Back to Journals » International Journal of Nanomedicine » Volume 14
Preparation Of Nanobubbles Modified With A Small-Molecule CXCR4 Antagonist For Targeted Drug Delivery To Tumors And Enhanced Ultrasound Molecular Imaging
Authors Peng Y, Zhu L ![]() , Wang L
, Wang L ![]() , Liu Y, Fang K, Lan M, Shen D, Liu D, Yu Z, Guo Y
, Liu Y, Fang K, Lan M, Shen D, Liu D, Yu Z, Guo Y
Received 29 March 2019
Accepted for publication 2 September 2019
Published 26 November 2019 Volume 2019:14 Pages 9139—9157
DOI https://doi.org/10.2147/IJN.S210478
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Mian Wang
Yanli Peng,1,2,* Lianhua Zhu,1,* Luofu Wang,3 Yu Liu,1 Kejing Fang,1 Minmin Lan,1,2 Daijia Shen,1 Deng Liu,1 Zhiping Yu,3 Yanli Guo1
1Department of Ultrasound, Southwest Hospital, Third Military Medical University (Army Medical University), Chongqing, People’s Republic of China; 2State Key Laboratory of Silkworm Genome Biology, Southwest University, Chongqing, People’s Republic of China; 3Department of Urology, Army Featured Medicine Center, Third Military Medical University (Army Medical University), Chongqing, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yanli Guo
Department of Ultrasound, Southwest Hospital, Third Military Medical University (Army Medical University), No 30 Gaotanyan Street, Shapingba District, Chongqing 400038, People’s Republic of China
Tel +86 236 876 5439
Fax +86 236 876 5439
Email [email protected]
Purpose: To construct nanobubbles (PTX-AMD070 NBs) for targeted delivery of paclitaxel (PTX) and AMD070, examine their performance in ultrasound molecular imaging of breast cancer and cervical cancer and their therapeutic effect combined with ultrasound targeted nanobubble destruction (UTND).
Materials and methods: PTX-AMD070 NBs were prepared via an amide reaction, and the particle size, zeta potential, encapsulation rate and drug loading efficiency were examined. Laser confocal microscopy and flow cytometry were used to analyze the targeted binding ability of PTX-AMD070 NBs to CXCR4+ MCF-7 cells and C33a cells. The effect of PTX-AMD070 NBs combined with UTND on cell proliferation inhibition and apoptosis induction was detected by CCK-8 assays and flow cytometry. The contrast-enhanced imaging features of PTX-AMD070 NBs and paclitaxel-loaded nanobubbles were compared in xenograft tumors. The penetration ability of PTX-AMD070 NBs in xenograft tissues was evaluated by immunofluorescence. The therapeutic effect of PTX-AMD070 NBs combined with UTND on xenograft tumors was assessed.
Results: PTX-AMD070 NBs showed a particle size of 494.3±61.2 nm, a zeta potential of −22.4±1.75 mV, an encapsulation rate with PTX of 53.73±7.87%, and a drug loading efficiency with PTX of 4.48±0.66%. PTX-AMD070 NBs displayed significantly higher targeted binding to MCF-7 cells and C33a cells than that of PTX NBs (P<0.05), and combined with UTND manifested a more pronounced effect in inhibiting cell proliferation and promoting apoptosis than other treatments. PTX-AMD070 NBs aggregated specifically in xenograft tumors in vivo, and significantly improved the image quality. Compared with other treatment groups, PTX-AMD070 NBs combined with UTND exhibited the smallest tumor volume and weight, and the highest degree of apoptosis and necrosis.
Conclusion: PTX-AMD070 NBs improved the ultrasound imaging effect in CXCR4+ xenograft tumors and facilitated targeted therapy combined with UTND. Therefore, this study provides an effective method for the integration of ultrasound molecular imaging and targeted therapy of malignant tumors.
Keywords: nanobubbles, ultrasound imaging, paclitaxel, AMD070, malignant tumor
Introduction
Malignant tumors have become one of the main threats to human life and health. By 2025, an estimated 19.3 million new cases will be diagnosed annually.1,2 Chemotherapy is currently one of the most common methods for cancer treatment, especially for tumors with metastatic tendency or having undergone metastasis.3,4 At present, chemotherapy is mostly achieved by means of systemic administration of drugs, but the chemotherapeutic agents used in clinics may not be delivered effectively and specifically at tumor tissues. This not only reduces the effectiveness of antitumor therapy but also has detrimental effects in normal tissues and causes cardiac, liver, and kidney toxicity.5–7 To improve the therapeutic effect of chemotherapeutic agents and minimize their impact on normal tissues, targeted delivery systems may be developed to enhance their intended release in tumor tissues. Lipid carriers are a commonly used targeted drug delivery system and have some unique advantages: 1) lipids are the main component of cell membranes, feature great biocompatibility and safety in vivo and do not manifest toxicity or immunogenicity. 2) Lipids can have different configurations and carry hydrophobic chemotherapeutic agents via noncovalent bonds to improve their aqueous solubility. 3) In the host, the chemotherapeutic drugs in lipid carriers can be released in a targeted fashion upon specific environmental metrics (pH, temperature, light, and ultrasound); the lipid surface can also be subject to polyethylene glycol (PEG) modification to avoid phagocytic ingestion, thereby prolonging the circulation time of the drugs in the body and enhancing their stability and biological activity. 4) Lipids are easy to modify and can be attached to specific ligands to form targeted carriers, thereby facilitating targeted delivery of chemotherapeutic drugs. 5) Lipids can also encapsulate gases to form ultrasound contrast agents and in turn greatly augment ultrasound imaging quality.8–13
Lipid ultrasound contrast agents have been widely used in the diagnosis of thrombus, tumors, inflammation, and other conditions. These materials allow ultrasound-assisted, targeted release of the drug or gene that they carry and in turn improve the therapeutic outcome. At present, ultrasound contrast agents are mostly at the micron scale, and thus, they cannot penetrate the endothelial space of tumor vessels to allow ultrasound molecular imaging or focused delivery of drugs or genes. Ultrasound contrast agents with a nanoscale particle size and strong permeability can pass through tumor blood vessels and accumulate in tumor extravascular tissues.14–17 Lipid nanobubbles (NBs) represent a common nanoscale ultrasound contrast agent with strong permeability and high stability that can not only enhance extravascular ultrasound imaging of tumors but also allow ultrasound-assisted focused delivery of drugs or genes to tumor parenchymal cells. Moreover, specific antibodies or ligands that target tumor tissue can be conjugated to the surface of drug- or gene-loaded NBs, thereby improving the aggregation ability of NBs in tumor extravascular tissue and in turn improving the outcome of extracellular ultrasound molecular imaging and targeted therapy.18–21
CXC chemokine receptor 4 (CXCR4) is excessively abnormally expressed on the membrane of most tumor cells. CXCR4 and its ligand CXCL12 form a CXCL12/CXCR4 axis, which plays a crucial role in tumor growth and metastasis. Correspondingly, blockage of this axis facilitates antitumor therapy. Therefore, NBs carrying CXCR4 antagonists can not only specifically bind to tumor cells to augment ultrasound imaging but can also inhibit tumor growth and metastasis.22–27 In light of this, herein, we conjugated AMD070, a small-molecule CXCR4 antagonist, to paclitaxel-carrying NBs, thereby generating a NB (PTX-AMD070 NBs) with dual therapeutic effects. The material was then subject to in vivo and in vitro studies in which we examined its binding to breast cancer and cervical cancer cells, its ability to enhance ultrasound imaging, its targeted release of paclitaxel, and the process and mechanism underlying AMD070-based tumor inhibition. This study provides a means to integrate ultrasound molecular imaging and targeted therapy by conjugating an ultrasound contrast agent with a drug carrier, which can be used for treatment of multiple tumor types.
Materials And Methods
Main Reagents
Fetal bovine serum, RPMI1640 medium, and trypsin were purchased from HyClone (Logan Utah, USA). Dipalmitoylphosphatidic acid, dipalmityl phosphatidylglycerol, and dipalmitoylphosphatidyl ethanolamine were purchased from CordenPharma (Corden Pharma, Liestal, Switzerland). Dipalmitoylphosphatidylcholine was purchased from Avanti Polar Lipids, Inc. (Alabaster, AL, USA). The crosslinking product between 1,2-distearoyl-phosphatidylethanolamine and polyethylene glycol conjugate-2000 was purchased from NANOCS (Boston, MA, USA). 1,1ʹ-dioctadecyl-3,3,3ʹ,3ʹ-tetramethylindocarbocyanine perchlorate (DiI), 4′,6-diamidino-2-phenylindole (DAPI), CCK-8 reagents and a TUNEL kit were purchased from Beyotime Biotechnology (Shanghai, China). Paclitaxel (PTX), carbodiimide (EDC), and N-hydroxysuccinimide (NHS) were purchased from Solarbio (Beijing, China). AMD070 was purchased from Absin Biotechnology (Shanghai, China). Rabbit anti-human CXCR4 monoclonal antibody, rat anti-mouse CD31 monoclonal antibody, FITC-labeled rabbit anti-rat secondary antibody, and Alexa Fluor 488-labeled goat anti-rabbit secondary antibody were purchased from Abcam (Cambs, UK). An Annexin V Apoptosis Detection Kit FITC kit was purchased from Thermo Fisher Scientific (Waltham, MA, USA). BALB/c-nu female nude mice were purchased from Huafukang Bioscience Co., Inc. (Beijing, China).
Expression Of CXCR4 In Tumor Cells
Human breast cancer MCF-7 cells (CXCR4 positive), human cervical cancer C33a cells (CXCR4 positive), and human MDA-MB-468 breast cancer cells (CXCR4 negative) were obtained from Shanghai cell bank of Chinese Academy of Sciences. The cells were cultured in RPMI1640 medium containing 10% fetal bovine serum, penicillin (100 μg/mL), and streptomycin (100 μg/mL) at 37°C in 5% CO2. MCF-7 cells, C33a cells and MDA-MB-468 cells grown to a logarithmic phase were inoculated into 24-well plates at a concentration of 1×105 cells/mL and incubated overnight. The cells were then fixed with 4% paraformaldehyde for 15 mins, before being blocked with 5% bovine serum albumin (BSA-PBS) for 1 hr at 37°C. The samples were incubated with rabbit anti-human CXCR4 monoclonal antibody overnight at 4°C and Alexa Fluor 488-labeled goat anti-rabbit secondary antibody for 2 hrs at room temperature. The wells were then rinsed with PBS and stained with DAPI for 5 mins, and the expression of CXCR4 in tumor cells was observed under a confocal laser microscope.
Synthesis And Detection Of DSPE-PEG2000-AMD070
DSPE-PEG2000-COOH dissolved in dimethyl sulfoxide (DMSO) was shaken and activated with supplementation of EDC and NHS (the molar ratio of DSPE-PEG2000-COOH:EDC:NHS was 1:2:2) at room temperature for 1 hr. Subsequently, AMD070 was added to the mixture to facilitate the reaction at room temperature for 1 hr. The DSPE-PEG2000-AMD070 conjugates were obtained by removing unreacted EDC, NHS, and AMD070 via dialysis (MWCO, 2000). AMD070, DSPE-PEG2000-COOH, and DSPE-PEG2000-AMD070 were dissolved in deuterated chloroform (CDCl3) (Shanghai Ziqi Laboratory Equipment Co., Ltd., Shanghai, China). The 1H NMR spectra of the samples were analyzed on a picoSpin 80 NMR spectrometer (Thermo Fisher Scientific, Waltham, MA, USA) to verify successful construction of DSPE-PEG2000-AMD070.
Preparation And Validation Of PTX-AMD070 NBs
DPPA, DPPC, DPPE, DPPG, DSPE-PEG2000-AMD070, and PTX (the mass ratio was 1:3:1:3:2:2) were suspended in a mixture of glycerol and PBS (1:9 v/v) in a specific proportion and dissolved overnight on a rotary shaker. The solution was transferred to a ciilin bottle, the air inside was replaced with C3F8 (Tianjin Institute of Nuclear Industry Physics and Chemical Engineering, Tianjin, China), and the solution was vortexed on an ST Amalgam Capsule Blender for 90 seconds. The vibrated solution was centrifuged at 1300 rpm for 3 min to remove the bottom lipid material and PTX that did not form bubbles and to obtain the supernatant mixture including NBs and microbubbles. Then the supernatant mixture was continuously centrifuged at 300 rpm for 3 min to remove supernatant microbubbles and to obtain PTX-AMD070 NBs. In addition, PTX NBs without AMD070 were prepared using the same method. The particle size and zeta potential of PTX-AMD070 NBs and PTX NBs were measured on a Malvern Zetasizer nano ZS90 detector. An appropriate amount of DiI was added to the targeted drug-loaded nanoparticles to generate DiI-labeled nanoparticles. The distribution of PTX-AMD070 NBs was observed under a light microscope and a confocal laser microscope, and the structure and morphology were studied under a transmission electron microscope. The agarose model was constructed as follows: 100 mL ultrapure water was used to dissolve 1 g agarose powder in 200 mL-beaker, and the beaker was placed in a microwave oven. After heating for 10 min, the solution cooled in room temperature, and the agarose model was constructed. A hole in the core of the model was made, which was used to add NBs. The ultrasound imaging intensity of PTX-AMD070 NBs of different concentrations (1.0×108, 2.0×107, 4.0×106, 8.0×105, 1.6×105, and 3.2×104/mL) was determined using a vevo2100 small animal ultrasound scanner in B mode (center frequency 21 MHz). The ultrasound imaging intensity of PTX-AMD070 NBs before and after blast of a high mechanical index was compared.
Encapsulation Rate And Drug Load Of PTX-AMD070 NBs
The encapsulation rate and drug load of PTX-AMD070 NBs were evaluated via High Performance Liquid Chromatography (HPLC). PTX-AMD070 NBs were irradiated using a WED-100 Ultrasonic Therapy Machine with a radiation power of 2.5 W/cm2 and a radiation time of 30 mins. The concentration of PTX was determined with an Agilent 1260 Infinity HPLC system (Agilent Technologies, Santa Clara, USA). The determination was carried out on an ZORBAX Eclipse Plus C18 analytical column (5 μm, 250×4.6mm) gradient eluting with mobile phases of acetonitrile (mobile phase A) and water (mobile phase B) at the flow rate of 1.0 mL/min. The column temperature was 40°C and the detection wavelength was 227 nm. The formulas for calculating drug encapsulation rate and drug load were as follows:


in which W1 represents the amount of drug carried by PTX-AMD070 NBs, W2 represents the total amount of drugs, and W3 stands for the total weight of drugs and lipids.
In Vitro PTX-AMD070 NB Binding Experiment
MCF-7 cells, C33a cells, and MDA-MB-468 cells were inoculated into 24-well plates at a concentration of 1×105/mL and cultured overnight. The cells were fixed in 4% paraformaldehyde for 15 mins and subsequently blocked in 5% BSA-PBS at 37°C for 1 hr. DiO was used to stain the cell membrane, and the cells were incubated with 30 µL DiI-labeled PTX-AMD070 NBs (1×107/mL) at 4°C for 2 hrs. After DAPI double staining, the binding of PTX-AMD070 NBs to cells was observed under a confocal laser microscope. Flow cytometry was used to quantitatively determine the cell affinity of PTX-AMD070 NBs using the following procedure: MCF-7 cells, C33a cells and MDA-MB-468 cells grown to a logarithmic phase were trypsinized and collected and the cell concentrations were adjusted to 1×106/mL. Each cell line was divided into three groups as follows: the first group was the blank control; the second group was treated with 1×107/mL PTX NBs; the third group was treated with the same concentration of PTX-AMD070 NBs. Cells were incubated at 37°C for 40 mins. The fluorescence signal of each group was analyzed via flow cytometry.
Anti-Proliferation Capacity Of PTX-AMD070 NBs
MCF-7 cells, C33a cells, and MDA-MB-468 cells were added to 96-well plates at a density of 1 × 104 cells/well and cultured overnight. Each well was supplemented with different concentrations of AMD070 and PTX and incubated at 37°C for 48 hrs. Ten microliters of CCK-8 reagent were added to each well, and the plate was incubated at 37°C for 2 hrs. The OD450 values of individual wells were measured with a multifunctional plate reader to determine the anti-proliferation activity of PTX and AMD070. Anti-proliferation activity was compared among different treatment groups in which the concentration of PTX was consistently 10 μM (control group, AMD070 group, PTX+AMD070 group, PTX-AMD070 NBs group, and PTX-AMD070 NBs+US group). The plates were irradiated using the WED-100 ultrasonic therapy machine at a radiation power of 1 W/cm2 and radiation time of 20 seconds. After 24 hrs of treatment, 10 μL CCK-8 reagent was added to each well, and the plates were incubated at 37°C for 2 hrs. The OD450 values were examined for different treatment groups.
Cell Apoptosis Detection
MCF-7 cells, C33a cells, and MDA-MB-468 cells were inoculated into 6-well plates at a density of 5×105 cells/well and incubated overnight. After treatment of each group (blank control group, AMD070 group, PTX group, PTX+AMD070 group, PTX-AMD070 NBs group, and PTX-AMD070 NBs+US group), the cells were cultured for an additional 48 hrs. The cells were collected, suspended in 200 μL binding buffer, supplemented with fluorescein isothiocyanate (FITC), and incubated at room temperature for 20 mins, followed by addition of pyridine iodide. The level of apoptosis in each group was determined by flow cytometry.
In Vivo Imaging Of PTX-AMD070 NBs
The mouse experiments in this study were approved by the Laboratory Animal Welfare and Ethics Committee of the Army Medical University, and the animals were cared for according to the guidelines of the Army Medical University for Animal Welfare. MCF-7 cells, C33a cells or MDA-MB-468 cells grown to a logarithmical phase were subcutaneously implanted into 4- to 5-week-old BALB/c-nu female nude mice at a concentration of 1×108/mL to construct the xenograft models. When the volume of a xenograft tumor reached a size of ca. 1 cm3, the tumor was examined with the vevo2100 small animal ultrasound scanner equipped with an S250 high-frequency probe. After the maximum transverse section of the transplanted tumor was displayed, the probe was immobilized and adjusted to a mode of contrast-enhanced ultrasound (at a central frequency of 18 MHz). Contrast-enhanced ultrasound images of the transplanted tumors were continuously collected after the nude mice were injected with 200 μL PTX-AMD070 NBs and PTX NBs (5x108/mL) via the caudal vein. When the ultrasound imaging intensity of transplanted tumors returned to the baseline level, ultrasound image acquisition was stopped; the residual NBs in animals were blasted by pressing the “Burst” button, before another type of NB was injected. The quantitative analysis software built into the vevo2100 scanner was employed to analyze the acquired images, thereby generating time-intensity curves of PTX-AMD070 NBs and PTX NBs and comparing the peak time, peak intensity, and area under the curves (AUCs) of the two NB types.
Distribution Of PTX-AMD070 NBs In Tissues
After the tumor-bearing nude mice were injected with 200 μL DiI-labeled PTX-AMD070 NBs (5x108/mL) via the caudal vein, the xenograft tissues and left thigh muscle tissues were collected and prepared into frozen sections. The sections were fixed with 4% paraformaldehyde, and rat anti-mouse CD31 monoclonal antibody (1:300) was added and incubated with sections overnight at 4°C. FITC-conjugated rabbit anti-rat secondary antibody was added and incubated with sections at room temperature in the dark for 3 hrs. The sections were then double stained with DAPI, and the distribution of PTX-AMD070 NBs in the tissue was observed under a confocal laser microscope.
In Vivo Therapeutic Effect
Nude mice were randomly divided into 6 groups (5 rodents/group), namely, a blank control group, AMD070 group, PTX group, PTX+AMD070 group, PTX-AMD070 NBs group, and PTX-AMD070 NBs+US group. The PTX dose for each nude mouse was 10 mg/kg. The same treatments volume was injected via the caudal vein in each treatment group. The animals underwent 6 treatment sessions, with one session every 3 days. Ultrasonic irradiation was carried out using the WED-100 ultrasonic therapy machine at a radiation power of 1.0 W/cm2 and a radiation time of 15 mins. Before the treatment, each mouse was weighed, and the long and short tumor diameters were measured with a Vernier caliper. After the treatment, the transplanted tumor in each nude mouse was removed, embedded in paraffin, sectioned, and stained with eosin and hematoxylin, before apoptosis in the tumor tissue was examined using a TdT-mediated dUTP nick-end labeling (TUNEL) assay.
Statistical Analyses
One-way ANOVA and paired sample t-tests were performed using Social Pack for Social Sciences 22.0. All the data are expressed as the mean±standard deviation. P<0.05 was considered statistically significant. GraphPad Prism 6.0 was used to plot the histograms and curves.
Results
Expression Of CXCR4 In Tumor Cells
After reaction with anti-CXCR4 monoclonal antibody and fluorescent secondary antibody, green fluorescence signals were observed in breast cancer MCF-7 cells and cervical cancer C33a cells but not in breast cancer MDA-MB-468 cells under a laser confocal microscope (Figure 1).
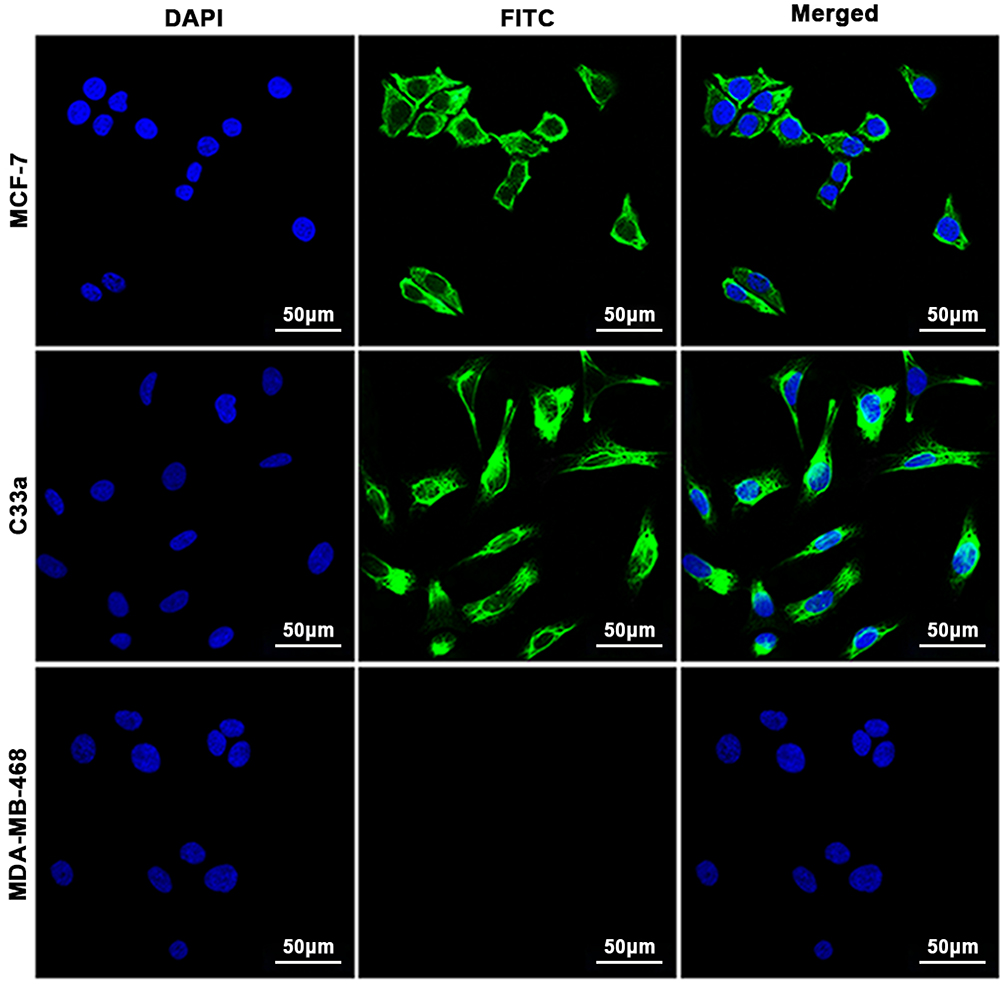 |
Figure 1 Cell immunofluorescence assay. CXCR4 was expressed in MCF-7 cells and C33a cells but not in MDA-MB-468 cells. The nuclei are stained blue, and cell membranes expressing CXCR4 are shown in green. |
Identification Of DSPE-PEG2000-AMD070
DSPE-PEG2000-AMD070 was synthesized by reacting the amino groups of AMD070 with the carboxyl groups of DSPE-PEG2000-COOH. The 4.81 ppm peak corresponds to the amino group of AMD070, whereas the 7.56–8.5 ppm peak represents the benzene ring of AMD070 (Figure 2A). The characteristic benzene peak ring was present in the DSPE-PEG2000-COOH spectrum (Figure 2B). The NMR spectrum of DSPE-PEG2000-AMD070 showed that there was no characteristic amino peak at 4.81 ppm, but the benzene ring peak at 7.56–8.5 ppm was present. In other words, there were benzene rings but no amino groups in DSPE-PEG2000-AMD070, indicating successful synthesis of DSPE-PEG2000-AMD070 (Figure 2C).
 |
Figure 2 Identification of DSPE-PEG2000-COOH. (A) The 1H NMR spectrum of AMD070. (B) The 1H NMR spectrum of DSPE-PEG2000-COOH. (C) The 1H NMR spectrum of DSPE-PEG2000-AMD070. |
Basic Characteristics Of PTX-AMD070 NBs
Under a light microscope and a laser confocal microscope, the PTX-AMD070 NBs were homogenously dispersed, similar particle size and no apparent aggregates (Figure 3A and B). Under the transmission electron microscope, the PTX-AMD070 NBs were spherical and had a smooth and sharp surface (Figure 3C). PTX NBs had a particle diameter of 472.9±60.3 nm and a polydispersity coefficient of 0.067 (Figure 3D); PTX-AMD070 NBs had a particle diameter of 494.3±61.2 nm and a polydispersity coefficient of 0.173 (Figure 3E). In addition, the zeta potential of PTX NBs was −21.1±1.05 mV, whereas that of PTX-AMD070 NBs was −22.4±1.75 mV. Of note, the high negative charge is conducive to maintaining good dispersion and stability of PTX-AMD070 NBs. In the in vitro agarose model, the PTX-AMD070 NB ultrasound imaging intensity was positively correlated with NB concentration (Figure 3F and G). After ultrasound exposure with a high mechanical index ultrasonic irradiation, the ultrasonic imaging intensity of PTX-AMD070 NBs displayed a significant reduction, indicating that PTX-AMD070 NBs could be destroyed by a high mechanical index ultrasonic wave (Figure 3H and I). Last, HPLC revealed that the encapsulation rate and drug load of PTX-AMD070 NBs were 53.73±7.87% and 4.48±0.66%, respectively.
 |
Figure 3 Basic characteristics of the nanobubbles. (A) Observation of PTX-AMD070 NBs under a light microscope. (B) Observation of DiI-labeled PTX-AMD070 NBs under a fluorescence microscope. (C) Observation of PTX-AMD070 NBs under a transmission electron microscope. (D) Spectrum of PTX NB particle sizes. (E) Spectrum of PTX-AMD070 NB particle sizes. (F) An ultrasound image of PTX-AMD070 NBs in vitro. (G) Quantitative analysis of imaging intensity of PTX-AMD070 NBs in vitro. (H) Ultrasound images of PTX-AMD070 NBs in vitro before and after ultrasonic blast. (I) Quantitative analysis of imaging intensity of PTX-AMD070 NBs before and after ultrasonic blast (*P<0.05). |
Ability Of PTX-AMD070 NBs To Bind Tumor Cells In Vitro
The binding ability of PTX-AMD070 NBs to three cell types was observed via laser confocal microscopy. A large number of DiI-labeled PTX-AMD070 NBs (red fluorescence) were observed around MCF-7 and C33a cell membranes (green fluorescence). In contrast, no DiI-labeled PTX-AMD070 NBs were found around MDA-MB-468 cells (Figure 4A). Flow cytometry showed that the affinity of PTX-AMD070 NBs to MCF-7 cells and C33a cells was significantly higher than that of PTX NBs to both cells (Figure 4B and C), while there was no significant difference in the affinity to MDA-MB-468 cells between PTX-AMD070 NBs and PTX NBs (Figure 4D). Quantitative analysis showed that the binding rates of PTX-AMD070 NBs to MCF-7 cells, C33a cells and MDA-MB-468 cells were 65.7±5.66%, 47.9±7.77%, and 1.5±0.51%, respectively, and those of PTX NBs were 2.19±1.41%, 2.72±2.27%, and 1.26±0.24%, respectively (Figure 4E).
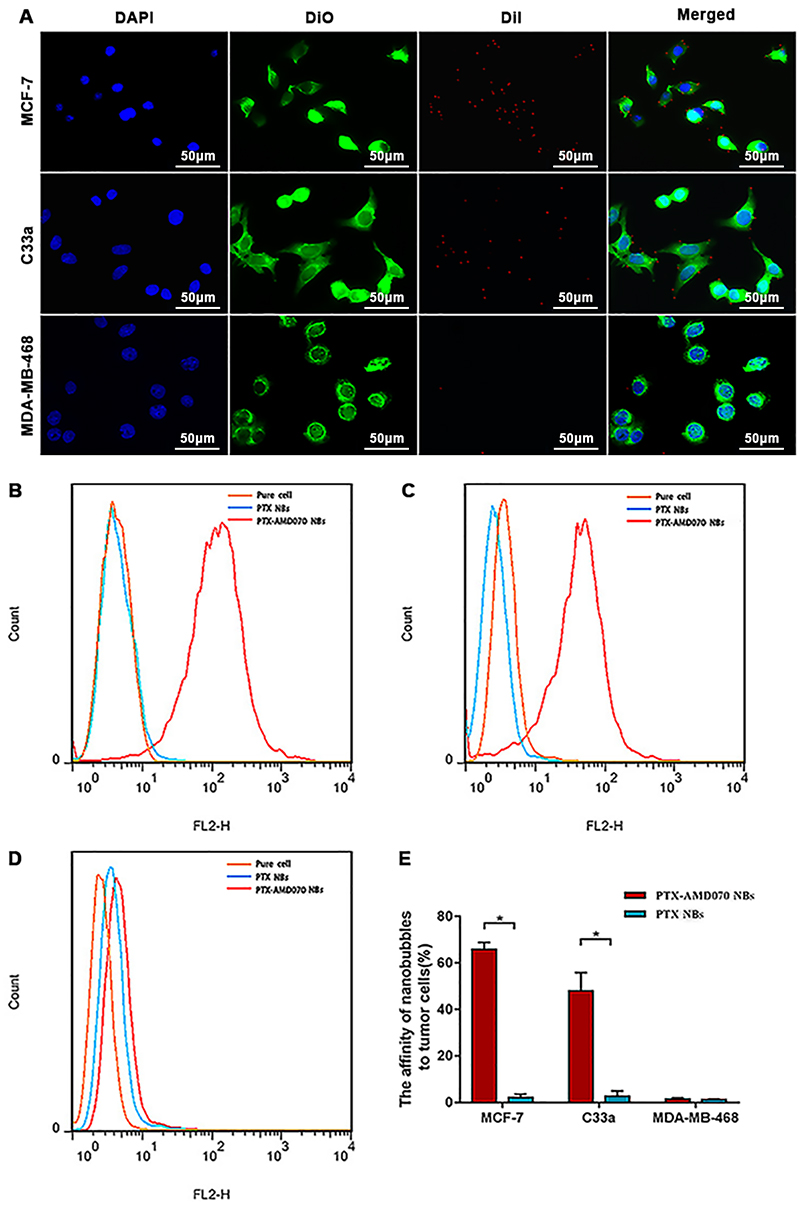 |
Figure 4 In vitro affinity of nanobubbles for cells. (A) Observation of DiI-labeled PTX-AMD070 NBs binding to MCF-7 cells, C33a cells, and MDA-MB-468 cells under a laser confocal microscope. The nuclei are stained blue; the cell membrane is stained with DiO (green); PTX-AMD070 NBs are stained with DiI (red). (B) Analysis of affinity between nanobubbles and MCF-7 cells determined by flow cytometry. (C) Analysis of affinity between nanobubbles and C33a cells determined by flow cytometry. (D) Analysis of affinity between nanobubbles and MDA-MB-46 cells determined by flow cytometry. (E) Quantitative analysis of affinity between the two types of nanobubbles and three types of cells. *There was significant difference between the cell binding ability of PTX-AMD070 NBs and PTX NBs (P<0.05). |
Assessment Of Antitumor Proliferation In Vitro
CCK-8 assays showed that the higher the AMD070 concentration, the greater the proliferation inhibition of MCF-7 cells and C33a cells; in contrast, the inhibitory effects of AMD070 at different concentrations on MDA-MB-468 cells were not significantly different. The results showed that AMD070 had significant inhibitory effects on CXCR4-expressing cells but not on the cells without CXCR4 (Figure 5A). The survival rates of the three tumor cell types gradually decreased with an increased PTX concentration. The IC50 values of PTX in MCF-7 cells, C33a cells, and MDA-MB-468 cells were 20.4 μM, 11.9 μM, and 27.9 μM, respectively (Figure 5A). CCK-8 assays were used to evaluate the anti-proliferation ability of the different treatments (Figure 5B). All three cell types exhibited the lowest survival rates when treated with PTX-AMD070 NBs+US, and the survival rates differed significantly from those in the PTX-AMD070 NBs group (P<0.05). In addition, the cell survival rate in the PTX AMD070 group was lower than that in the PTX-AMD070 NBs group (P<0.05). In MCF-7 cells and C33a cells, there was a significant difference in cell survival between the PTX+AMD070 group and PTX group (P<0.05). However, in MDA-MB-468 cells, there was no significant difference between the PTX+AMD070 group and PTX group (P>0.05). The results indicate that AMD070 manifested a prominent synergism with PTX to enhance its inhibition of CXCR4-positive tumor cells.
 |
Figure 5 Examination of tumor proliferation suppression ability in vitro using CCK-8 assays. (A) Tumor proliferation suppression ability of AMD070 and PTX at different concentrations. *The survival rate of MCF-7 cells was significantly different from that of cells without drug treatment (P<0.05). #The survival rate of C33a cells was significantly different from that of cells without drug treatment (P<0.05). ▲The survival rate of MDA-MB-468 cells was significantly different from that cells without drug treatment (P<0.05). (B) Tumor proliferation suppression outcome in different treatment groups. *There was a significant difference in cell survival rate between the two groups (P<0.05). Abbreviations: NBs, nanobubbles; PTX, paclitaxel; US, ultrasound. |
Analysis Of Cell Apoptosis In Vitro
An annexin V-FITC/PI double staining kit was used to analyze cell apoptosis in the different treatment groups. The total apoptosis rates of MCF-7 cells and C33a cells in the different treatment groups were in an ascending order as follows: Control<AMD070<PTX-AMD070 NBs<PTX<AMD070+PTX<PTX-AMD070 NBs +US (Figure 6A–C). The apoptosis rates in the PTX+AMD070 groups were higher than those in the PTX groups and AMD070 groups, indicating a synergistic effect between the two compounds. The apoptosis rates in the PTX+AMD070 groups were higher than those in the PTX-AMD070 NBs groups, indicating that the release of PTX and AMD070 in PTX-AMD070 NBs was low and slow. The PTX-AMD070 NBs+US groups showed the highest apoptosis rates, which were significantly different from those in the PTX-AMD070 NBs groups (P<0.05). The results showed that ultrasound irradiation significantly enhanced the ability of PTX-AMD070 NBs to promote apoptosis. In contrast, MDA-MB-468 cells exhibited comparable apoptosis rates between the PTX group (19.00±0.48%) and the PTX+AMD070 group (19.22±0.35%) (P>0.05). The results indicate that there was no synergism between AMD070 and PTX in promoting apoptosis of CXCR4-negative MDA-MB-468 cells (Figure 6D).
 |
Figure 6 Apoptosis of tumor cells detected by flow cytometry. (A) Apoptosis rates of MCF-7 cells, C33a cells, and MDA-MB-468 cells in the different treatment groups. (B–D) Quantitative comparison of apoptosis rates of MCF-7 cells, C33a cells, and MDA-MB-368 cells between different treatment groups; *There was significant difference in apoptosis rate between the two groups (P<0.05). Abbreviations: EA, early apoptosis; LA, late apoptosis; TA, total apoptosis. |
Effect Of Ultrasound Molecular Imaging In Vivo
The effects of PTX-AMD070 NBs and PTX NBs on ultrasound molecular imaging in three types of xenograft tumors were analyzed and compared. The contrast-enhanced images of PTX-AMD070 NBs and PTX NBs in xenograft tumors of MCF-7 cells, C33a cells and MDA-MB-468 cells are shown in Figure 7A. The time-intensity curves revealed that in the CXCR4-positive xenografts of MCF-7 and C33a cells, the ultrasound intensity of PTX-AMD070 NBs was significantly higher than that of PTX NBs (P<0.05) (Figure 7B and C). On the other hand, in the CXCR4-negative xenografts of MDA-MB-468 cells, there was no significant difference in ultrasound intensity between PTX-AMD070 NBs and PTX NBs (P>0.05) (Figure 7D). In xenografts of MCF-7 and C33a cells, the peak intensity and area under the curve (AUC) of PTX-AMD070 NBs were significantly higher than those of PTX NBs (P<0.05), whereas in xenografts of MDA-MB-468 cells, there was no significant difference in peak intensity and AUC between PTX-AMD070 NBs and PTX NBs (P>0.05) (Table 1). Last, there was no significant difference in peak time between PTX-AMD070 NBs and PTX NBs in all three types of xenografts (P>0.05) (Table 1).
 |
Figure 7 Ultrasound images of nanobubbles in xenografts. (A) Ultrasound images of nanobubbles in the three types of xenografts. The red dotted circle represents the xenograft area. (B–D) The time-intensity curves of the nanobubbles in the MCF-7, C33a, and MDA-MB-468 xenografts. |
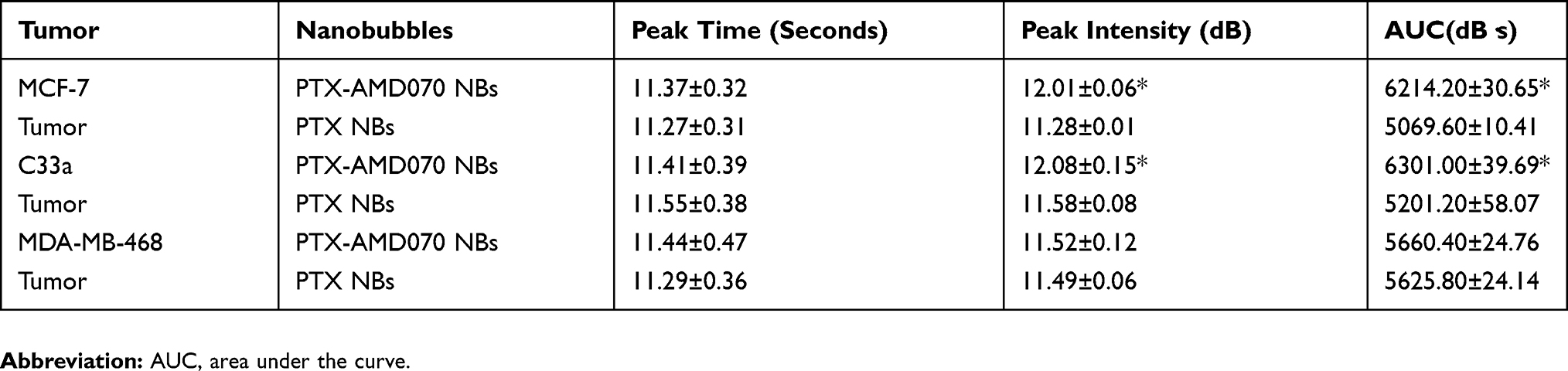 |
Table 1 Ultrasonic Imaging Parameters Of PTX-AMD070 NBs And PTX NBs In Xenograft Tumors |
Distribution Of PTX-AMD070 NBs In Tissues
Fluorescence immunohistochemistry was employed to examine the distribution of PTX-AMD070 NBs in xenograft and left thigh muscle tissues. DiI-labeled PTX-AMD070 NBs were distributed in the intravascular and extravascular spaces of MCF-7, C33a and MDA-MB-468 xenografts. However, PTX-AMD070 NBs were only present in blood vessels of thigh muscle tissues, and not in the extravascular space (Figure 8).
 |
Figure 8 Distribution of PTX-AMD070 NBs in tissues. Distribution of PTX-AMD070 NBs in MCF-7 xenografts (upper panel), in C33a xenografts (middle and upper panel), in MDA-MB-468 xenografts (middle and lower panel), and in thigh muscle tissue (lower panel). The nuclei are stained blue; the tumor blood vessels are shown in green; the DiI-labeled PTX-AMD070 NBs are red. |
In Vivo Therapeutic Effect Of PTX-AMD070 NBs
After the in vivo treatment, the mouse body weight, tumor volume ratio, tumor mass and tumor inhibition rate in different treatment groups were compared. For nude mice bearing MCF-7 and C33a xenografts, the most effective outcome in suppressing tumor growth was in the PTX-AMD070 NBs+US group (Figure 9A). During the course of treatment, the weight of mice bearing MCF-7 and C33a xenografts in all groups except the blank control group showed continuous growth, with the PTX-AMD070 NBs+US group manifesting the highest increase (Figure 9B and C). On posttreatment day 18, the tumor volume ratios of the nude mice bearing MCF-7 and C33a xenografts in different groups exhibited the following descending order: blank control group>AMD070 group>PTX group>PTX+AMD070 group>PTX-AMD070 NBs>PTX-AMD070 NBs+US group. During treatment, the volume ratio of the xenografts in the PTX-AMD070 NBs group continuously decreased (Figure 9D and E). After treatment, the PTX-AMD070 NBs+US group showed a tumor mass significantly smaller than that of other groups and a tumor inhibition rate significantly higher than that of other groups (P<0.05) (Table 2). After combining the tumor volume ratio and tumor inhibition rate results, the therapeutic outcome in different treatment groups was as follows: AMD070 group < PTX group < PTX+AMD070 group < PTX-AMD070 NBs < PTX-AMD070 NBs+US group, indicating that with ultrasound exposure, PTX-AMD070 NBs could release PTX and AMD070 to target the xenograft tumors, thereby significantly augmenting the therapeutic effect against transplanted tumors.
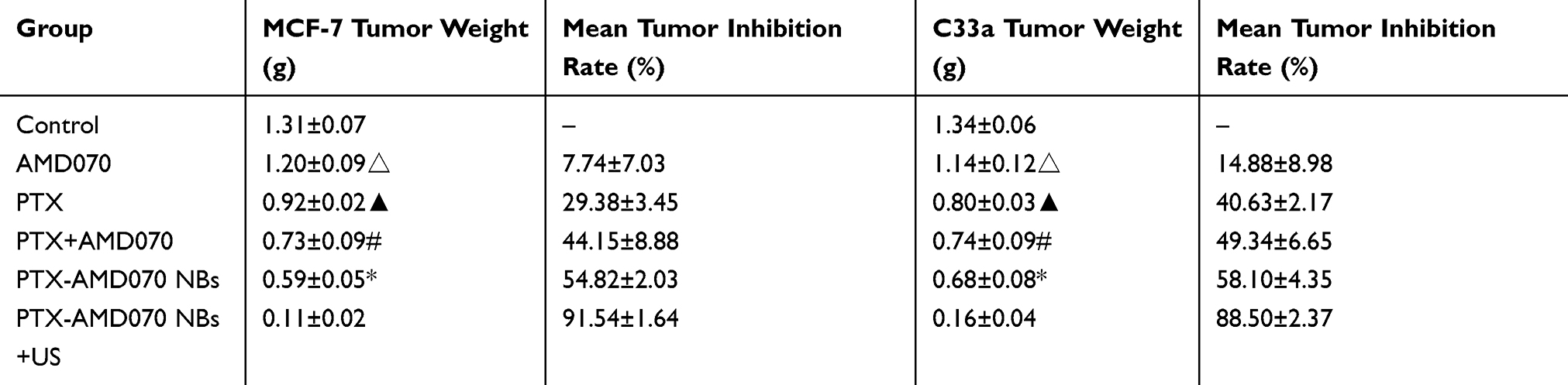 |
Table 2 Tumor Mass And Tumor Inhibition Rate In Different Treatment Groups |
Tissue sections of MCF-7 and C33a xenografts were microscopically observed. HE staining revealed that cell necrosis was present in all the treatment groups and was the most prominent in the PTX-AMD070 NBs+US group (Figure 10). Moreover, TUNEL staining revealed that apoptosis in MCF-7 and C33a xenografts was the most severe in the PTX-AMD070 NBs+US group (Figure 10).
 |
Figure 9 The curative effect in different treatment groups. (A) Xenografts in the tumor-bearing nude mice in different treatment groups. The red circles represent the xenograft areas. Scale, 1 cm. (B) Weight changes of nude mice bearing MCF-7 xenografts in different treatment groups. (C) Weight changes of nude mice bearing C33a xenografts in different treatment groups. (D) Tumor volume changes in nude mice bearing MCF-7 xenografts in different treatment groups. (E) Tumor volume changes in nude mice bearing C33a xenografts in different treatment groups. |
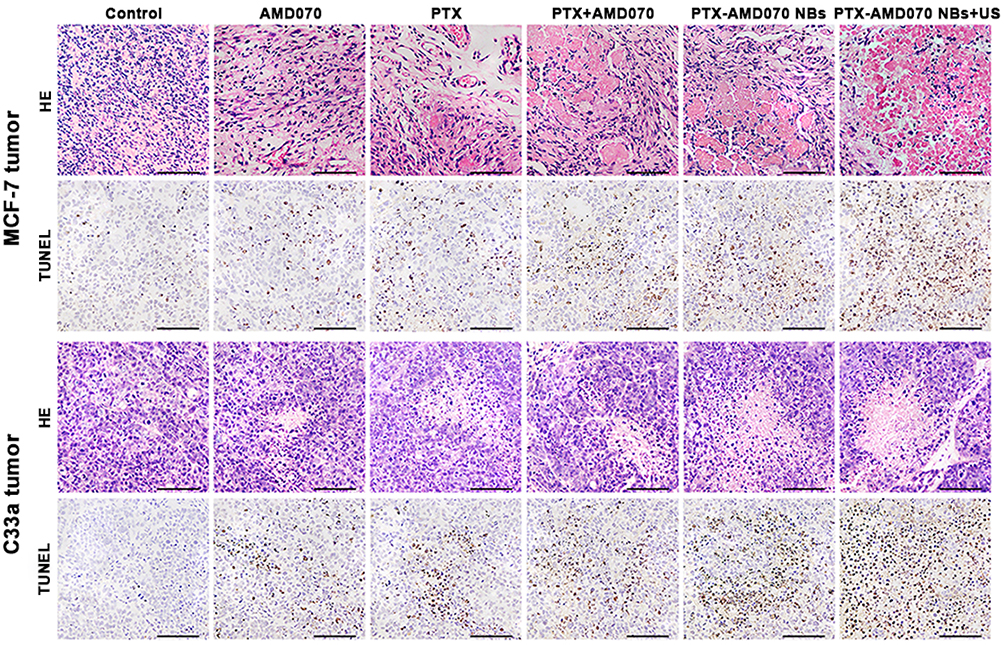 |
Figure 10 H&E staining and TUNEL staining of xenograft tissues. Scale, 100 μm. |
Discussion
Malignant tumors are characterized by extraordinary growth, invasion and metastasis abilities and thus pose a serious threat to patient life and health. The current chemotherapeutic drugs often have a variety of disadvantages, such as strong side effects, short term of curation, poor specificity, and propensity to induce drug resistance.28–30 With the rapid development of nanotechnology, targeted drug delivery systems provide a new approach for tumor chemotherapy. Such systems allow targeted release of chemotherapeutic drugs, thereby augmenting the concentrations of chemotherapeutic drugs in tumor tissues and boosting the curative effects. In recent years, rapid development in contrast-enhanced ultrasonography technology has ushered in more effective ultrasound contrast agents, which can not only improve ultrasound imaging of tumors and correspondingly their early diagnosis but also may function as a carrier of drug- or gene-therapy to promote treatment efficacy.31,32 However, because ultrasound contrast agents are typically at the micrometer scale, it is difficult to employ them for ultrasound molecular imaging outside tumor blood vessels and targeted delivery of drugs or genes to tumor parenchyma cells. Hence, integration of tumor diagnosis and therapy via development of ultrasound contrast agents capable of carrying therapeutic drugs/genes, passing through tumor blood vessels, and binding to tumor cells is a pivotal step. Integration of nanotechnology and ultrasound contrast agents can diminish the particle size of contrast agents and improve their aggregation in the extravascular space in tumor tissue, thereby facilitating extravascular ultrasound molecular imaging and targeted delivery of drugs to tumor parenchymal cells.33,34
CXCR4 is a G protein-coupled receptor that is highly expressed in various tumor cells and is closely related to tumor prognosis. CXCR4 interacts with its cognate ligand SDF-1 to encourage tumor cell proliferation and invasion. As such, CXCR4 is a specific target for tumor therapy. Tumor growth and metastasis can be suppressed by blocking the interaction between CXCR4 and SDF-1.35–37 Thus far, several CXCR4-targeted drugs have been developed, among which AMD070, a small-molecule antagonist of the receptor, has entered the second phase of clinical trials. This compound boasts several advantages, including low molecular weight, strong permeation ability, and high aqueous solubility. A large body of experimental evidence has demonstrated that AMD070 not only specifically binds to CXCR4-positive cells but also effectively inhibits tumor cell proliferation and metastasis.38–40 PTX is a microtubule stabilizer and is widely used as a chemotherapy agent against breast cancer, cervical cancer, and other tumors in the clinic. However, its antitumor therapeutic performance is reduced by certain issues, such as strong side effects, poor aqueous solubility, and inadequate specificity.41–43
Hence, herein, we generated targeted drug-loaded NBs, namely, PTX-AMD070 NBs, which incorporated AMD070, a small-molecule CXCR4 antagonist, and PTX, a commonly used chemotherapeutic drug. The goal of this study was to study the specific binding of AMD070 and PTX to various tumor cells, targeted ultrasound imaging contrast enhancement, and the therapeutic effect of concurrent use of the NBs with UTND. PTX-AMD070 NBs have the advantages of small particle size, good targeting specificity, and high encapsulation rate. PTX-AMD070 NBs were shown here to bind specifically to CXCR4-positive MCF-7 cells and C33a cells but not to CXCR4-negative MDA-MB-468 cells. The results indicate that PTX-AMD070 NBs exhibit specific binding to CXCR4-positive tumor cells. AMD070 can inhibit the proliferation of CXCR4-positive tumor cells, and the inhibitory effect is positively correlated with concentration. In comparison, PTX AMD070 treatment exhibited significantly higher proliferation inhibition and apoptosis promotion effects than PTX-AMD070 NBs treatment. The results indicated that in an external static environment, free PTX and AMD070 are more likely to diffuse and enter cells than PTX-AMD070 NBs, thereby leading to greater tumor suppression in vitro. Furthermore, PTX-AMD070 NBs+US treatment generated an outcome of proliferation inhibition and apoptosis promotion that was clearly superior to those observed with other treatments, indicating that integration of UTND and PTX-AMD070 NBs significantly augments the anti-tumor effects of AMD070 and PTX. This is the result of two factors. First, the cavitation effect of ultrasound can enhance the permeability of tumor cell membranes, which is advantageous for PTX entry into tumor cells, where it binds tubulin and promotes tumor suppression. Second, ultrasound irradiation results in targeted destruction of PTX AMD070 NBs, thereby providing targeted release of AMD070 and PTX around tumor cells, which is beneficial for the uptake of AMD070 and PTX by tumor cells and thus boosts their anti-tumor activities.
PTX-AMD070 NBs significantly improved the quality of ultrasound images of xenografts in nude mice. The NB material generated superior contrast-enhanced images in MCF-7 and C33a xenografts compared with PTX NBs, evidenced by greater peak intensity and AUCs. On the other hand, there was no significant difference between PTX-AMD070 NBs and PTX NBs in enhancement of ultrasound imaging in MDA-MB-468 xenografts. These results indicate that PTX-AMD070 NBs, which carry a small-molecule CXCR4 antagonist, specifically bind to tumor tissues expressing this receptor. As a consequence, the NBs can accumulate in tumor tissues with positive CXCR4 expression, which results in an increase in the number of particles in CXCR4-positive tumor tissue sections and in turn significantly enhances the ultrasound imaging quality. In other words, CXCR4 can be adopted as a specific target for ultrasound molecular imaging of multiple malignant tumors. PTX-AMD070 NBs carrying AMD070 enhanced ultrasound imaging of CXCR4-positive tumor tissues, which is beneficial for early diagnosis of malignant tumors. Fluorescence immunohistochemistry confirmed that DiI-labeled PTX-AMD070 NBs were distributed in intravascular and extravascular spaces in MCF-7, C33a, and MDA-MB-468 xenografts, suggesting that PTX-AMD070 NBs could penetrate the vascular system of the transplanted tumor tissue and enter the interstitial space in the transplanted tumor. Thus, our results provide the pathomorphological basis for PTX-AMD070 NBs to achieve ultrasound molecular imaging of extravascular tumor parenchymal cells and targeted drug delivery to tumor parenchymal cells.
The therapeutic effect observed in the PTX-AMD070 NBs group was better than that in the PTX AMD070 group, which was in contrast with the corresponding results of the in vitro cell assays. The main reasons were as follows. First, in the dynamic environment in vivo, free AMD070 and PTX were rapidly metabolized, whereas PTX-AMD070 NBs could release the drugs slowly and persistently in tumor tissues. Second, due to the activity of AMD070 on the surface of PTX-AMD070 NBs, the concentration of PTX in tumor tissue was increased and their therapeutic effect increased. This in conjunction with the tumor-suppression effects of AMD070 facilitates dual antitumor treatment. The therapeutic effect in the PTX-AMD070 NBs+US group was significantly better than that in the other groups, which was consistent with the in vitro antitumor assay results. The results demonstrate that concurrent application of UTND and PTX-AMD070 NBs could significantly augment the anti-tumor activities of AMD070 and PTX. Further analysis of xenograft sections revealed that the PTX-AMD070 NBs+US group exhibited necrosis and apoptosis rates in xenograft tissues significantly greater than those in the other groups. The results suggest that UTND combined with PTX-AMD070 NBs can significantly improve the therapeutic effects on MCF-7 and C33a cell xenografts by inhibiting cell proliferation and promoting apoptosis in the tumor.
In summary, the drug-loaded NBs with small and highly effective targeted delivery were synthesized in this study. AMD070, a small-molecule CXCR4 antagonist, not only has the ability to specifically bind many types of tumor cells but also has the ability to inhibit tumor cell proliferation. Combining the anti-tumor effect of AMD070 with that of PTX may facilitate early diagnosis and targeted therapy for many tumor types. PTX-AMD070 NBs can pass through tumor blood vessels and specifically bind to tumor parenchymal cells, thereby improving ultrasound molecular imaging of tumor parenchymal cells. Importantly, under ultrasound irradiation, the nanoparticles can provide targeted release of AMD70 and PTX, which achieves dual anti-tumor treatment. Nevertheless, there are some limitations in this study. First, steps are needed to further optimize the preparation technology for the targeted drug-loaded NBs, increase the drug loading capacity, and assess the safety of combined application of UTND and the NBs. Second, in-depth studies are needed to elucidate the molecular mechanisms by which targeted drug-loaded NBs carrying the small molecule antagonist AMD070 specifically bind to tumor cells, block the CXCL12/CXCR4 axis, release PTX, and suppress tumor growth and metastasis.
Conclusion
In this study, AMD070, a small-molecule CXCR4 antagonist, and PTX, a tumor chemotherapeutic drug, were conjugated to NBs with a lipid shell to construct PTX-AMD070 NBs, drug-loaded NBs targeting CXCR4. PTX-AMD070 NBs feature several advantages, including small particle size, high specificity, and enhanced ultrasound imaging, and can facilitate targeted release of PTX and AMD070 under ultrasound irradiation. Thus, PTX-AMD070 NBs achieve the dual inhibition of tumor growth and integrate diagnosis and treatment of malignant tumors. This study not only provides targeted drug-loaded NBs that are highly effective in integrating diagnosis and therapy for early treatment of many cancer types but also offers a new method and detailed research basis for ultrasonic molecular imaging and targeted therapy against malignant tumors mediated by NBs.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No 81 77071355, No 81771856, No 81571732), Municipal Social and Rural (Science & Technology) Innovation Project of Chongqing (No cstc2016shmszx130080).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–E386. doi:10.1002/ijc.29210
2. Borras JM, Lievens Y, Barton M, et al. How many new cancer patients in Europe will require radiotherapy by 2025? An ESTRO-HERO analysis. Radiother Oncol. 2016;119(1):5–11. doi:10.1016/j.radonc.2016.02.016
3. Miller KD, Siegel RL, Lin CC, et al. Cancer treatment and survivorship statistics, 2016. CA: Cancer J Clin. 2016;66(4):271–289. doi:10.3322/caac.21349
4. Cats A, Jansen EPM, van Grieken NCT, et al. Chemotherapy versus chemoradiotherapy after surgery and preoperative chemotherapy for resectable gastric cancer (CRITICS): an international, open-label, randomised phase 3 trial. Lancet Oncol. 2018;19(5):616–628. doi:10.1016/S1470-2045(18)30132-3
5. Tacar O, Sriamornsak P, Dass CR. Doxorubicin: an update on anticancer molecular action, toxicity and novel drug delivery systems. J Pharm Pharmacol. 2013;65(2):157–170. doi:10.1111/j.2042-7158.2012.01567.x
6. Moslehi JJ. Cardiovascular toxic effects of targeted cancer therapies. N Engl J Med. 2016;375(15):1457–1467. doi:10.1056/NEJMra1100265
7. Curigliano G, Cardinale D, Dent S, et al. Cardiotoxicity of anticancer treatments: epidemiology, detection, and management. CA: Cancer J Clin. 2016;66(4):309–325. doi:10.3322/caac.21341
8. Bozzuto G, Molinari A. Liposomes as nanomedical devices. Int J Nanomed. 2015;10:975. doi:10.2147/IJN.S68861
9. Jiang L, Li L, He X, et al. Overcoming drug-resistant lung cancer by paclitaxel loaded dual-functional liposomes with mitochondria targeting and pH-response. Biomaterials. 2015;52:126–139. doi:10.1016/j.biomaterials.2015.02.004
10. Schwendener RA. Liposomes as vaccine delivery systems: a review of the recent advances. Ther Adv Vaccines. 2014;2(6):159–182. doi:10.1177/2051013614541440
11. Barenholz Y. Amphipathic weak base loading into preformed liposomes having a transmembrane ammonium ion gradient: from the bench to approved DOXIL. In: Gregoriadis G, editor. Liposome Technol. Boca Raton: CRC Press; 2016:25–50.
12. Deshpande PP, Biswas S, Torchilin VP. Current trends in the use of liposomes for tumor targeting. Nanomedicine. 2013;8(9):1509–1528. doi:10.2217/nnm.13.118
13. Eloy JO, de Souza MC, Petrilli R, et al. Liposomes as carriers of hydrophilic small molecule drugs: strategies to enhance encapsulation and delivery. Colloids and Surf B. 2014;123:345–363. doi:10.1016/j.colsurfb.2014.09.029
14. Liu HL, Fan CH, Ting CY, et al. Combining microbubbles and ultrasound for drug delivery to brain tumors: current progress and overview. Theranostics. 2014;4(4):432. doi:10.7150/thno.8074
15. Christensen-Jeffries K, Browning RJ, Tang MX, et al. In vivo acoustic super-resolution and super-resolved velocity mapping using microbubbles. IEEE T Med Imaging. 2015;34(2):433–440. doi:10.1109/TMI.2014.2359650
16. Unger E, Porter T, Lindner J, Grayburn P. Cardiovascular drug delivery with ultrasound and microbubbles. Adv Drug Delivery Rev. 2014;72:110–126. doi:10.1016/j.addr.2014.01.012
17. Jeon M, Song W, Huynh E, et al. Methylene blue microbubbles as a model dual-modality contrast agent for ultrasound and activatable photoacoustic imaging. J Biomed Opt. 2014;19(1):016005. doi:10.1117/1.JBO.19.1.015008
18. Lohse D, Zhang X. Surface nanobubbles and nanodroplets. Rev Mod Phys. 2015;87(3):981. doi:10.1103/RevModPhys.87.981
19. Yu L, Yang JF, Lou XW. Formation of CoS2 nanobubble hollow prisms for highly reversible lithium storage. Angew Chem Int Ed. 2016;55(43):13422–13426. doi:10.1002/anie.201606776
20. Lohse D, Zhang X. Pinning and gas oversaturation imply stable single surface nanobubbles. Phys Rev E. 2015;91(3):031003. doi:10.1103/PhysRevE.91.031003
21. Zhu X, Verzicco R, Zhang X, Lohse D. Diffusive interaction of multiple surface nanobubbles: shrinkage, growth, and coarsening. Soft Matter. 2018;14(11):2006–2014. doi:10.1039/c7sm02523h
22. Teicher BA, Fricker P. CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin Cancer Res. 2010;16(11):2927–2931. doi:10.1158/1078-0432.CCR-09-2329
23. Xu C, Zhao H, Chen H, et al. CXCR4 in breast cancer: oncogenic role and therapeutic targeting. Drug Des Dev Ther. 2015;9:4953.
24. Zhao H, Guo L, Zhao H, et al. CXCR4 over-expression and survival in cancer: a system review and meta-analysis. Oncotarget. 2015;6(7):5022.
25. Scala S. Molecular pathways: targeting the CXCR4-CXCL12 axis-untapped potential in the tumor microenvironment. Clin Cancer Res. 2015;21(19):4278–4285. doi:10.1158/1078-0432.CCR-14-0914
26. Wester HJ, Keller U, Schottelius M, et al. Disclosing the CXCR4 expression in lymphoproliferative diseases by targeted molecular imaging. Theranostics. 2015;5(6):618. doi:10.7150/thno.11251
27. Philipp-Abbrederis K, Herrmann K, Knop S, et al. In vivo molecular imaging of chemokine receptor CXCR4 expression in patients with advanced multiple myeloma. EMBO Mol Med. 2015;7(4):477–487. doi:10.15252/emmm.201404698
28. Zhong Y, Meng F, Deng C, et al. Ligand-directed active tumor-targeting polymeric nanoparticles for cancer chemotherapy. Biomacromolecules. 2014;15(6):1955–1969. doi:10.1021/bm5003009
29. He W, Wang Q, Srinivasan B, et al. A JNK-mediated autophagy pathway that triggers c-IAP degradation and necroptosis for anticancer chemotherapy. Oncogene. 2014;33(23):3004. doi:10.1038/onc.2013.256
30. Martins I, Raza SQ, Voisin L, et al. Anticancer chemotherapy and radiotherapy trigger both non-cell-autonomous and cell-autonomous death. Cell Death Dis. 2018;9(7):716. doi:10.1038/s41419-018-1111-y
31. Jain A, Tiwari A, Verma A, et al. Ultrasound-based triggered drug delivery to tumors. Drug Delivery Transl Res. 2018;8(1):150–164. doi:10.1007/s13346-017-0448-6
32. Paefgen V, Doleschel D, Kiessling F. Evolution of contrast agents for ultrasound imaging and ultrasound-mediated drug delivery. Front Pharmacol. 2015;6:197. doi:10.3389/fphar.2015.00197
33. Zhao YZ, Lin Q, Wong HL, et al. Glioma-targeted therapy using Cilengitide nanoparticles combined with UTMD enhanced delivery. J Controlled Release. 2016;224:112–125. doi:10.1016/j.jconrel.2016.01.015
34. Liu Z, Zhang J, Tian Y, et al. Targeted delivery of reduced graphene oxide nanosheets using multifunctional ultrasound nanobubbles for visualization and enhanced photothermal therapy. Int J Nanomed. 2018;13:7859. doi:10.2147/IJN
35. Hunter ZR, Xu L, Yang G, et al. The genomic landscape of Waldenström macroglobulinemia is characterized by highly recurring MYD88 and WHIM-like CXCR4 mutations, and small somatic deletions associated with B-cell lymphomagenesis. Blood. 2014;123(11):1637–1646. doi:10.1182/blood-2013-09-525808
36. Qin L, Kufareva I, Holden LG, et al. Crystal structure of the chemokine receptor CXCR4 in complex with a viral chemokine. Science. 2015;347(6226):1117–1122. doi:10.1126/science.1261064
37. Guo F, Wang Y, Liu J, et al. CXCL12/CXCR4: a symbiotic bridge linking cancer cells and their stromal neighbors in oncogenic communication networks. Oncogene. 2016;35(7):816. doi:10.1038/onc.2015.139
38. Kinouchi M, Uchida D, Kuribayashi N, et al. AMD070, a novel orally bioavailable CXCR4 inhibitor, inhibits the metastases of oral cancer via SDF-1/CXCR4 system[abstract]. Am Assoc Cancer Res. 2016;76(suppl14):4117.
39. Kuribayashi N, Uchida D, Kinouchi M, et al. Effect of a novel orally bioavailable CXCR4 inhibitor, AMD070, on the metastasis of oral cancer cells[abstract]. Am Assoc Cancer Res. 2018;78(suppl13):4191.
40. Morimoto M, Matsuo Y, Koide S, et al. Enhancement of the CXCL12/CXCR4 axis due to acquisition of gemcitabine resistance in pancreatic cancer: effect of CXCR4 antagonists. BMC Cancer. 2016;16(1):305. doi:10.1186/s12885-016-2340-z
41. Wilke H, Muro K, Van Cutsem E, et al. Ramucirumab plus paclitaxel versus placebo plus paclitaxel in patients with previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (RAINBOW): a double-blind, randomised phase 3 trial. Lancet Oncol. 2014;15(11):1224–1235. doi:10.1016/S1470-2045(14)70420-6
42. Stathis A, Hess D, von Moos R, et al. Phase I trial of the oral smoothened inhibitor sonidegib in combination with paclitaxel in patients with advanced solid tumors. Invest New Drugs. 2017;35(6):766–772. doi:10.1007/s10637-017-0454-z
43. Shitara K, Özgüroğlu M, Bang YJ, et al. Pembrolizumab versus paclitaxel for previously treated, advanced gastric or gastro-oesophageal junction cancer (KEYNOTE-061): a randomised, open-label, controlled, phase 3 trial. Lancet. 2018;392(10142):123–133. doi:10.1016/S0140-6736(18)31257-1
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.