Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Preparation, pharmacokinetics and biodistribution of baicalin-loaded liposomes
Authors Wei Y, Guo J, Zheng X, Wu J, Zhou Y, Yu Y, Ye Y, Zhang L, Zhao L, Zou Y, Zheng W, Liu H
Received 16 April 2014
Accepted for publication 24 May 2014
Published 1 August 2014 Volume 2014:9(1) Pages 3623—3630
DOI https://doi.org/10.2147/IJN.S66312
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Yumeng Wei,1,* Jianmin Guo,2,* Xiaoli Zheng,2,* Jun Wu,3 Yang Zhou,1 Yu Yu,4 Yun Ye,1 Liangke Zhang,4 Ling Zhao1
1School of Pharmacy, 2Institute of Basic Medical Sciences, Luzhou Medical College, Luzhou City, Sichuan Province, People’s Republic of China; 3Department of Clinical Pharmacy and Outcomes Sciences, College of Pharmacy, University of South Carolina, Greenville, SC, USA; 4School of Pharmacy, Chongqing Medical University, Chongqing, People’s Republic of China
*These authors contributed equally to this work
Abstract: Baicalin (BA) is a major constituent of Scutellaria baicalensis Georgi, a medicinal herb. Previous pharmacokinetic studies of BA showed its low oral bioavailability. The aim of the present study was to develop a novel BA-loaded liposome (BA-LP) to enhance oral bioavailability. BA-LP, composed of BA, Tween® 80, Phospholipon® 90H, and citric acid at weight ratio of 96/50/96/50, respectively, was prepared by the effervescent dispersion technique and characterized in terms of morphology, size, zeta potential, encapsulation efficiency, and the in vitro release. Pharmacokinetics and biodistribution studies were carried out in rats after oral administration of BA-LP and a carboxymethyl cellulose suspension containing BA (BA-CMC) as a control. BA-LP exhibited a spherical shape by transmission electron microscopy observation. BA-LP had a mean particle size of 373±15.5 nm, zeta potential of -20.1±0.22 mV, and encapsulation efficiency of 82.7%±0.59%. The BA-LP showed a sustained-release behavior, and the in vitro drug-release kinetic model fit well with the Weibull distribution equation: lnln (1/(1-Q)) =0.609 lnt -1.230 (r=0.995). The oral bioavailability and the peak concentration of the BA-LP was threefold and 2.82-fold that of BA-CMC, respectively. The in vivo distribution results indicated that drug concentrations were significantly increased in the liver, kidney, and lung in the case of BA-LP, which were 5.59-fold, 2.33-fold, and 1.25-fold higher than those of BA-CMC, respectively. In conclusion, the study suggested that BA-LP might be a potential oral drug delivery system to improve bioavailability of BA.
Keywords: nanoliposomes, bioavailability, nanoliposomes, in vitro release, bioavailability, in vivo evaluation
Introduction
The dry roots of Scutellaria baicalensis have been frequently used in traditional Chinese medicine for the treatment of inflammation, fever, ulcer, and cancer.1–3 In recent years, a large number of pharmacological studies have shown that flavonoids were the main active constituents in S. baicalensis. 3–5 At present, more than 60 flavonoids have been isolated from S. baicalensis and identified. Baicalin (BA) is one of main active flavonoids of S. baicalensis Georgi. Chemically, BA (Figure 1) is a 7-D-glucuronic acid, 5,6-dihydroxy flavone.6 Though BA has been widely used in a number of therapeutic areas because of its antioxidant, anti-inflammatory, antibacterial, and anticancer effects,6–8 its poor solubility (the maximum solubility is reported to be 91 μg/mL in water at 20°C) and low oral bioavailability (the absolute bioavailability was 2.2% after oral administration of BA in rats) severely limit its clinical application.6,9,10
 | Figure 1 Chemical structure of baicalin. |
In order to overcome the shortcoming of BA, the development of novel dosages and formulations of BA has attracted an increasing attention in the pharmaceutical field in recent years. These have included microspheres,11 microcapsules,12 nanoemulsions,13 inclusion complexes,14,15 solid lipid nanoparticles,16 BA-polyvinylpyrrolidone coprecipitate, and a BA-phospholipid complex.17,18 Indeed, the studies have shown an increased solubility and improvement of oral bioavailability in the preclinical phase. However, the currently marketed preparations, including BA capsules and tablets, have to be administered at a large dose to obtain effective drug plasma concentration, due to low oral bioavailability.13 Therefore, it is necessary for us to develop a novel drug delivery system to improve the oral bioavailability of BA.
Liposomes, a versatile and effective nanometer-scale drug delivery system, seem to be one of the most promising of the candidate agents and are nontoxic, biocompatible, and biodegradable.19–21 They can carry hydrophilic and lipophilic as well as amphoteric drug molecules entrapped either in the core or in the liposome bilayer. It has been demonstrated that liposomes can increase drug solubility and stability as well as provide good drug loading for both hydrophilic and lipophilic drugs.22–28 Thus, a number of studies showed the liposome carrier could improve the oral bioavailability of poorly bioavailable drugs and change the in vivo distribution of entrapped drugs.29–32 For example, Wang et al developed long-circulating nanoliposomes of quercetin, prepared by the emulsified evaporation–low temperature solidification method, using glyceryl behenate and Tween® 80 as carriers. The researchers showed that the oral absorption of the long-circulating quercetin liposomes in the liposomal formulation, in mice, was better than that of quercetin suspension.33 Elsewhere, it was shown that the oral bioavailability of curcumin liposomes was significantly improved after oral administration in rats.34 Finally, puerarin liposomes were prepared by film dispersion ultrasonication. The results of pharmacokinetic studies showed the relative availability of puerarin liposome suspension to the puerarin solution was 168%, which indicated that use of liposome as a drug carrier could enhance the oral absorption of puerarin in rats.35
Therefore, the objective of this study was to develop a novel BA-loaded nanoliposome (BA-LP) using the effervescent dispersion technique, to improve its oral bioavailability, and to characterize BA-LP in terms of morphology, size, zeta potential, encapsulation efficiency, and the in vitro release. Pharmacokinetics and biodistribution studies were carried out in rats after oral administration of BA-LP and a carboxymethyl cellulose (CMC) suspension containing BA (BA-CMC), as reference preparation.
Materials and methods
Materials
Baicalin was purchased from Mianyang Dongfangyuan Bio-Technology Co., Ltd. (Mianyang, People’s Republic of China). Rutin (internal standard) was obtained from Chengdu Mansite Pharmaceutical Co., Ltd. (Chengdu, People’s Republic of China). Phospholipon® 90H (HSPC) was from Nattermann-Phospholipid GmbH (Cologne, Germany); citric acid was from Hunan Dongting Citric Acid Co., Ltd. (Yueyang, People’s Republic of China); carbonic acid monosodium salt (NaHCO3) was from Hunan Huari Medicine Co., Ltd. (Changzhi, People’s Republic of China). Tween 80 (injection grade) was from Beijing Huiyou Chemical Co., Ltd. (Beijing, People’s Republic of China); dehydrated alcohol was from Qingdao Xingguang Chemical Co., Ltd. (Qingdao, People’s Republic of China); acetonitrile and methanol (high-performance liquid chromatography [HPLC]-grade) was from Merck KGaA (Darmstadt, Germany); ammonium acetate was from YSCHEM Co., Ltd. (People’s Republic of China); and 0.9% sodium chloride injection was obtained from Sichuan Kelun Pharmaceutical Co., Ltd. (Chengdu, People’s Republic of China). Ultrapure water used in this study was prepared using a water purification system.
Animals
Sprague-Dawley rats of either sex (200–250 g) were from the Laboratory Animal Center of Luzhou Medical College (Luzhou, Sichuan, People’s Republic of China) and were maintained at the temperature of 20°C±2°C on a 12-hour light–dark cycle, with relative humidity of 50%–60% and with free access to food and water. They were fasted for 12 hours before oral administration. All procedures involving rats were approved by the Luzhou Medical College Animal Ethical Experimentation Committee.
Preparation of BA-LP
BA-LP was prepared by the effervescent dispersion technique as described in our previous research, with modification.23,24 Briefly, BA (96 mg), HSPC (96 mg), Tween 80 (50 mg), and citric acid (50 mg) were dissolved in 5 mL of ethanol. The solution was filtered through a 0.22 μm hydrophobic membrane and then added into 10 mL of NaHCO3 water solution (0.35%, w/v) at 5°C–8°C while the system was stirred, using a half-moon paddle stirrer at 1,000 rpm, for about 1 hour. The mixture solution was heated to 25°C while continually stirred until it became less opaque. The BA-LP solution was collected and stored at 2°C–8°C for further study.
Physicochemical characterization of BA-LP
The morphologies of BA-LP were examined by transmission electron microscopy (TEM). The BA-LP samples were diluted with ultrapure water and dropped on a Formvar®-coated copper grid and then were air-dried for 1 minute at room temperature before negative staining with phosphotungstic acid solution (2%, w/v).
The particle size and zeta potential of BA-LP was measured with a Malvern Zetasizer Nano ZEN3600 analyzer (Malvern Instruments Ltd, Malvern, UK). The samples of BA-LP were diluted to the appropriate concentration with ultrapure water before measurement.
Entrapment efficiency of BA-LP
The BA content of BA-LP was detected with HPLC. An HPLC assay was carried out using a reverse-phase C18 column (Inertsil® ODS-SP; 4.6×250 mm, 5 μm particle size; GL Sciences, Tokyo, Japan) with a guard column (Phenomenex C18, 4.0×3.0 mm, 5 μm particle size; Phenomenex Torrance, CA, USA) on a Dionex Ultimate® 3000 series HPLC system (Thermo Fisher Scientific Inc, Waltham, MA, USA) (with a LPG-3400SD pump; VWD-3100 ultraviolet-visible [UV-Vis] [spectrophotometer] detector; WPS-3000 auto injector; and TCC-3000 column oven). The methanol, acetonitrile, and 0.4% (v/v) aqueous phosphoric acid (7.5:7.5:85, v/v) was chosen as a mobile phase at the detection wavelength of 278 nm. The flow rate was 1.0 mL/min at 35°C, and the injection volume was 20 μL.
The entrapment efficiency of BA-LP was measured using the dialysis method, as described in our previous study.27 The BA-LP sample was diluted with ultrapure water, and then each 1 mL of BA-LP was dialyzed against 250 mL of phosphate-buffered saline (PBS) (pH 7.4) for 8 hours. After dialysis, free drug was determined by HPLC, as established in this study. An aliquot of 0.2 mL of BA-LP was vortex-mixed with 0.8 mL of methanol for 5 minutes and then was filtrated through a 0.22 μm hydrophobic Millipore membrane (Taizhou Luqiao Sijia Biochemical Plastic Factory, Zhejiang, People’s Republic of China), and the total drug contents in the BA-LP sample was determined by HPLC. The entrapment efficiency was calculated by the following equation:
|
|
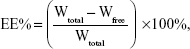
where EE% was the entrapment efficiency, Wfree was the amount of free BA in the BA-LP sample, and Wtotal was the total amount of BA in BA-LP sample.
Stability in gastrointestinal media
To investigate the stability of BA-LP in gastrointestinal environment, the liposomal formulation was incubated in simulated gastric fluid (SGF) (pH 1.2) and simulated intestinal fluid (SIF) (pH 6.8). The former was prepared by dissolving pepsin (Zhengzhou Hongxiang Chemical Co., Ltd., Zhengzhou, People’s Republic of China) (200 mg) in 10 mL of water containing 0.7 mL concentrated HCl, after which NaCl (200 mg) was added, and the solution volume was made up to 100 mL with water. The pH value of the final solution was adjusted to 1.2 with concentrated HCl. The latter was prepared by dissolving KH2PO4 (Zhengzhou Tianbang Chemical Co., Ltd., Zhengzhou, People’s Republic of China) (680 mg) in water (20 mL), followed by addition of 7.7 mL of NaOH (0.2 M) and pancreatin (1 g). The solution volume was adjusted up to 100 mL with water, and the pH value was adjusted to 6.8 with NaOH, followed by addition of sodium taurocholate (Wuhan Boxing Bio-Technology Co., Ltd., Wuhan, People’s Republic of China) (6 mM), to investigate the effect of bile salts. Then, 1 mL of BA-LP suspension was introduced into the SGF or SIF (9 mL), followed by its incubation for 3 and 6 hours in the gastrointestinal media. The effect of gastrointestinal environment on the stability of BA-LP was evaluated by measuring the particle size and entrapment efficiency.
In vitro release studies
The in vitro release studies of BA-LP vs BA solution were performed using the dialysis bag method (molecular weight cutoff [MWCO] 8,000–14,000 Da), with 100 mL of PBS (pH 7.4) as a release medium. Briefly, 1 mL of BA-LP and free-BA solution as control containing BA 7.5 mg/mL, respectively, was placed into dialysis bags and tightly sealed. And then, the test bags were immersed in release medium at a stirring rate of 100 rpm and at 37°C. At predetermined time points, 0.5 mL dissolution media was withdrawn, which was complemented with 0.5 mL of fresh release medium, at 37°C, to maintain the same volume. The sample solution was centrifuged at 10,000 rpm for 10 minutes, and the supernatant was then injected into the HPLC system. The in vitro release behaviors were plotted and fitted using different release dynamic models.
Pharmacokinetics and tissue distribution studies
The rats used in this study were randomly divided into four groups with five animals each. Groups 1 and 2 received the oral BA-LP at a dose of 100 mg/kg of BA. Groups 3 and 4 received the same oral dose in the form of BA-CMC as a control. For the pharmacokinetics studies, 0.3 mL of whole-blood samples from groups 1 and 3 was collected at 0.083, 0.25, 0.5, 1.0, 1.5, 2, 3.0, 5.0, 8.0, 12, and 24 hours following the oral administration and immediately centrifuged at 5,000 rpm for 10 minutes to obtain plasma samples. The supernatant plasma (0.1 mL) was transferred to new tubes and stored at −20°C until analysis. For the tissue distribution studies, at 30 minutes after the oral administration, the whole-blood samples were collected, after which the rats were sacrificed, by cervical dislocation, to obtain tissue samples. Heart, liver, spleen, lung, kidney, stomach, and brain were immediately removed and washed with 0.9% saline solution; then, redundant liquid was wiped using an absorbent paper and the specimens were weighed, and homogenized, and were frozen at −20°C until analyzed by HPLC.
The plasma or tissue homogenate samples (0.1 mL) were spiked with 50 μL of rutin internal standard solution (80.0 μg/mL in methanol) and 50 μL of ammonium acetate buffer (pH 3.5; 1 M) and vortexed for 1 minute in a 1.5 mL polypropylene tube, and then mixed with 0.6 mL of acetonitrile by vortex-mixing for 3 minutes. After centrifugation at 8,000 rpm for 10 minutes, the clear supernatant was collected and evaporated to dryness under a nitrogen gas stream at 40°C. The residue was then dissolved with 200 μL of mobile phase, and the solution was centrifuged at 10,000 rpm for 10 minutes. Then, 20 μL of the clear supernatant was injected into the HPLC system.
Data analysis
The plasma drug concentration–time curve was analyzed using the software program DAS 2.0 (Drug and Stastistics) was provided from Anhui Provincial Center for Drug Clinical Evaluation http://www.drugsina.com/. A noncompartmental model was chosen to investigate the pharmacokinetic parameters, including the area under the plasma drug concentration–time curve (AUC0–t), the mean residence time, the elimination half-life, apparent volume of distribution (Vz), clearance rate (CLz), and the peak concentration (Cmax).
All results were expressed as the mean ± standard deviation (SD), and statistically significant differences between the pharmacokinetic data of BA-LP and BA-CMC, as control, were evaluated using a two-tailed t-test. The level of significant difference was set at P<0.05.
Results
Preparation and characterization of BA-LP
On the basis of the single factor experiments, formulation and preparation technology were optimized with encapsulation efficiency as the index. The optimization formulation was composed of Tween 80 (50 mg), BA (96 mg), citric acid (50 mg), and HSPC (96 mg), based on the orthogonal experiment. The image from the TEM showed that BA-LP had a round shape and was 300–400 nm in size. The particle size of BA-LP measured by the Malvern ZEN 3600 was 373±15.5 nm, which was consistent with the results obtained from TEM. The zeta potential of BA-LP was −20.3±0.52 mV. The entrapment efficiency of BA-LP was 82.7%±0.59%.
Stability in simulated gastrointestinal fluid
The effect of SGF on the stability of BA-LP is summarized in Table 1. BA-LP was found to be unstable in SGF (pH 1.2) and SIF (pH 6.8). The particle size and entrapment efficiency of BA-LP were significantly affected (P<0.05) when exposed to SGF. In the case of SIF, no significant difference in particle size was observed, while significant change in entrapment efficiency occurred (P<0.05).
 | Table 1 Stability of BA-LP in simulated gastrointestinal fluid |
In vitro release of BA from BA-LP
In the in vitro release study, dialysis bag method was chosen for separation of free BA from BA-LP, and the results are shown in Figure 2 and Table 2. BA solution (BA was dissolved in 0.1% NaOH, then HCl was used to adjust to pH 7.3) released rapidly and was almost completed within 8 hours. However, the BA-LP released 35%±0.65%, 68%±1.01% and 82%±1.67% of BA within 2, 8, and 20 hours of dialysis, respectively.
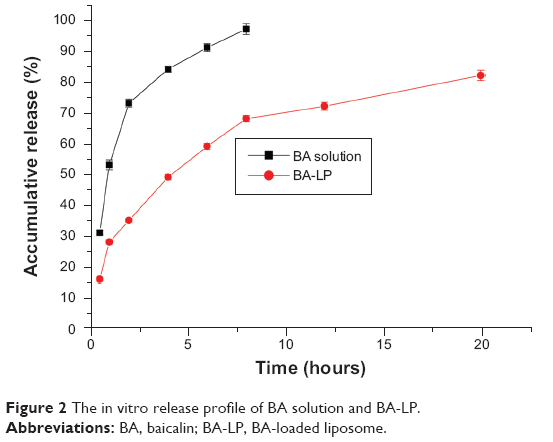 | Figure 2 The in vitro release profile of BA solution and BA-LP. |
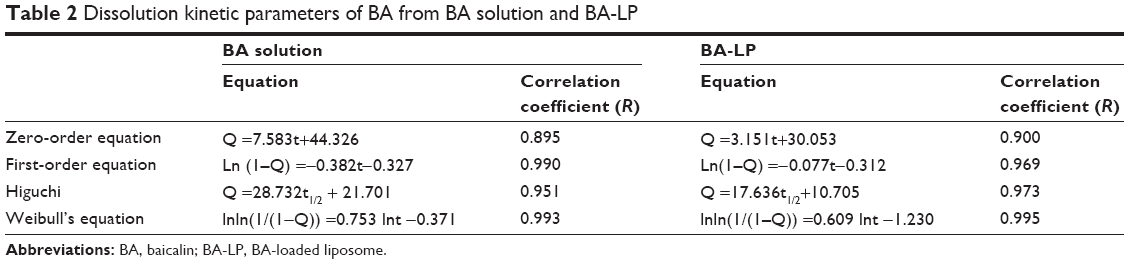 | Table 2 Dissolution kinetic parameters of BA from BA solution and BA-LP |
Pharmacokinetics study
To investigate the pharmacokinetic behavior of BA in the form of liposomes and in a CMC suspension, each formulation, at a dose of 100 mg/kg of BA, was orally administered to rats. The plasma drug concentration–time profiles of BA, after oral administration of BA solution and BA-LP, are shown in Figure 3. The plasma drug concentration–time data of BA was analyzed with a noncompartmental model, and the results of the pharmacokinetic parameters are shown in Table 3.
 | Figure 3 The plasma concentration–time profile of BA after oral administration of BA solution and BA-LP to rats, at a dose of 100 mg/kg. |
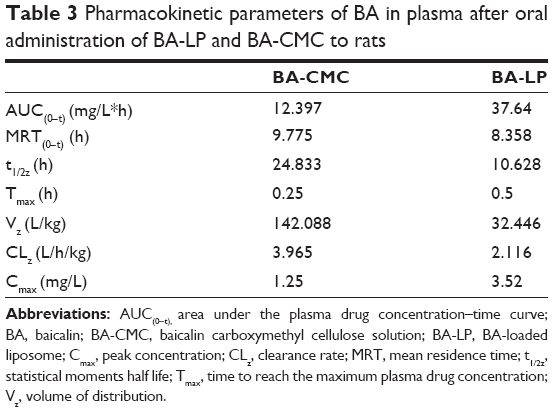 | Table 3 Pharmacokinetic parameters of BA in plasma after oral administration of BA-LP and BA-CMC to rats |
Tissue distribution study
The in vivo tissue distribution of BA was evaluated after oral administration of the BA-CMC suspension and BA-LP at a dose of 100 mg/kg to rats, and the results are shown in Figure 4.
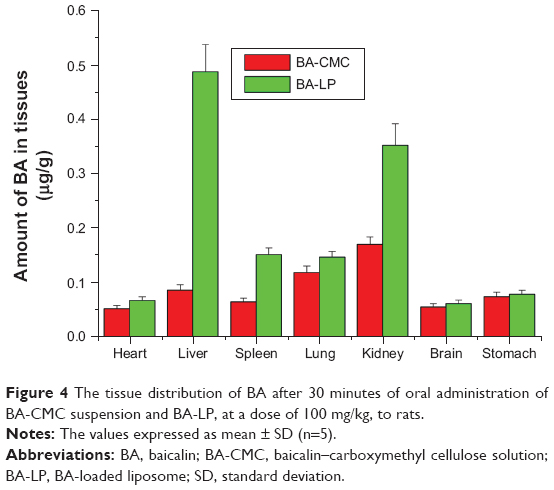 | Figure 4 The tissue distribution of BA after 30 minutes of oral administration of BA-CMC suspension and BA-LP, at a dose of 100 mg/kg, to rats. |
Discussion
Among the various new drug delivery dosage formats and technologies, the liposomal formulation is considered to be a relatively nontoxic technology that can encapsulate both lipophilic and hydrophilic drugs.22–28 Furthermore, the liposomes can improve the oral bioavailability of drugs poorly absorbed through the gastrointestinal tract and alter the in vivo distribution of the drugs encapsulated into liposomes, to increase the therapeutic index.22,29–32 Currently, liposomes have been considered one of the most promising drug delivery carriers. Several preparation technologies for liposome development have been studied in recent years, including the film hydration method, ethanol injection method, and high-pressure homogenization.22 However, each of the aforementioned technologies has its own shortcomings, such as the need for the use of toxic organic solvent or high operative temperature. Therefore, it is necessary to develop a novel preparation technology to obtain liposomes.
BA is a promising compound that has potential use in the treatment of several diseases, either administered alone or by a combined use with another agents.6 However, due to its low hydrophilicity and poor absorption following oral administration, the low oral bioavailability of BA limits its therapeutic efficacy and clinical application.13,16 Therefore, on the basis of our previous studies,23,24 the novel effervescent dispersion technique was used to develop BA-LP for the first time.
Three batches of BA-LP were prepared using the optimization formulation, to investigate the production method. The resulting BA-LP was round in shape, with a particle size of 373±15.5 nm. The zeta potential of BA-LP was −20.3±0.52 mV, which indicated that there were a great number of negative charges on the surface of BA-LP. It was found from the previous studies that the higher the absolute value of the zeta-potential (more than 15 mV), the stronger is the electrostatic repulsion between particles, which makes these liposome particles more difficult to congregate and more stable in the dispersal system.26,36 The entrapment efficiency of BA-LP was 82.7%±0.59%, which was higher than that (40.4%–60.1%) of previous studies.37,38 Chen et al and Liu et al developed BA liposomes, prepared by the reverse-phase evaporation method, with entrapment efficiency of 40.4% and 60.1%, respectively.37,38 This suggested that different formulations and preparation technology for liposomes development could result in significant differences in entrapment efficiency.
In order to investigate the stability of BA-LP in the gastrointestinal tract, the liposomal formulation was incubated with SGF (pH 1.2) and SIF (pH 6.8). BA-LP was quite unstable when exposed to SGF. The excess of hydrogen ion in the SGF could diffuse in the inner water phase of the BA-LP, which resulted in aggregation and destabilization of the liposomes.30 When exposed to SIF, BA-LP was unstable in the case of entrapment efficiency. It was suggested that the constituents of SIF, like bile salts, acted as surfactants and increased the solubilization of the lipid, which also led to destabilization of the liposomal formulation.39
The in vitro release studies were performed in PBS (pH 7.4). From Figure 2, the first 2 hours was an initial burst-release phase, which could be related to the release of the drug adsorbed on the surface of liposomes. Thereafter, the release rate became slow, demonstrating a typical sustained and prolonged drug-release behavior. From Table 2, the in vitro drug-release kinetic model of BA-LP fit well with the Weibull distribution equation: lnln (1/(1−Q)) =0.609 lnt −1.230 (r=0.995). Therefore, it was speculated that the sustained-release property of BA-LP can enhance the absorption of BA in the gastrointestinal tract.
The results of the pharmacokinetic studies exhibited a biphasic absorption phenomenon, shown in Figure 3, namely, the first absorption peak appeared at about 0.25 hours, then formed the second absorption peak at about 1.25 hours, both of which were shorter than those (0.5 and 5 hours, respectively) of previous study.16 On the basis of the results of reported studies,6,16 it is suggested that the enterohepatic recycling of BA played an important role in the absorption of BA through the gastrointestinal tract: Thus, BA itself was poorly absorbed from the rat gastrointestinal tract, where it was hydrolyzed to its aglycone moiety, namely baicalein, by intestinal bacteria; then the baicalein was absorbed rapidly and converted back to BA in the systemic circulation, resulting in the second absorption peak.40
The plasma drug concentration–time data of BA was analyzed with a noncompartmental model, and the results of pharmacokinetic parameters are shown in Table 3. Significant differences in the pharmacokinetic parameters of BA were observed between BA-CMC and BA-LP. The Cmax of BA-LP was 2.82-fold that of the BA-CMC suspension (P<0.05). The AUC0−t of BA in the form of BA-LP was significantly increased and was 3.04-fold that of BA-CMC. The increased Cmax and AUC0−t showed an increased oral absorption. However, compared with BA-CMC, apparent Vz, and CLz of BA were reduced in the case of BA-LP, which might result in a decrease in the chance of liver metabolism, to improve the oral bioavailability of BA.41 In addition, the increased oral bioavailability of BA in the form of BA-LP might be attributed to the combination of several effects.42–44 First, the particle size of BA-LP played a key role in the nanoparticle absorption rate – in general, the decreased particle size may improve the drug dissolution rate by increasing the surface area, to increase absorption in the gastrointestinal tract. Second, the HSPC materials might improve the permeation of BA through the gastrointestinal membrane, to increase oral absorption. Third, the bioadhesive character of BA-LP could increase the affinity between the nanoliposomes and gastrointestinal membrane, therefore, the extended residence time at the site of absorption would help to increase the uptake of BA.
It can be seen from the in vivo distribution at 30 minutes after administration that for BA-CMC, drug concentration was relatively high in the kidney and lung but low in the heart and the brain; for BA-LP, drug concentration was significantly increased in the liver, spleen, and lung, to 5.59-fold, 2.33-fold, and 1.25-fold that of BA-CMC in the liver, spleen and lung, respectively. Thus, the tissue distribution behavior of BA was significantly altered in the case of BA-LP after oral administration in comparison with BA-CMC suspension, in rats. However, the mechanism of biodistribution after oral administration needs to be clarified by further studies. Baicalin has shown various strong pharmacological activities, including anti-inflammatory, antitumoral, antiradical, antiproliferative, cardioprotective, and so on. Importantly, it shows no, or very little, toxicity to normal epithelial and normal peripheral blood and myeloid cells.5 In addition, Scutellaria barbata is widely used as an antitumor agent in China, and the chloroform fraction has been shown to have the strongest cytotoxicity on cancer cell lines, with a lower cytotoxic effect on a normal liver cell line.4 Similarly, although the drug concentration of BA-LP was significantly increased in the liver and lung, it was safe for the liver and lung to some extent. It is speculated BA-LP would be beneficial for the treatment of liver and lung diseases.
Conclusion
A novel BA-LP formulation was successfully developed with the effervescent dispersion technique. BA-LP had high encapsulation efficiency and showed a sustained-release behavior that fit well with the Weibull distribution equation. Compared with BA-CMC suspension, the oral bioavailability and tissue distribution of BA-LP was significantly improved. The novel liposome formulation of BA could be a potential treatment for liver, kidney, and lung diseases.
Acknowledgments
This study was financially supported of by the National Natural Science Foundation of the People’s republic of China (grant numbers 81101678, 81341124, and 31201566), the Science and Technology Support Project of Sichuan Province (grant numbers 2013SZZ006 and 2014SZ0071), the Key Program of the Scientific Research Foundation of the Education Department of Sichuan Province (grant numbers 11ZZ024, 12ZZ020, and 12ZB066), the Scientific Research Foundation of the Administration of Traditional Chinese of Sichuan Province (grant number 2012-F-026), the Scientific Research Foundation of the Health Bureau of Sichuan Province (grant numbers 130270 and 130269), and the Key Program of the Scientific Research Foundation of Bureau of Science and Technology of Luzhou City (grant numbers 2013LZLY-K80, 2013LZLY-J10, 2013-S-47[17/20], 2011-S-32[1/4]) and Produce-Learn-Research Projects of Luzhou Medical College (grant number 2012CXY-01).
Disclosure
The authors report no conflicts of interest in this study.
References
Kim AR, Kim SN, Jung IK, Kim HH, Park YH, Park WS. The inhibitory effect of Scutellaria baicalensis extract and its active compound, baicalin, on the translocation of the androgen receptor with implications for preventing androgenetic alopecia. Planta Med. 2014;80(2–3):153–158. | ||
Du Z, Wang K, Tao Y, Chen L, Qiu F. Purification of baicalin and wogonoside from Scutellaria baicalensis extracts by macroporous resin adsorption chromatography. J Chromatogr B Analyt Technol Biomed Life Sci. 2012;908:143–149. | ||
Haranaka R, Hasegawa R, Nakagawa S, Sakurai A, Satomi N, Haranaka K. Antitumor activity of combination therapy with traditional Chinese medicine and OK432 or MMC. J Biol Response Mod. 1988;7(1):77–90. | ||
Li-Weber M. New therapeutic aspects of flavones: the anticancer properties of Scutellaria and its main active constituents Wogonin, Baicalein and Baicalin. Cancer Treat Rev. 2009;35(1):57–68. | ||
Yu J, Liu H, Lei J, Tan W, Hu X, Zou G. Antitumor activity of chloroform fraction of Scutellaria barbata and its active constituents. Phytother Res. 2007;21(9):817–822. | ||
Srinivas NR. Baicalin, an emerging multi-therapeutic agent: pharmacodynamics, pharmacokinetics, and considerations from drug development perspectives. Xenobiotica. 2010;40(5):357–367. | ||
Shen YC, Chiou WF, Chou YC, Chen CF. Mechanisms in mediating the anti-inflammatory effects of baicalin and baicalein in human leukocytes. Eur J Pharmacol. 2003;465(1–2):171–181. | ||
Shieh DE, Liu LT, Lin CC. Antioxidant and free radical scavenging effects of baicalein, baicalin and wogonin. Anticancer Res. 2000; 20(5A):2861–2865. | ||
Xing J, Chen X, Zhong D. Absorption and enterohepatic circulation of baicalin in rats. Life Sci. 2005;78(2):140–146. | ||
Zhou X, Liu D, Liu H, et al. Effect of low molecular weight chitosans on drug permeation through mouse skin: 1. Transdermal delivery of baicalin. J Pharm Sci. 2010;99(7):2991–2998. | ||
Yan JJ, Li AP, Li XR. [Study on techniques of preparing baicalin gelatin microspheres]. Chinese Journal of Information on Traditional Chinese Medicine. 2009;16(1):30–32. Chinese. | ||
Wang YM, Yue HK, Chang M, Zhang J, Qi L. [Study on the preparation of baicalin sustained-release microcapsule]. Journal of Anhui Agricultural Sciences. 2011;39(20):12086–12088. Chinese. | ||
Zhao L, Wei Y, Huang Y, He B, Zhou Y, Fu J. Nanoemulsion improves the oral bioavailability of baicalin in rats: in vitro and in vivo evaluation. Int J Nanomedicine. 2013;8:3769–3779. | ||
Yang L, Song WZ, Zhang Y. Baicalin-β-cyclodextrin inclusion compound preparation. J Henan University. 2013;32(3):179–183. | ||
Li JX, Han LL, Chao JB. Preparation and characterization of the inclusion complex of baicalein with γ-cyclodextrin: an antioxidant ability study. J Incl Phenom Macrocycl Chem. 2012;73(1–4):247–254. | ||
Hao J, Wang F, Wang X, et al. Development and optimization of baicalin-loaded solid lipid nanoparticles prepared by coacervation method using central composite design. Eur J Pharm Sci. 2012;47(2):497–505. | ||
Li B, Wen M, Li W, He M, Yang X, Li S. Preparation and characterization of baicalin-polyvinylpyrrolidone coprecipitate. Int J Pharm. 2011;408(1–2):91–96. | ||
Li N, Je YJ, Yang M, Jiang XH, Ma JH. Pharmacokinetics of baicalin-phospholipid complex in rat plasma and brain tissues after intranasal and intravenous administration. Pharmazie. 2011;66(5):374–377. | ||
Dicheva BM, Koning GA. Targeted thermosensitive liposomes: an attractive novel approach for increased drug delivery to solid tumors. Expert Opin Drug Deliv. 2014;11(1):83–100. | ||
Liu M, Li M, Sun S, et al. The use of antibody modified liposomes loaded with AMO-1 to deliver oligonucleotides to ischemic myocardium for arrhythmia therapy. Biomaterials. 2014;35(11):3697–3707. | ||
Dai Y, Zhou R, Liu L, Lu Y, Qi J, Wu W. Liposomes containing bile salts as novel ocular delivery systems for tacrolimus (FK506): in vitro characterization and improved corneal permeation. Int J Nanomedicine. 2013;8:1921–1933. | ||
Ramos-Cabrer P, Campos F. Liposomes and nanotechnology in drug development: focus on neurological targets. Int J Nanomedicine. 2013;8: 951–960. | ||
Wei Y, Xue Z, Ye Y, Wang P, Huang Y, Zhao L. Pharmacokinetic and tissue distribution of paclitaxel in rabbits assayed by LC-UV after intravenous administration of its novel liposomal formulation. Biomed Chromatogr. 2014;28(2):204–212. | ||
Wei Y, Xue Z, Ye Y, Huang Y, Zhao L. Paclitaxel targeting to lungs by way of liposomes prepared by the effervescent dispersion technique. Arch Pharm Res. 2014;37(6):728–737. | ||
Wei Y, Zhao L. Passive lung-targeted drug delivery systems via intravenous administration. Pharm Dev Technol. 2014;19(2):129–136. | ||
Zhao L, Wei Y, Li W, et al. Solid dispersion and effervescent techniques used to prepare docetaxel liposomes for lung-targeted delivery system: in vitro and in vivo evaluation. J Drug Target. 2011;19(3):171–178. | ||
Zhao L, Ye Y, Li J, Wei YM. Preparation and the in vivo evaluation of paclitaxel liposomes for lung targeting delivery in dogs. J Pharm Pharmacol. 2011;63(1):80–86. | ||
Zhao L, Wei YM, Zhong XD, et al. PK and tissue distribution of docetaxel in rabbits after i.v. administration of liposomal and injectable formulations. J Pharm Biomed Anal. 2009;49(4):989–996. | ||
Jain S, Kumar D, Swarnakar NK, Thanki K. Polyelectrolyte stabilized multilayered liposomes for oral delivery of paclitaxel. Biomaterials. 2012;33(28):6758–6768. | ||
Jain S, Patil SR, Swarnakar NK, Agrawal AK. Oral delivery of doxorubicin using novel polyelectrolyte-stabilized liposomes (layersomes). Mol Pharm. 2012;9(9):2626–2635. | ||
Niu M, Lu Y, Hovgaard L, et al. Hypoglycemic activity and oral bioavailability of insulin-loaded liposomes containing bile salts in rats: the effect of cholate type, particle size and administered dose. Eur J Pharm Biopharm. 2012;81(2):265–272. | ||
Gradauer K, Barthelmes J, Vonach C, et al. Liposomes coated with thiolated chitosan enhance oral peptide delivery to rats. J Control Release. 2013;172(3):872–878. | ||
Wang G, Chang MQ, Yang GY, Zeng N, Ye F. Oral absorption of long circulating nano-liposomes of quercetin in mice. Herald of Medicine. 2011;30(10):1266–1269. | ||
Kong XY, Du YQ, Li L, et al. Plasma miR-216a as a potential marker of pancreatic injury in a rat model of acute pancreatitis. World J Gastroenterol. 2010;16(36):4599–4604. | ||
Gu YZ, Zhou W, Zhai GX. [Preparation of puerarin liposome and its oral absorption in rat]. Zhong Yao Cai. 2007;30(8):970–973. Chinese. | ||
Dong Y, Feng SS. Methoxy poly(ethylene glycol)-poly(lactide) (MPEG-PLA) nanoparticles for controlled delivery of anticancer drugs. Biomaterials. 2004;25(14):2843–2849. | ||
Chen XQ, Yang RX. Preparation of baicalin liposome and determination of its entrapment efficiency. GuangZhou Chemical Ind Techno. 2009;37(6):138–139. | ||
Liu ZF, Yang ZH, Zhang SH, Liu WL. Study on preparation and main drug content determination of baicalin liposomes. Modern Med Health. 2010;16:2411–2413. | ||
Rowland RN, Woodley JF. The stability of liposomes in vitro to pH, bile salts and pancreatic lipase. Biochim Biophys Acta. 1980;620(3): 400–409. | ||
Akao T, Kawabata K, Yanagisawa E, et al. Baicalin, the predominant flavone glucuronide of scutellariae radix, is absorbed from the rat gastrointestinal tract as the aglycone and restored to its original form. J Pharm Pharmacol. 2000;52(12):1563–1568. | ||
Varshosaz J, Tabbakhian M, Mohammadi MY. Formulation and optimization of solid lipid nanoparticles of buspirone HCl for enhancement of its oral bioavailability. J Liposome Res. 2010;20(4):286–296. | ||
Hu K, Cao S, Hu F, Feng J. Enhanced oral bioavailability of docetaxel by lecithin nanoparticles: preparation, in vitro, and in vivo evaluation. Int J Nanomedicine. 2012;7:3537–3545. | ||
Duchêne D, Ponchel G. Bioadhesion of solid oral dosage forms, why and how? Eur J Pharm Biopharm. 1997;44(1):15–23. | ||
Venkatesan N, Uchino K, Amagase K, Ito Y, Shibata N, Takada K. Gastro-intestinal patch system for the delivery of erythropoietin. J Control Release. 2006;111(1–2):19–26. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.