversion to estrogen account in part for development of pubic/axillary hair. Müllerian derivatives and female external genitalia are well differentiated, but without significant estrogen exposure remain small and infantile in size. A small percentage (3%–5%) of adults with Turner syndrome will menstruate spontaneously and exhibit breast development. In these cases, 45,X/46,XX mosaicism should be suspected. If pregnancy is desired, hormone replacement therapy can be initiated to increase uterine size, followed by assisted reproductive technology, namely in vitro fertilization with an oocyte donor. The success rate of clinical pregnancies using this approach is approximately 50% per cycle without an increased risk of chromosomal abnormalities in the offspring.9 However, coexisting cardiac abnormalities associated with Turner syndrome may increase the risk of pregnancy for the mother. Because the maternal risk in individuals with Turner syndrome is unpredictable and there is a risk of unanticipated maternal cardiac decompensation, the American Society for Reproductive Medicine discourages this type of approach to achieve pregnancy.
X isochromosome
Normal division of the centromere occurs in the longitudinal plane. When the centromere splits abnormally in the transverse plane, the resulting chromosome pair contains structurally identical arms with identical genes. The isochromosome for the long arm (q) is the most common X structural abnormality. These patients have streak gonads and tend to have the Turner stigmata.7
Microdeletions on the X chromosome undetectable by conventional karyotyping are also found in women with POI. Although there are regions on the short (p) and long arms (q) of the X chromosome that are designated ovarian genes (POF1 and POF2, respectively), women with premature ovarian failure have been noted to have alterations outside these designated areas. A more detailed list of known gene mutations are, listed in Table 2 and 3.
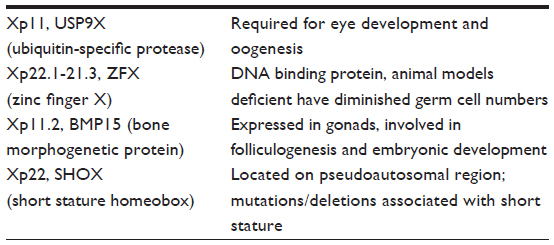 | Table 2 Proposed gene mutations on Xp associated with primary ovarian insufficiency |
 | Table 3 Proposed gene mutations on Xq associated with primary ovarian insufficiency |
FMR1 gene
Fragile X syndrome is an inherited X-linked dominant disorder that is a leading cause of inherited cognitive disability. The degree of cognitive disability is typically more severe in males although both males and females can be affected.10 The Fragile X mental retardation 1 (FMR1) gene is mapped on the X chromosome at position q27.3. The fragile site of the X chromosome contains a trinucleotide (CGG) repeat in the 5′ region of the gene. In normal variants, the trinucleotide repeat ranges from six to 55 repeats. When the repeat expands to 55–200, the individuals are considered premutation carriers, and those above 200 are considered affected.10 It has been shown that females carrying a premutation have up to a 23% rate of POI and experience earlier menopause by approximately 5 years.10 In female premutation carriers, the ovarian dysfunction depends on CGG repeat size, although the relationship is not linear. The underlying molecular mechanisms of how Fragile X causes POI are unknown, although there is a hypothesis that the FMR1 premutation may have a toxic RNA gain-of-function effect on ovarian follicle dynamics.10
46,XX associated with POI
XX gonadal dysgenesis not associated with phenotypic anomalies are most commonly inherited in an autosomal recessive fashion. There is variance in phenotypic penetration noted among siblings.11–13 It has been challenging to identify the specific autosomal genes responsible for various forms of XX gonadal dysgenesis. There are sporadic cases associated with reciprocal autosomal translocations that have not been easily reproduced. Mouse models suggest alterations at the following gene regions may impact POI: 4p11–q12 (tyrosine kinase receptor), 12q22 (mast cell growth factor), 9q33 (nuclear receptor factor), 6p21.3 (DNA mismatch repair), and 18q21.3 (cell death repressor protein).14
Syndromic ovarian failure
Studies of individuals with cerebellar ataxia with XX gonadal dysgenesis have shown variance in the manifestation of mental retardation, cataracts, and neurosensory deafness, suggesting a single mutant gene is unlikely to explain all causes.15
Gonadal dysgenesis and multiple malformation syndromes
Other variants presenting with XX gonadal dysgenesis have associated somatic features, including microcephaly, arachnodactyly, epibulbar dermoids, short stature, metabolic acidosis, blepharophimosis-ptosis-epicanthus, Malouf syndrome (dilated cardiomyopathy, mental retardation, blepharoptosis), and limb-mammary syndrome (ectrodactyly, ectodermal dysplasia, cleft lip/palate).16–18
Metabolic abnormalities associated with POI
A number of inherited enzymatic pathway disorders have been associated with ovarian follicular dysfunction leading to POI. Galactose 1-phosphate uridylyltransferase deficiency (galactosemia) was one of the first to be characterized.19 The gene encoding this enzyme is located on 9p and the pathogenesis is believed to involve toxic accumulation of galactose during infancy. Phenotypic features of this disorder involve defects in the ocular, renal, and hepatic systems.19 Because galactosemia requires treatment in childhood to prevent mental retardation, it is unlikely to be diagnosed in an otherwise healthy adult presenting only with POI.
Carbohydrate-deficient glycoprotein deficiency
This condition is most commonly caused by a missense mutation located on chromosome 16p13, a phosphomannomutase gene mutation (PMM1). The enzyme defect results in accumulation of mannose-6-phosphate which cannot be converted to mannose-1-phospate, and is associated with a wide variety of neurologic abnormalities, including ataxia, hyperreflexia, hypotonia, joint contractions, and epilepsy.20
17 α-hydroxylase/17,20 desmolase deficiency
Defects in the sex steroid biosynthetic pathway can lead to predictable consequences depending on the enzyme deficiency. When 17 α-hydroxylase/17,20-lyase is deficient, pregnenolone cannot be converted to 17 α-hydroxypregnenolone. This leads to a reduced ability to produce cortisol, androstenedione, testosterone, and estrogens. This condition in XX genetic females will be associated with normal appearing external genitalia, but affected individuals fail to undergo secondary sexual development at puberty. The ovaries do respond to exogenous gonadotropins. The disorder is a result of an autosomal recessive mutation isolated to a single gene, ie, CYP17 located on 10q24–25.21
Aromatase mutations
Deficiency of aromatase activity prevents conversion of androgens to estrogens and is often associated with clitoral hypertrophy and primary amenorrhea in females.22 Treatment with exogenous estrogen will induce the growth spurt, breast development, and menstrual cycles.
Other disorders leading to ovarian follicular dysfunction
Other disorders leading to ovarian follicular dysfunction include mutations in hormone receptors and downstream post-receptor signaling. Although extremely rare, mutations in the structure, metabolism, or action of gonadotropins can cause POI. Mutations in the FSH receptor, first described in a Finnish population, will cause elevation of FSH levels. These patients often present with primary or secondary amenorrhea and elevated FSH levels, but will have normal-appearing ovarian follicles visualized on transvaginal ultrasound.23 Savage syndrome describes individuals with gonadotropin post-receptor defects. These women present with elevated luteinizing hormone and FSH levels; however, their ovaries contain multiple immature ovarian follicles. These women have normal karyotypes and are resistant to exogenous gonadotropin stimulation.23
Autoimmune-associated causes of POI
POI can be associated with a variety of autoimmune disorders, most commonly thyroiditis.23 There is also a strong association between POI and autoimmune polyendocrine syndrome. This condition may include hypothyroidism, adrenal insufficiency, hypoparathyroidism, and type 1 diabetes mellitus.23 Other associated autoimmune disorders include dry eye syndrome, myasthenia gravis, rheumatoid arthritis, systemic lupus erythematosus, and congenital thymic aplasia. Currently, there are no standardized tests to identify the presence of ovarian autoantibodies but it is now possible to screen for antiadrenal antibodies.23
Infectious causes of POI
Mumps oophoritis has been directly linked to POI, although data are scarce. It should be suspected in women presenting with mumps parotitis and lower abdominal pain. More recently, there has been suggestion that human immunodeficiency virus (HIV) infection (or antiviral therapy) can lead to POI. A prospective study in 2010 evaluated 78 HIV-positive women and found that these women tended to have abnormal levels of antral follicular count (63%), FSH (36%), inhibin B (57%), and anti-Müllerian hormone (AMH, 23%) relative to the normal population.24
Iatrogenic causes of POI
Medical treatment for neoplastic conditions can be associated with POI. Chemotherapy and radiotherapy are well documented causes of POI. Both significantly reduce ovarian function. In general, the rate of follicular atresia is directly related to level of exposure. Chemotherapy induces apoptosis of mature ovarian follicles. Histologic examination of ovarian tissue after treatment will often show cortical fibrosis, vascular damage, and decreased follicular numbers.25 Vinca alkaloids, anthracycline antibiotics, and antimetabolites appear to be low risk for inducing gonadotoxicity. Alkylating agents (ie, cyclophosphamide) carry a much higher risk and are cytotoxic even if the ovary is in a resting state. These agents will lead to POI in approximately 40% of women. Suppression of luteinizing hormone and FSH secretion with a gonadotropin-releasing hormone agonist prior to chemotherapy appears to reduce ovarian damage,26 whereas administration of oral contraceptives appears to have little protective effect.
Direct radiation to the pelvis for treatment of Hodgkin lymphoma accelerates atresia in ovarian follicles. Even with pelvic shielding, it is difficult to protect ovarian function which is highly sensitive to radiation. One study concluded that 26% of women with total abdominal radiation for approximately 3.5 years developed POI by age 23 years.27 Surgical transposition of the ovaries outside the pelvis, well away from the radiation field, is effective at minimizing radiation exposure. For women using this approach, assisted reproductive technology procedures are often required to achieve fertility.
Surgical causes of POI
Aside from surgical menopause due to bilateral oophorectomy, surgical removal of the uterus can result in earlier menopause in some women. This is likely explained by inadvertent damage to the ovarian blood supply during surgery. One study noted the average age of menopause in women who had undergone isolated hysterectomies was 45.0±4.0 years. This is significantly lower than the average age of menopause in women who have not undergone this surgery.28,29
Racial differences in ovarian reserve
Recent evidence supports the notion that ovarian reserve differs among varying racial/ethnic groups. Seifer et al provided evidence of racial differences in ovarian reserve as measured by AMH.30 This study compared AMH levels at two time points in a racially diverse, multicenter cohort study of HIV-infected women (median age 37.5 years and 43.3 years). Black women demonstrated average AMH levels approximately 25.2% lower than those of whites after controlling for age, smoking, body mass index, and HIV status. Additionally, AMH levels in Hispanic women were 24.6% lower than those in white women, although this difference did not reach statistical significance.30
Diagnosis and evaluation
Current studies have failed to determine specific biomarkers or signs/symptoms of POI that will accurately predict when menopause will occur. Patients may present with a shortening or increase in the intermenstrual cycle interval, menstrual irregularities including oligomenorrhea, dysfunctional uterine bleeding, or amenorrhea. Women may note symptoms of estrogen deficiency such as vasomotor symptoms, mood disturbances, and atrophic vaginitis, although these later symptoms are usually delayed. Patients can also experience long-term consequences of hypoestrogenism which include osteoporosis, accelerated cardiovascular aging, and neurocognitive disorders.1
Unfortunately, standardized diagnostic criteria for POI have yet to be established. It is advisable to perform a complete history and physical examination to exclude secondary causes of amenorrhea. These conditions include pregnancy, polycystic ovarian syndrome, hypothalamic amenorrhea, chronic medical illness secondary to poorly controlled diabetes or celiac disease, lifestyle habits (extreme exercise, poor caloric intake), hypothalamic or pituitary lesions, hyperprolactinemia, hypothyroidism, and hyperthyroidism. The screening history should also focus on a family history of early menopause, previous ovarian/pelvic surgery, as well as chemotherapy or radiation therapy which may identify a cause. The clinician should probe for a personal or family history of autoimmune disorders (eg, thyroid disorders, diabetes, Addison’s disease, vitiligo, systemic lupus, rheumatoid arthritis, celiac disease), fragile X syndrome, or intellectual disability.4
The physical examination should focus on body habitus, evidence of normal secondary sexual characteristics, as well as evidence of vaginal atrophy secondary to hypoestrogenism. For women with 3 consecutive months of amenorrhea, laboratory testing should include human chorionic gonadotropin, FSH, thyroid-stimulating hormone, prolactin, and estradiol levels. If serum estradiol is low and FSH is elevated, these values should be repeated in 1 month. If FSH remains elevated, additional testing, including a peripheral karyotype, fragile X screen, antiadrenal antibodies, serum calcium, and a morning cortisol, should be obtained (see Figure 1 and Table 4). Should a Y chromosome be identified, the patient should be counseled regarding gonadal removal because these individuals have an increased potential for malignancy.
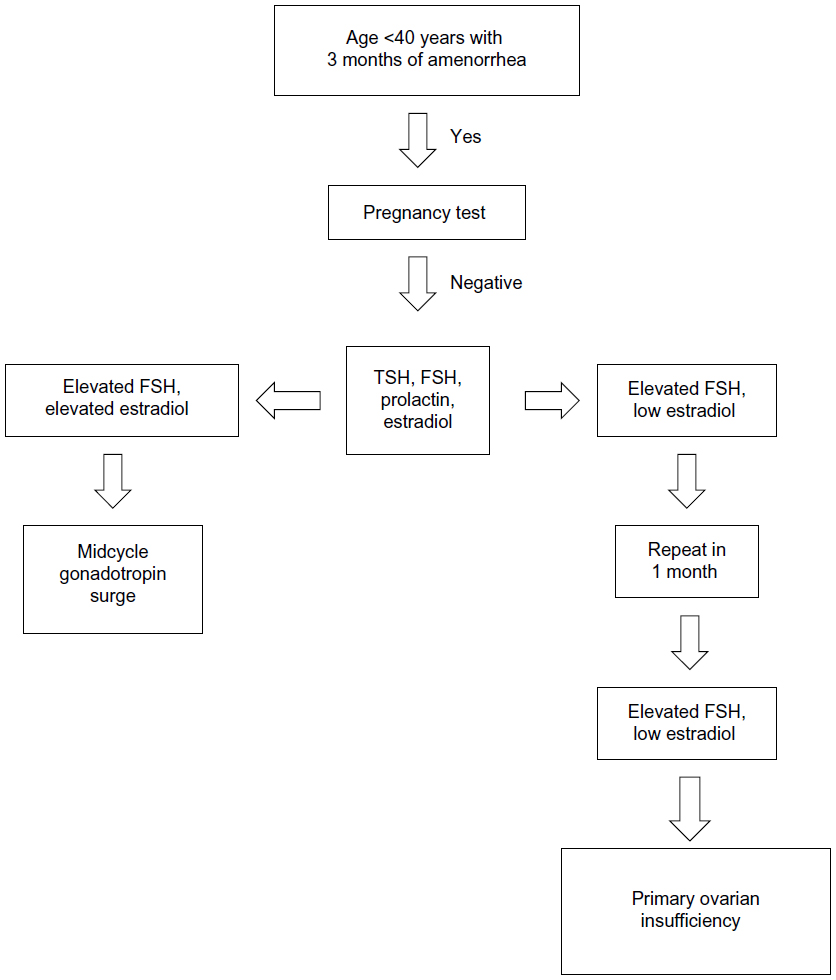 | Figure 1 Laboratory testing to confirm diagnosis of primary ovarian insufficiency. |
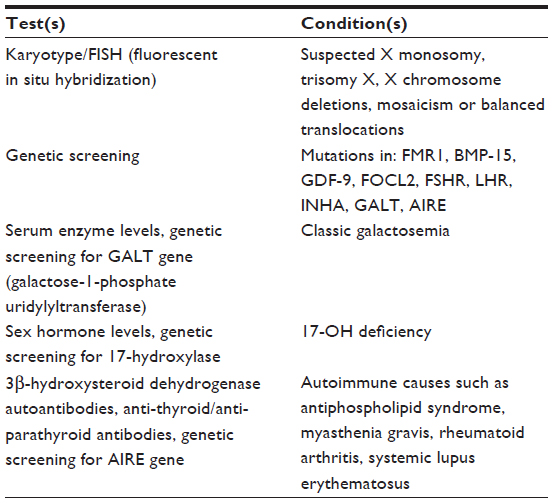 | Table 4 Diagnostic testing for conditions associated with primary ovarian insufficiency |
For those patients with an estradiol level >50 pg/mL and/or intermittent uterine bleeding, there may be some evidence for residual ovarian function. For those desiring fertility, low-dose hormone replacement (ie, estradiol 2 mg orally or estradiol 50 μg/day transdermally with cyclic progesterone 200 mg/day for 12 days each month) will allow ovulation to occur and should be effective in treating hypoestrogenic symptoms. For women on this type of regimen, failure to have a withdrawal bleed during progesterone treatment may indicate pregnancy.
Because it is possible to have periodic estrogen production in POI, administering a progesterone withdrawal test may not be particularly helpful. Other diagnostic tests include transvaginal ultrasound imaging of the ovaries. Findings of a normal ovarian size/volume and presence of a high antral ovarian follicle count makes the diagnosis of POI less likely. For those women with diminished ovarian function, options to assess ovarian reserve include cycle day 3 FSH levels, AMH, inhibin B, and transvaginal ultrasound-determined antral follicle count. Elevated FSH levels drawn on day 3 imply a poor ovarian reserve.31,32
As a consequence of decreased estrogen levels, women with POI often do not achieve peak bone density and may experience loss of bone mass. If hormone therapy is initiated and the woman has not experienced fractures, it is not necessary to do bone mineral density testing.
Management of POI
A multidimensional approach should be undertaken to manage POI (see Table 5). This includes initiation and management of hormone replacement therapy, contraception or fertility management, psychosocial support, and annual screening to assess thyroid function, adrenal function, and routine preventive health care.
 | Table 5 Treatment of primary ovarian insufficiency |
Hormone replacement therapy
Hormone replacement therapy (HRT) is advised not only to provide relief from menopause-related symptoms (ie, vasomotor symptoms and vaginal atrophy) but also to maintain bone density and reduce the risk of cardiovascular disease.33 Starting estrogen doses should be equivalent to the mid-follicular phase menstrual cycle estrogen concentrations, ie, providing the equivalent of 50–100 μg transdermal estradiol daily.30,31 To reduce the risk of endometrial hyperplasia, 5–10 mg of medroxyprogesterone acetate should be given for 12 days of the month, provided that the uterus is present. Although there is no clinical trial to lend support to an optimal length of time HRT should continue for women with POI, it is generally recommended that this regimen be continued at least until the average age of natural menopause (age 51 years).32 The risks of HRT are low and do not appear to be significantly different from those women with ongoing ovarian function at a perimenopausal age. If HRT is contraindicated or declined, weight-bearing exercises, increased calcium and vitamin D intake, and avoidance of tobacco and alcohol should be recommended. It must be emphasized that these later strategies have been shown to be inadequate at maintaining bone density in the reproductive-aged population. In addition, bone density should be monitored in those women who are not on hormone therapy.
Follow-up visits should be scheduled at 6–12-month intervals with periodic testing of thyroid-stimulating hormone, calcium, and cortisol levels. If antiadrenal antibodies are present, an adrenocorticotropic hormone stimulation test to assess adrenal reserve or referral to an endocrinologist is advised.
Contraception/fertility management
It is important to establish plans for conception soon after the diagnosis of POI is made. There is the distinct possibility of spontaneous and unpredictable ovulation. For those who are not interested in achieving a pregnancy, contraception should be offered. Combined oral contraceptives have the added benefit of providing HRT but intrauterine devices or barrier methods are also acceptable alternatives. For those opting for an intrauterine device or barrier methods, supplemental HRT should be advised.
Fertility preservation
Ideally, counseling regarding future childbearing options will be with a specialist in fertility prior to initiation of chemotherapy or radiation treatment. Women undergoing these treatments are in a unique position in that there is a potential for fertility preservation. Options for fertility preservation include ovarian hyperstimulation with oocyte retrieval followed by oocyte or embryo cryopreservation, ovarian tissue cryopreservation, or ovarian suppression with a gonadotropin-releasing hormone agonist. It should be noted that not all of these options are available for women at all centers, and some techniques such as ovarian tissue cryopreservation are still considered experimental. To date, the fertility treatment with the highest success rate has been in vitro fertilization with use of donor oocytes. This has been associated with a live birth rate of approximately 30%–40% per embryo transferred. Other alternatives include cryopreservation of embryos and or oocytes for use in future in vitro fertilization cycles. However, the current evidence indicates inferiority in pregnancy success rates when compared with use of donor oocytes. This is due to our limited ability to reliably stimulate mature follicles from stored ovarian tissue, frozen oocytes, and the likelihood that follicles harvested from women with underlying POI would be of lower quality. Embryo donation and adoption are other alternatives that should be discussed.
Psychologic support
Women undergoing POI may experience significant psychologic disturbances.34 Some will experience a range of emotions, and providers should offer support regarding infertility, altered self-image, and sexual dysfunction.35 Patients may benefit from referral to a psychologist and support groups, such as the International Premature Ovarian Failure Association.35
Summary
POI represents a continuum from impaired ovarian function with intermittent ovulation to premature menopause as the defining end point, characterized by permanent loss of ovarian function. Because of the potential long-term consequences of hypoestrogenism, including cardiovascular disease, neurocognitive decline, menopausal symptoms, osteoporosis, and infertility, it is important to establish a correct diagnosis expeditiously and identify associated medical conditions. Management should reflect a comprehensive approach, including providing hormone replacement therapy, fertility management, and physical or emotional support. The need for long-term follow-up of this population with preventive maintenance therapy and periodic surveillance cannot be overemphasized.
Disclosure
The authors report no conflicts of interest in this work.
References
McKinlay SM, Brambilla DJ, Posner JG. The normal menopause transition. Maturitas. 1992;14:103–115. | |
Miro F, Parker SW, Aspinall LJ, Coley J, Perry PW, Ellis JE. Sequential classification of endocrine stages during reproductive aging in women: the FREEDOM study. Menopause. 2005;12:281–290. | |
Coulam CB, Bustillo M, Schulman JD. Empty follicle syndrome. Fertil Steril. 1986;46:1153–1155. | |
Nelson LM. Clinical practice. Primary ovarian insufficiency. N Engl J Med. 2009;360:606–614. | |
Rafique S, Sterling E, Lawrence N. A new approach to primary ovarian insufficiency. Obstet Gynecol Clin N Am. 2012;39:567–586. | |
Persani L, Rossetti R, Cacciatore C. Genes involved in human premature ovarian failure. J Mol Endocrinol. 2010;45:257–279. | |
Simpson JL. Gonadal dysgenesis and abnormalities of the human sex chromosomes: current status of phenotypic-karyotypic correlations. Birth Defects Orig Artic Ser. 1975;11:23–59. | |
Willard HF. The sex chromosomes and X chromosome inactivation. In: Scriver CR, Beaudet AL, Sly WS, et al, editors. The Metabolic and Molecular Bases of Inherited Disease. New York, NY, USA: McGraw-Hill; 2001;8. | |
Foudila T, Soderstrom-Antilla V, Hovatta O. Turner’s syndrome and pregnancies after oocyte donation. Hum Reprod. 1999;14:532–535. | |
Willemsen R, Levenga J, Oostra B. CGG repeat in the FMR1 gene: size matters. Clin Genet. 2011;80:214–225. | |
Simpson JL. Gonadal dysgenesis and sex chromosome abnormalities. Phenotypic/karyotypic correlations. In: Vallet HL, Peter IH, editors. Genetic Mechanisms of Sexual Development. New York, NY, USA: Academic Press; 1994. | |
Simpson JL, Christakos AC, Horwith M, Silverman FS. Gonadal dysgenesis associated with apparently normal chromosomal complements. Birth Defects. 1971;12:215–218. | |
Boczkowski K. Pure gonadal digenesis and ovarian dysplasia in sisters. Am J Obstet Gynecol. 1970;106:626–628. | |
Simpson JL, Rajkovic A. Ovarian differentiation and gonadal failure. Am J Med Genet. 1999;89:186–200. | |
Skre H, Bassöe HH, Berg K, Frövig AG. Cerebellar ataxia and hypergonadotropic hypogonadism in two kindreds. Chance concurrence, pleiotropism or linkage? Clin Genet. 1976;9:234–244. | |
Maximilian C, Ionesca B, Bucur A. [Two sisters with major gonadal dysgenesis, dwarfism, microcephaly, arachnodactyly, and normal karyotype 46, XX]. J Genet Hum. 1970;18:365–378. French. | |
Quayle SA, Copeland KC. 46,XX gonadal dysgenesis with epibulbar dermoid. Am J Med Genet. 1991;40:75–76. | |
Pober BR, Zemel S, Hisama FM. 46, XX gonadal dysgenesis, short stature and recurrent metabolic acidosis in two sisters. Am J Hum Genet. 1998;63:A117. | |
Kaufman FR, Kogut MD, Donnell GN, Goebelsmann U, March C, Koch R. Hypergonadotropic hypogonadism in female patients with galactosemia. N Engl J Med. 1981;304:994–998. | |
Bjursell C, Stibler H, Wahlström J, et al. Fine mapping of the gene for carbohydrate-deficient glycoprotein syndrome, type 1 (CDG1): linkage disequilibrium and founder effect in Scandinavian families. Genomics. 1997;39:247–253. | |
Yanase T. 17α-hydroxylase/17,20-lyse defects. J Steroid Biochem Mol Biol. 1995;53:153–157. | |
Mullis PE, Yoshimura N, Kuhlmann B, Lippuner K, Jaeger P, Harada H. Aromatase deficiency in a female who is compared heterozygote for two new point mutations in the P450arom gene: impact of estrogens on hypergonadotropic hypogonadism, multicystic ovaries, and bone densitometry in childhood. J Clin Endocrinol Metab. 1997;82:1739–1745. | |
Rebar R. Evaluation of amenorrhea, anovulation, and abnormal bleeding. Available from: http://www.endotext.org/chapter/evaluation-of-amenorrhea-anovulation-and-abnormal-bleeding/?singlepage=true. Accessed October 10, 2013. | |
Ohl J, Partisani M, Demangeat C, Binder-Foucard F, Nisand I, Lang JM. [Alterations of ovarian reserve tests in human immunodeficiency virus (HIV)-infected women.] Gynecol Obstet Fertil. 2010;38:313–317. French. | |
Clowse ME, Behera MA, Anders CK, et al. Ovarian preservation by GnRH agonists during chemotherapy; a meta-analysis. J Womens Health (Larchmt). 2009;18:311–319. | |
Fleischer R, Vollenhoven B, Weston G. The effects of chemotherapy and radiotherapy on fertility in premenopausal women. Obstet Gynecol Surv. 2011;66:248–254. | |
De Moraes-Ruehsen M, Jones G. Premature ovarian failure. Fertil Steril. 1967;18:440–461. | |
Farquhar CM, Sadler L, Harvery SA, Stewart AW. The association of hysterectomy and menopause: a prospective cohort study. BJOG. 2005;112:956–962. | |
Siddle N, Sarrel P, Whitehead M. The effect of hysterectomy on the age at ovarian failure: identification of a subgroup of women with premature loss of ovarian function and literature review. Fertil Steril. 1987;47:94–100. | |
Seifer DB, Golub ET, Lambert-Messerlian G, et al. Variations in serum Mullerian inhibiting substance between white, black and Hispanic women. Fertil Steril. 2009;92:1674–1678. | |
Rebar RW. Premature ovarian failure. Obstet Gynecol. 2009;113:1355–1363. | |
Parker WH, Broder MS, Chang E, et al. Ovarian conservation at the time of hysterectomy and long-term health outcomes in the Nurses’ Health Study. Obstet Gynecol. 2009;113:1027–1037. | |
Rivera CM, Grossardt B, Rhodes D, et al. Increased cardiovascular mortality after early bilateral oophorectomy. Menopause. 2009;16:15–23. | |
van der Stege JG, Groen H, van Zadelhoff SJ, et al. Decreased androgen concentrations and diminished general and sexual well-being in women with premature ovarian failure. Menopause. 2008;15:23–31. | |
International Premature Ovarian Failure Association. Alexandria, VA, USA. Available from: http://www.pofsupport.org. Accessed August 10, 2013. | |
Simpson J. Ovarian dysgenesis and premature ovarian failure caused by X chromosomal abnormalities. The Global Library of Women’s Medicine; 2011. Available from: http://www.glowm.com/section_view/heading/Ovarian%20Dysgenesis%20and%20Premature%20Ovarian%20Failure%20Caused%20by%20X%20Chromosomal%20Abnormalities/item/353. Accessed August 10, 2013. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.