Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Preparation and evaluation of tilmicosin-loaded hydrogenated castor oil nanoparticle suspensions of different particle sizes
Authors Chen X, Wang T, Lu M, Zhu L, Wang Y, Zhou W
Received 8 December 2013
Accepted for publication 22 January 2014
Published 27 May 2014 Volume 2014:9(1) Pages 2655—2664
DOI https://doi.org/10.2147/IJN.S58898
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Xiaojin Chen, Ting Wang, Mengmeng Lu, Luyan Zhu, Yan Wang, WenZhong Zhou
Department of Preventitive Veterinary Medicine, College of Veterinary Medicine, China Agricultural University, Beijing, People's Republic of China
Abstract: Three tilmicosin-loaded hydrogenated castor oil nanoparticle (TMS-HCO-NP) suspensions of different particle sizes were prepared with different polyvinyl alcohol surfactant concentrations using a hot homogenization and ultrasonic technique. The in vitro release, in vitro antibacterial activity, mammalian cytotoxicity, acute toxicity in mice, and stability study were conducted to evaluate the characteristics of the suspensions. The in vitro tilmicosin release rate, antibacterial activity, mammalian cytotoxicity, acute toxicity in mice, and stability of the suspensions were evaluated. When prepared with polyvinyl alcohol concentrations of 0.2%, 1%, and 5%, the mean diameters of the nanoparticles in the three suspensions were 920±35 nm, 452±10 nm, and 151±4 nm, respectively. The three suspensions displayed biphasic release profiles similar to that of freeze-dried TMS-HCO-NP powders, with the exception of having a faster initial release. Moreover, suspensions of smaller-sized particles showed faster initial release, and lower minimum inhibitory concentrations and minimum bactericidal concentrations. Time-kill curves showed that within 12 hours, the suspension with the 151 nm particles had the most potent bactericidal activity, but later, the suspensions with larger-sized particles showed increased antibacterial activity. None of the three suspensions were cytotoxic at clinical dosage levels. At higher drug concentrations, all three suspensions showed similar concentration-dependent cytotoxicity. The suspension with the smallest-sized particle showed significantly more acute toxicity in mice, perhaps due to faster drug release. All three suspensions exhibited good stability at 4°C and at room temperature for at least 6 months. These results demonstrate that TMS-HCO-NP suspensions can be a promising formulation for tilmicosin, and that nanoparticle size can be an important consideration for formulation development.
Keywords: tilmicosin, solid lipid nanoparticle suspension, particle size, release rate, antibacterial activity, toxicity
Introduction
Tilmicosin is a semisynthetic macrolide antibiotic developed by Lilly Research Laboratories (Indianapolis, IN, USA),1 which has potent and broad-spectrum antimicrobial activity and rapid accumulation in bovine macrophages and mammary gland epithelial cells.2,3 Following intravenous or subcutaneous administration, tilmicosin rapidly passes from the blood into milk and is eliminated slowly from the milk.2 Thus, tilmicosin can be used for the treatment of bovine mastitis; however, tilmicosin can also cause acute cardiac toxicity.2 Cattle can tolerate doses as high as 50 mg/kg if the drug is administered subcutaneously, but they have toxic reactions consistent with cardiovascular damage when injected intravenously, even at doses as low as 5 mg/kg.4 Pigs also exhibit a toxic reaction to tilmicosin (increased respiration, emesis, and convulsions) at 10 mg/kg when administered intramuscularly.4
Since tilmicosin is a Lewis base with two tertiary amines, it is highly hydrophobic in basic conditions, with almost negligible solubility above pH 10. In acid conditions, tilmicosin can form salts, which increase its aqueous solubility.5,6 Currently, soluble tilmicosin phosphate is used in clinics; however, this form exhibits problems of low potency, poor absorption, and low systemic bioavailability. Given these disadvantages, the development of a novel delivery system for tilmicosin is warranted.
Our previous studies demonstrated that a lyophilized powdered form of tilmicosin-loaded hydrogenated castor oil solid lipid nanoparticles (TMS-HCO-NP) is a promising formulation for improving the pharmacological and toxicological characteristics of tilmicosin, enhancing the therapeutic efficacy and decreasing tilmicosin toxicity.7–9 A pharmacokinetics study examining what occurs following its subcutaneous administration in mice demonstrated that this formulation shows significantly longer systemic circulation, greater bioavailability, and lower maximum serum concentration than native tilmicosin.8 A subsequent mouse mastitis study indicated that the TMS-HCO-NP formulation has enhanced antibacterial activity and therapeutic efficacy at a lower dose and frequency than tilmicosin alone.7 A lower peak tilmicosin concentration may account for the reduced acute toxicity of TMS-HCO-NP.9
Solid lipid nanoparticles (SLN) are colloidal drug delivery systems that can be prepared as either aqueous dispersions (suspensions) or dry powders. Although liquid dispersions of SLN can also be converted into dry powders for stable storage by spray-drying or lyophilization,10,11 suspensions are still preferred due to great ease of preparation and low cost.12,13 Production cost is an important driver for developing veterinary clinical applications,14 and SLN suspensions can be produced on a large scale and can be used directly.15,16
Particle size has become an important developmental consideration in the field of nanobiology, particularly during the development of new drug delivery systems.17,18 Formulation methods that decrease the size of the drug particles to submicron sizes have shown promise for enhancing the dissolution of poorly soluble drugs.19–23 According to the Noyes–Whitney equation, smaller particle sizes equate to a larger effective surface area which, in turn, increases the dissolution rate.24 Accordingly, there are examples in which decreasing drug particle size improves bioavailability.19 The size of the drug particles at the microscopic level can affect biodistribution; in some cases, it is as important as the chemical composition of the drug.26
Factors that influence SLN particle size include the SLN preparation method, as well as the type and amount of surfactant and lipid used. In this study, several TMS-HCO-NP formulations were prepared by hot homogenization and ultrasonication, and their particle sizes were controlled by varying the surfactant concentration. The physical, biological, and chemical characteristics of TMS-HCO-NP suspensions with different particle sizes were evaluated. The results provide an enhanced understanding of the optimization process of nanoparticles, and the resulting formulations are promising alternatives for improving tilmicosin delivery.
Materials and methods
Materials
Hydrogenated castor oil (HCO) was obtained from Tongliao Tonghua Castor Chemical Co, Ltd (Inner Mongolia, People’s Republic of China). The tilmicosin (93.1%, weight/weight) was produced by Shandong Jiulong Hisince Pharmaceutical Co., Ltd (Jinan, Shandong Province, People’s Republic of China). Polyvinyl alcohol (PVA) (87%–90% hydrolyzed, average molecular weight: 30,000–70,000) was purchased from Sigma-Aldrich (St Louis, MO, USA). MTT (3-[4,5-Dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) was purchased from Sigma-Aldrich. Chloroform was of analytical reagent grade and bought from Beijing Chemical Works (Beijing, People’s Republic of China). Other chemicals and reagents not specified in the text were of analytical grade or equivalent. The baby hamster kidney cell line (BHK-21) was from the American Type Culture Collection (Manassas, VA, USA).
Animals and bacteria
Imprinting control region Kunming mice of each sex (20–22 g) were obtained from Vital River Laboratory Animal Technology Co, Ltd (Beijing, People’s Republic of China). The animals were housed at room temperature (RT) under natural day and night cycles with free access to water and food. They were kept for 1 week before use. All experimental protocols concerning the handling of mice were conducted in accordance with the requirements of the Institutional Animal Care and Use Committee at China Agricultural University (Beijing, People’s Republic of China).
Staphylococcus aureus (clinical strain) was isolated from herds of cows shedding the microbe (Lvhe-Dairy, Sanyuan, Beijing, People’s Republic of China). Bacteria were streaked from glycerol-frozen stocks onto agar plates and incubated overnight at 37°C. A single bacterial colony from the fresh plates was inoculated in the Mueller–Hinton broth (MHB) and grown at 37°C in a shaking incubator at 180 rpm to an OD600 of 0.6 (in the period of the logarithmic phase), which corresponds to 2.0×108 colony-forming units (cfu)/mL confirmed by plating serial dilutions. For the in vitro study, the bacteria were diluted in broth.
Preparation of TMS-HCO-NP suspensions
Tilmicosin-loaded SLN suspensions were formulated by hot homogenization and ultrasonic technique, as described previously.8 Briefly, 0.3 g of tilmicosin was mixed with 2.7 g of HCO in a 100 mL tube and heated in a boiling water bath. After the lipid was melted and the drug was dissolved in the melted lipid, 30 mL of 0.2%, 1%, or 5% PVA solution preheated in a boiling water bath was poured into the lipid phase and stirred at 150 rpm for 3 minutes to form an oil in water emulsion. The emulsion was sonicated for 30 minutes (VCX 750 Vibra-Cell™; Sonics and Materials, Inc., Newtown, CT, USA) using the 13 mm microprobe with an amplitude of 35% to form a nanoemulsion. The nanoemulsion was quickly poured into 30 mL of cold water to obtain a nanoparticle suspension. The control SLN was prepared similarly without adding tilmicosin.
Determination of mean diameter, polydispersity index, and zeta potential
The mean diameter (MD), polydispersity index (PDI), and zeta potential (ZP) of the SLN were determined by photon correlation spectroscopy using Zetasizer Nano ZS90 (Malvern Instruments, Malvern, UK) at 25°C. The samples of SLN suspension were diluted with distilled water by 10–20 times for the particle size and PDI analysis, and by 100–200 times for ZP determination to get optimum kilo counts per second of 20–400 for measurements.
Determination of drug loading
The nanoparticles were collected by centrifugation at 12,000 rpm (Centrifuge 5810 R; Eppendorf AG, Hamburg, Germany) for 60 minutes at 4°C and washed three times with distilled water. The SLN were resuspended in distilled water and lyophilized for 48 hours (LGJ-12 Freeze Dryer; Beijing Songyuan Huaxing Technology Development Co, Ltd, Beijing, People’s Republic of China). The control (blank) SLN without tilmicosin was prepared similarly.
To determine the drug content in the SLN, a weighed amount of freeze-dried SLN was dissolved in chloroform and the solution was analyzed directly at 291 nm using an ultraviolet spectrophotometer.7 The control nanoparticles were treated similarly as blanks for the measurements. The assay was repeated three times using different samples from independent preparations. Drug loading (DL) is defined as follows:
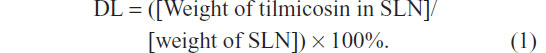
Microscopic analysis
The SLN suspension was diluted with distilled water by fivefold, and 2 μL of the dilution was placed onto a microscope slide. The nanoparticles were dried at RT for 5 minutes and photomicrographs of the SLN were taken using an inverted optical microscope (Olympus 1X71; Olympus Corporation, Tokyo, Japan).
Scanning electron microscopy
The surface morphology of the nanoparticles was characterized by scanning electron microscope (SEM) (Quanta 200F; FEI Company, Hillsboro, OR, USA). Briefly, 5 mg of the samples was suspended in 1 mL of distilled water, and 2 μL of the suspension was placed on a cover slip and dried at RT. The samples were coated with gold using an ion sputter pump and examined.
In vitro release
TMS-HCO-NP suspensions were diluted (1:1) with either a 0.9% (weight/volume) sodium chloride (NaCl) solution or 0.1 mol/L of hydrogen chloride (HCl). The diluted suspension (2 mL, donor solution) was added in a dialysis bag (molecular weight: 8000–14,000) and dialyzed against 38 mL 0.9% (w/v) NaCl solution or 0.1 M HCl (receiver solution) in a 50 mL tube at RT under magnetic stirring at 60 rpm. Lyophilized TMS-HCO-NP powder and native tilmicosin (with same amount of drug as the suspension) suspended or dissolved in 1 mL of the 0.9% NaCl solution or 0.1 M HCl were used as controls. At fixed time points, samples (2 mL) were withdrawn from the receiver solution and the same amount of fresh medium was added to maintain a constant volume. The blank nanoparticle formulation without tilmicosin was treated similarly and used as blanks for the measurements. The experiments were carried out in triplicate.
In vitro antibacterial activity
Minimal inhibitory concentration (MIC) and minimum bactericidal concentration (MBC) were determined by the broth dilution technique with MHB using an inoculum of 5×105 cfu/mL, as recommended by the Clinical and Laboratory Standards Institute.25 Briefly, 1 mL serial dilutions of the TMS-HCO-NP suspension were mixed with 1 mL of the bacterial culture (1×106 cfu/mL, in MHB) in a 4 mL sterile centrifuge tube (the final tilmicosin concentrations ranged from 2.0–34.0 μg/mL) and the mixture was incubated at 37°C in an incubator (Taicang Experimental Equipment Factory, Taicang, Jiangsu, People’s Republic of China) with shaking at 130 rpm. The MIC was defined as the lowest concentration inhibiting visible growth after incubation at 37°C for 12 hours.27 The MIC of native tilmicosin and released tilmicosin was measured in the same way. All experiments were carried out in triplicate. The MBC is measured by subculturing the broths used for MIC determination onto fresh agar plates. The MBC was the lowest concentration of the drug that results in killing 99.9% of the bacteria being tested.
Sustained antibacterial studies were conducted by mixing 1×106 cfu of the bacterial organisms with different tilmicosin formulations in 2 mL of the MHB medium in a 4 mL tube (final drug concentration: 3.0 μg/mL). The mixtures were incubated at 37°C in an incubator with shaking at 130 rpm. At fixed time points (12 hours, 24 hours, 36 hours, and 48 hours) serial dilutions of the mixtures (100 μL) were inoculated on Luria broth agar plates (Beijing Aoboxing Bio-Tech Co., Ltd, Beijing, People’s Republic of China) at 37°C. The colonies were counted when they could be observed by the naked eye. Growth graphs were plotted by calculating the number of colonies in 1 mL of the mixture versus the incubating time (hours). All experiments were carried out in triplicate.
Cytotoxicity
Cytotoxicity of TMS-HCO-NP suspensions was evaluated in the BHK-21 cell line using the MTT assay. The cells were seeded into a 96-well microplate at a density of 1×104 cells per well in 0.1 mL of Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum and antibiotics, and they were cultured in a humidified 5% CO2 incubator at 37°C. After 24 hours, the medium was replaced with 100 μL of DMEM complete medium containing tilmicosin (final drug concentration: 32 μg/mL, 64 μg/mL, 128 μg/mL, 256 μg/mL, and 512 μg/mL). Cytotoxicity of the blank nanoparticle suspensions of the same volume was also conducted. After 24 hours of coincubation, cell viability was assessed by MTT assay. Typically, 5 mg/mL of MTT in PBS was added to each well reaching a final MTT concentration of 0.5 mg/mL and incubated for 4 hours. Then, the supernatants were removed and the formazan crystals were dissolved in 150 μL of dimethyl sulfoxide. Aliquots were drawn from each well, and the absorbance at 490 nm was determined by a microplate reader (Bio-Rad 680; Bio-Rad Laboratories, Hercules, CA, USA). Cells without adding SLN were taken as control and set to 100% viability. The cell viability (%) was calculated using the following formula:
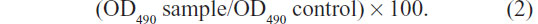
Acute toxicity
Mortality rates were used to evaluate the acute toxicity of the suspensions. A preliminary lethal test demonstrated that the suspension with the smallest-sized nanoparticle was the most toxic, and its absolute lethal dose was 550 mg/kg body weight (BW) of tilmicosin. This dose was used for the experiment.
Sixty mice were randomly divided into six groups, with five male and five female animals in each group. The nanoparticle suspensions were lyophilized and resuspended in a 0.9% NaCl solution to concentrate the drug to 27.5 mg/mL. A total of 500 μL of the concentrated suspension was subcutaneously administered on the dorsum of each mouse. The concentrated blank nanoparticle suspension with the smallest particle size and naked tilmicosin suspended in 1% PVA was administered in the control groups. Clinical symptoms and mortality rates were recorded continuously for 24 hours.
Stability studies
Stability studies of SLN were performed after the samples were stored at 4°C and at RT for 3 months and 6 months. The values of the MD, PDI, ZP, and DL were measured for the evaluation of the physical stability of the nanoparticle suspensions.
Results
Characteristics of TMS-HCO-NP
Optical microscopy and SEM images showed that the nanoparticles in the three suspensions were well dispersed and spherically shaped with smooth surfaces (Figure 1A and B). The mean particle sizes decreased with increases in PVA concentration. Further analysis by photon correlation spectroscopy showed that the PVA concentration significantly affected both the particle size and DL of the nanoparticles in the suspensions (Table 1). As PVA concentration increased from 0.2% to 5%, the particle size and DL decreased from 920 nm to 151 nm, and 7.73% to 3.76%, respectively. The PDIs of the three nanoparticle suspensions were not significantly different. The ZPs of the smallest nanoparticle suspension were slightly lower than that of the other two suspensions.
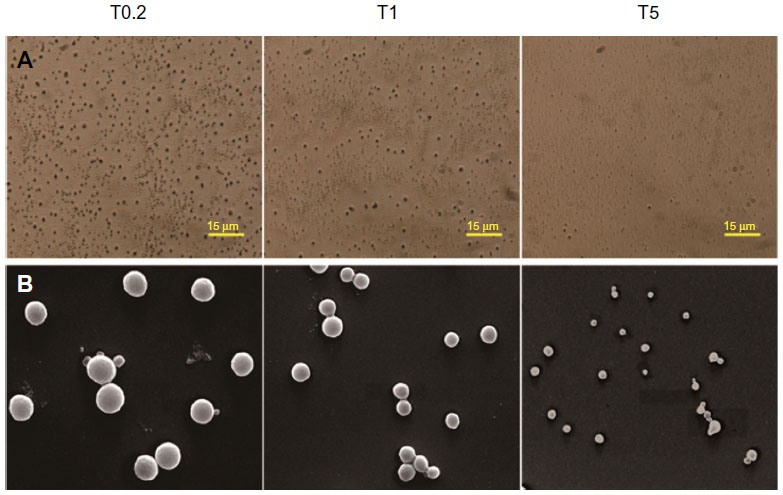 | Figure 1 Photographs of TMS-HCO-NP. |
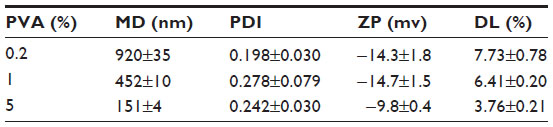 | Table 1 Characteristics of TMS-HCO-NP (mean ± standard deviation; n=3) |
In vitro release
The in vitro release profile of the TMS-HCO-NP suspensions compared to native tilmicosin is summarized as the cumulative percentage release of tilmicosin (Figure 2). The three TMS-HCO-NP suspensions displayed similar release patterns to that of freeze-dried TMS-HCO-NP powders. Drug release was rapid during the first 2 hours, followed by a relatively slower release phase lasting for 24 hours, after which a more gradual slow release was observed. Notably, the suspensions showed much faster initial release than the powders, and suspensions with smaller-sized particles showed faster initial release. During the first 2 hours in the NaCl solution (0.9% w/v), the cumulative releases of the three nanoparticle suspensions (particle sizes 900 nm, 450 nm and 150 nm) were 13.38%±1.50%, 24.04%±1.89%, and 29.35%±3.64%, respectively, compared to 2.80%±0.74%, 7.87%±0.34% and 14.37%±0.56% for the freeze-dried powders. The native tilmicosin control exhibited a rapid release with 45.65%±2.65% of drug released in 2 hours, and the release was almost complete (93.26%±0.48%) after 12 hours. Release studies in 0.1 M HCl showed similar trends to those in the NaCl solution, but the overall release was faster (Figure 2).
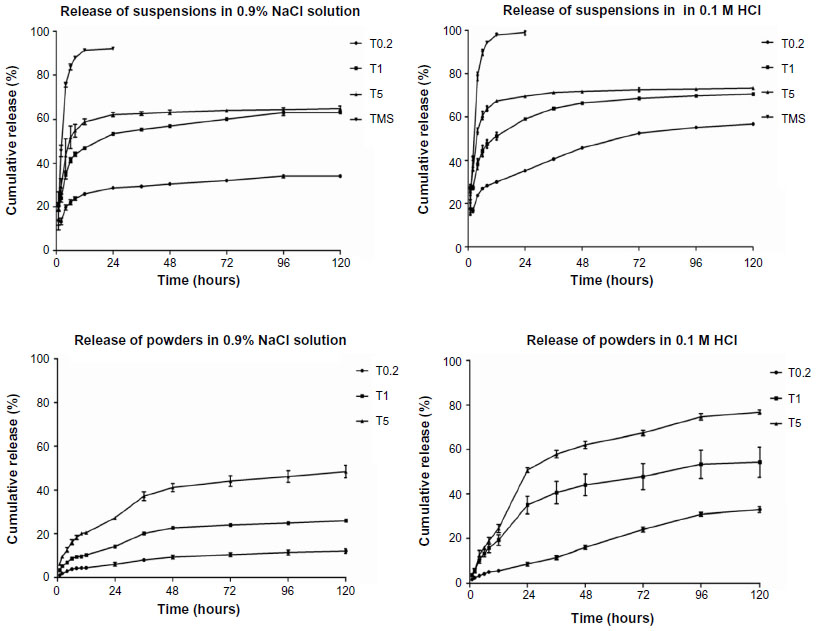 | Figure 2 In vitro release of different tilmicosin formulations (mean ± standard deviation; n=3). |
In vitro antibacterial activity
The results of in vitro antibacterial activity assessments are shown in Table 2. The MIC and MBC of native tilmicosin were 3.0 μg/mL and 12.0 μg/mL, respectively. The tilmicosin released from the TMS-HCO-NP suspensions had the same MIC and MBC values as that of the native tilmicosin, indicating that the antibacterial activity of the drug was not changed by the preparation and release processes. The results showed that the smaller the particle size, the lower the MIC and MBC values of the TMS-HCO-NP suspensions. The MIC and MBC values of the suspension with the smallest-sized particles decreased to the same levels as that shown by native tilmicosin.
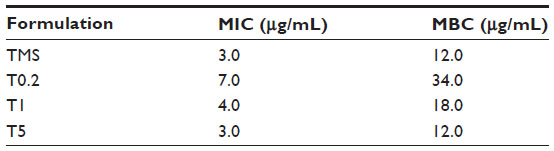 | Table 2 MIC and MBC of the tilmicosin formulations |
Next, the time-kill curves for the three TMS-HCO-NP suspensions and native tilmicosin were compared (Figure 3). Within the first 12 hours, suspensions of the smallest nanoparticles had the most potent antibacterial activity. The number of bacteria colonies in the cultures with the three suspensions increased from an initial density of 5.0×105 cfu/mL to densities of 4.0×106 cfu/mL, 4.5×107 cfu/mL, and 1.4×108 cfu/mL at 12 hours (particle size ranging from small to large) and 7.3×106 cfu/mL in the culture with the native drug. From 12–24 hours, the culture with the smallest-sized particle grew more rapidly, as it showed a fast cfu increase (from 4.0×106 cfu/mL to 1.2×108 cfu/mL) with a rate comparable to the growth of the culture with native tilmicosin (from 7.3×106 cfu/mL to 1.5×108 cfu/mL), whereas cultures with the other two suspensions grew more slowly (from 4.5×107 cfu/mL to 1.9×108 cfu/mL for 450 nm-sized particle and 1.4×108 cfu/mL to 2.7×108 cfu/mL for 900 nm-sized particle, respectively). Bacterial densities for all cultures increased slowly between 24–36 hours with no significant differences observed among the formulations.
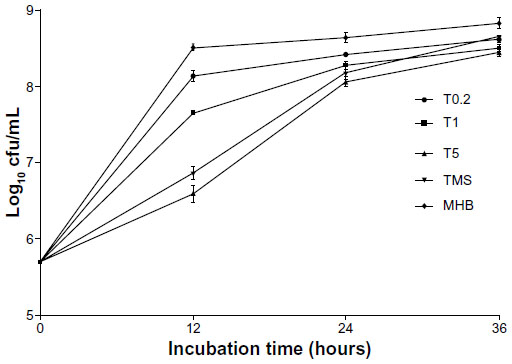 | Figure 3 In vitro time-kill curve (mean ± standard deviation; n=3). |
Cytotoxicity
Mammalian cytotoxicity for the three drug-loaded nanoparticle suspensions was assessed with a BHK-21 cell line, with drug concentrations ranging from 32–512 μg/mL (Figure 4). None of the three suspensions showed cytotoxic activity at 32 μg/mL. Higher doses led to gradually increasing cytotoxicity, but no significant differences were observed among the different formulations. The tilmicosin control showed very low cytotoxicity.
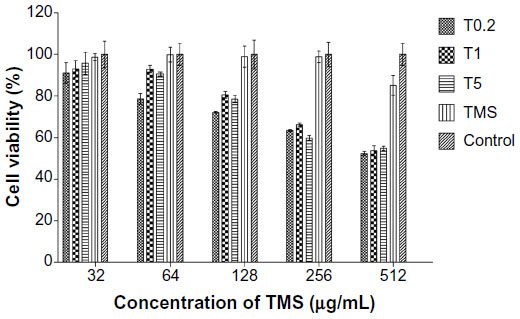 | Figure 4 Cytotoxicity of TMS-HCO-NP suspensions on the BHK-21 cell line. |
Acute toxicity
Acute toxicity was assessed for each of the formulations in mice following a single subcutaneous dose (Table 3). Treatment with the suspension containing the smallest-sized particles led to 100% mortality within 12 hours. Importantly, treatment with native tilmicosin suspended in 1% PVA also caused 100% mortality, whereas treatment with the drug-free nanoparticle control (smallest particle size) did not cause any mortality. Treatment with suspensions containing larger-sized particles did not cause mortality, although the treatment demonstrated significant clinical signs of toxicity, such as transient reversible action retardation, anorexia, and gloomy spirit. The spirit and behavior of the surviving mice were gradually recovered.
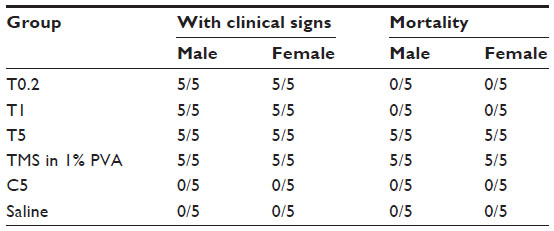 | Table 3 The ratio of animals with clinical signs and their mortality after treatment |
Stability
The three suspensions exhibited good stability at 4°C and RT during 6 months of storage. Slight increases in the MDs and PDIs and slight decreases in the DL and ZPs were observed (Table 4).
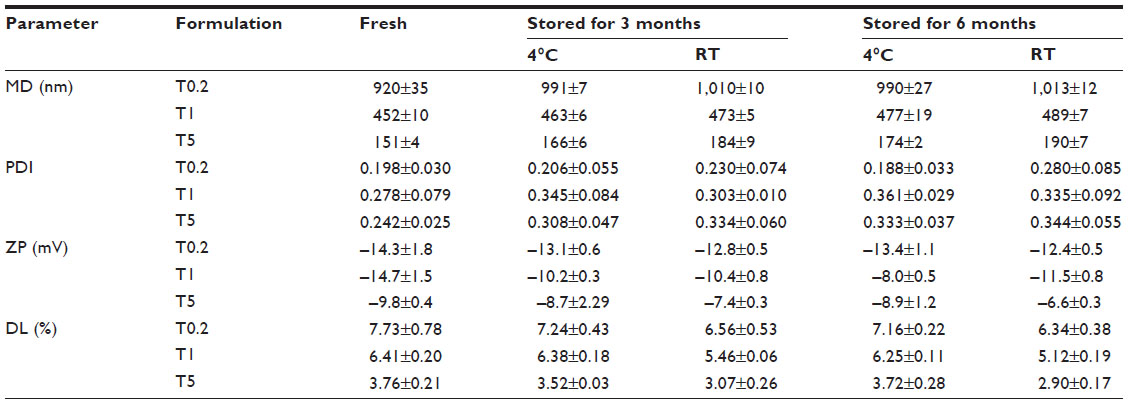 | Table 4 Characteristics of fresh and stored TMS-HCO-NP suspensions (mean ± standard deviation, n=3) |
Discussion
Tilmicosin-loaded HCO nanoparticle suspensions were prepared using a homogenization and ultrasonication method. Optical and SEM micrographs demonstrated that the surface morphology of the nanoparticles resembled a globular structure, where the shape did not change with different PVA concentrations. Varying the PVA concentration changed the particle size, but it had no significant impact on surface roughness or on the appearance of the nanoparticles.
The finding that a higher concentration of PVA was required to prepare suspensions with smaller particles sizes is consistent with a number of previous reports, where the findings indicated that higher PVA concentrations resulted in smaller nanoparticles.28,29 Moreover, adequate ultrasonication time is essential for preparing nanoparticles with a narrow particle size distribution. Notably, longer ultrasonication time did not result in decreased particle sizes when lower PVA concentrations were used. In this study, 30-minute ultrasonication was sufficient to obtain SLNs with lower PDI values. The calculation of PDI accounts for the mean particle size, the refractive index of the solvent, the measurement angle, and the variance of distribution. Lower PDI values indicate high homogeneity in the particle population, whereas high PDI values suggest a broad size distribution or different size populations. The PDI values were very similar among nanoparticles of the three suspensions prepared, thereby suggesting that PVA concentration does not significantly affect the PDI value.
The ZP values were slightly higher for 450 nm and 900 nm nanoparticles compared to the values for 150 nm nanoparticles. The higher PVA concentration used to prepare the smaller nanoparticles may lead to an increase in PVA density at the oil–water interface of the emulsion droplet, which could increase the thickness on the particle surface.29,30 A PVA layer at the surface of the nanoparticles can potentially shield the surface charge of the matrix and drugs.31 Thus, it is reasonable to expect that less shielding, and therefore a higher ZP, would be observed from nanoparticles prepared with 0.2% PVA and 1% PVA.
The lower DL observed for the smaller nanoparticles could be due to the increased diffusion of tilmicosin into the aqueous phase during the preparation of the nanoparticle suspension or during washing of the nanoparticles prior to lyophilization. As the particle size decreases, its surface area increases, leading to an exponential increase in the surface exposure of drug molecules, which would, in turn, provide more opportunity for drug–solvent interactions to occur during preparation and washing.
The in vitro release profiles of the three nanoparticle suspensions were biphasic and quite similar in shape to that of the freeze-dried powders. More initial burst release was observed with the suspensions than with the powders. This may be due to unincorporated tilmicosin in the suspensions, as reported previously.32 In our study, smaller particle sizes resulted in faster release rates for both a suspension and a powder. The largest nanoparticles had the slowest drug release rates despite having the highest DL, demonstrating that nanoparticle size significantly impacts the release rate. This is quite understandable since smaller particle sizes lead to a larger surface area per unit mass or volume, and the increased surface area provides more opportunities for drug–solvent interactions to occur. Thus, tilmicosin on the surface of smaller nanoparticles should show an initial burst when suspended. During the slow release phase, release rates for the three nanoparticle suspensions were not significantly different, suggesting that they had similar sustained release properties. Additionally, faster release was observed in 0.1 M HCl for the suspensions and powders, which is consistent with the higher solubility of tilmicosin in acidic solutions.
In vitro antibacterial activity analyses demonstrated that suspensions with larger nanoparticles had higher MIC and MBC values. Since the MIC and MBC are a function of the drug concentration, these results are consistent with the in vitro release data. Notably, the MIC and MBC values for the suspension with the 150 nm nanoparticles were the same as those for native tilmicosin, suggesting that the nanoparticles enhanced the antibacterial activity of tilmicosin, since the drug does not completely release from the nanoparticles upon dissolution.
Time-kill analyses demonstrated that during the first 12 hours, the suspensions with the 900 nm and 450 nm nanoparticles did not slow growth as much as the 150 nm nanoparticles or the native drug. The slower drug release observed during that time period could account for this difference. As with the MIC and MBC values, the bactericidal activity was slightly higher for 150 nm nanoparticles than for native tilmicosin.
The clinical dose of tilmicosin administered by subcutaneous injection was 10–20 mg/kg. In our in vitro analyses of cytotoxicity, none of the suspensions altered BHK-21 cell proliferation at concentrations up to 32 μg/mL. At higher concentrations (64–512 μg/mL of tilmicosin), all three suspensions showed similar concentration-dependent cytotoxic activity. This could be due to the low inherent cytotoxicity of native tilmicosin since the main differences among the three suspensions are found in the drug release speed. Alternatively, the cytotoxicity at high concentrations could be due to a high lipid concentration, since the ratio of lipid to tilmicosin in the nanoparticles is 9:1. Similar results have been reported for doxorubicin- and paclitaxel-loaded SLNs, which were more cytotoxic than the drug alone.33,34
Mortality, LD50, and clinical symptoms are often used as a basis for classifying chemicals into various categories based on toxicity.35 Our previous work showed that lyophilized TMS-HCO-NP with a mean particle size of 334±44 nm was not lethal to mice at a dose of 5 g/kg BW (equivalent to 540 mg/kg BW of tilmicosin), which suggests that TMS-HCO-NP could be classified as a low-toxic substance according to the toxicity categorization standards.9 This low toxicity is most likely due to good biocompatibility and biodegradability of the HCO and PVA. HCO can be metabolized into fatty acids and glycerin in vivo,36 and PVA can be degraded into acetic acid by oxidase and hydrolase enzymes. The oral LD50 of PVA is in the range of 15–20 g/kg.37 In the current study, the mortality rate associated with TMS-HCO-NP was assessed in mice. In the present study, the absolute LD of a suspension of 150 nm nanoparticles was 550 mg/kg BW, which was comparable to the lethality observed for a control group treated with native tilmicosin suspended in 1% PVA. At an equivalent bioactive dose, suspensions of larger nanoparticles were not lethal. The drug-free suspension of 150 nm nanoparticles was also not lethal. These results suggest that the lethality of the 150 nm suspension may be due to faster drug release, thereby increasing the concentration of the drug in the animal.
The stability of the nanoparticle suspensions is normally assessed by measuring the MD, PDI, ZP, and DL capacity after storage.38,39 It has been demonstrated that the stability of some SLN suspensions decreases as the storage temperature is increased.40,41 In the present study, all of the TMS-HCO-NP suspensions were stable at both 4°C and RT for at least 6 months. It is possible that the presence of the PVA surfactant might enhance the suspension stability, as has been previously observed.42
Conclusion
Hot homogenization and ultrasonication is a feasible method for preparing tilmicosin-loaded SLN suspensions. The nanoparticle size of the suspension can be controlled by varying the surfactant concentration. Particle size significantly affects sustained release properties, antibacterial activity, and in vivo toxicity of the suspensions. Due to their simple preparation and cost savings, TMS-HCO-NP suspensions with appropriate particle sizes can be potentially used in veterinary medicine to enhance the therapeutic efficacy while minimizing side effects during the treatment of domestic animal diseases, such as bovine mastitis and porcine contagious pleuropneumonia.
Acknowledgment
This work was supported by the Fundamental Research Funds for the Central Universities.
Disclosure
The authors report no conflicts of interest in this work. The authors alone are responsible for the content and writing of the paper.
References
Ose EE. In vitro antibacterial properties of EL-870, a new semi-synthetic macrolide antibiotic. J Antibiot (Tokyo). 1987;40(2):190–194. | |
Ziv G, Shem-Tov M, Glickman A, Winkler M, Saran A. Tilmicosin antibacterial activity and pharmacokinetics in cows. J Vet Pharmacol Ther. 1995;18(5):340–345. | |
Ramadan A. Pharmacokinetics of tilmicosin in serum and milk of goats. Res Vet Sci. 1997;62(1):48–50. | |
Backstrom L, McDonald J, Collins MT, Chung WB, Shyrock TR, Ose EE. Efficacy of tilmicosin, and a combination of tylosin and sulfamethazine, for control of swine atrophic rhinitis involving infection with toxigenic Pasturella multocida type D. Swine Health and Production. 1994;2(4):11–14. | |
FAO. Residue of some veterinary drugs in animals and foods: Tilmicosin, FAO/WHO Expert Committee on Food Additives. 1996. | |
Xu Z, Wang J, Shen W, Cen P. Study on the extraction equilibrium of tilmicosin between the aqueous and butyl acetate phases. Chem Eng Commun. 2006;193(4):427–437. | |
Wang XF, Zhang SL, Zhu LY, et al. Enhancement of antibacterial activity of tilmicosin against Staphylococcus aureus by solid lipid nanoparticles in vitro and in vivo. Vet J. 2012;191(1):115–120. | |
Han C, Qi CM, Zhao BK, et al. Hydrogenated castor oil nanoparticles as carriers for the subcutaneous administration of tilmicosin: in vitro and in vivo studies. J Vet Pharmacol Ther. 2009;32(2):116–123. | |
Xie S, Wang F, Wang Y, et al. Acute toxicity of tilmicosin-loaded hydrogenated castor oil-solid lipid nanoparticles. Part Fibre Toxicol. 2011;8:33. | |
Freitas C, Müllerä RH. Spray-drying of solid lipid nanoparticles (SLN TM). Eur J Pharm Biopharm. 1998;46(2):145–151. | |
Wissing SA, Kayser O, Müller RH. Solid lipid nanoparticles for parenteral drug delivery. Adv Drug Deliv Rev. 2004;56(9):1257–1272. | |
Müller RH, Mäder K, Gohla S. Solid lipid nanoparticles (SLN) for controlled drug delivery – a review of the state of the art. Eur J Pharm Biopharm. 2000;50(1):161–177. | |
Chattopadhyay P, Shekunov BY, Yim D, Cipolla D, Boyd B, Farr S. Production of solid lipid nanoparticle suspensions using supercritical fluid extraction of emulsions (SFEE) for pulmonary delivery using the AERx system. Adv Drug Deliv Rev. 2007;59(6):444–453. | |
Morley PS, Apley MD, Besser TE, et al; American College of Veterinary Internal Medicine. Antimicrobial drug use in veterinary medicine. J Vet Intern Med. 2005;19(4):617–629. | |
Mehnert W, Mäder K. Solid lipid nanoparticles: production, characterization and applications. Adv Drug Deliv Rev. 2001;47(2–3):165–196. | |
Matschke C, Isele U, van Hoogevest P, Fahr A. Sustained-release injectables formed in situ and their potential use for veterinary products. J Control Release. 2002;85(1–3):1–15. | |
Tallury P, Payton K, Santra S. Silica-based multimodal/multifunctional nanoparticles for bioimaging and biosensing applications. Nanomedicine (Lond). 2008;3(4):579–592. | |
Smith AM, Duan H, Mohs AM, Nie S. Bioconjugated quantum dots for in vivo molecular and cellular imaging. Adv Drug Deliv Rev. 2008;60(11):1226–1240. | |
Kawabata Y, Wada K, Nakatani M, Yamada S, Onoue S. Formulation design for poorly water-soluble drugs based on biopharmaceutics classification system: basic approaches and practical applications. Int J Pharm. 2011;420(1):1–10. | |
Müller RH, Keck CM. Twenty years of drug nanocrystals: where are we, and where do we go? Eur J Pharm Biopharm. 2012;80(1):1–3. | |
Müller RH, Gohla S, Keck CM. State of the art of nanocrystals – special features, production, nanotoxicology aspects and intracellular delivery. Eur J Pharm Biopharm. 2011;78(1):1–9. | |
Merisko-Liversidge E, Liversidge GG. Nanosizing for oral and parenteral drug delivery: a perspective on formulating poorly-water soluble compounds using wet media milling technology. Adv Drug Deliv Rev. 2011;63(6):427–440. | |
Merisko-Liversidge EM, Liversidge GG. Drug nanoparticles: formulating poorly water-soluble compounds. Toxicol Pathol. 2008; 36(1):43–48. | |
Noyes AA, Whitney WR. The rate of solution of solid substances in their own solutions. J Am Chem Soc. 1897;19(12):930–934. | |
Tomazic-Jezic VJ, Merritt K, Umbreit TH. Significance of the type and the size of biomaterial particles on phagocytosis and tissue distribution. J Biomed Mater Res. 2001;55(4):523–529. | |
Clinical and Laboratory Standards Institute. Methods for Dilution Antimicrobial Susceptibility Test for Bacteria That Grow Aerobically; Approved Standards - Seventh Edition. Clinical and Laboratory Standards Institute document M7-A7 [ISBN 1-56238-587-9]. Clinical and Laboratory Standards Institute, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898 USA, 2006. | |
Well M, Naber KG, Kinzig-Schippers M, Sörgel F. Urinary bactericidal activity and pharmacokinetics of enoxacin versus norfloxacin and ciprofloxacin in healthy volunteers after a single oral dose. Int J Antimicrob Agents. 1998;10(1):31–38. | |
Zambaux MF, Bonneaux F, Gref R, et al. Influence of experimental parameters on the characteristics of poly(lactic acid) nanoparticles prepared by a double emulsion method. J Control Release. 1998;50(1–3):31–40. | |
Lee SC, Oh JT, Jang MH, Chung SI. Quantitative analysis of polyvinyl alcohol on the surface of poly(D, L-lactide-co-glycolide) microparticles prepared by solvent evaporation method: effect of particle size and PVA concentration. J Control Release. 1999;59(2):123–132. | |
Sahoo SK, Panyam J, Prabha S, Labhasetwar V. Residual polyvinyl alcohol associated with poly (D,L-lactide-co-glycolide) nanoparticles affects their physical properties and cellular uptake. J Control Release. 2002;82(1):105–114. | |
Redhead HM, Davis SS, Illum L. Drug delivery in poly(lactide-co-glycolide) nanoparticles surface modified with poloxamer 407 and poloxamine 908: in vitro characterisation and in vivo evaluation. J Control Release. 2001;70(3):353–363. | |
Misra R, Acharya S, Dilnawaz F, Sahoo SK. Sustained antibacterial activity of doxycycline-loaded poly(D,L-lactide-co-glycolide) and poly(epsilon-caprolactone) nanoparticles. Nanomedicine (Lond). 2009;4(5):519–530. | |
Miglietta A, Cavalli R, Bocca C, Gabriel L, Gasco MR. Cellular uptake and cytotoxicity of solid lipid nanospheres (SLN) incorporating doxorubicin or paclitaxel. Int J Pharm. 2000;210(1–2):61–67. | |
Serpe L, Catalano MG, Cavalli R, et al. Cytotoxicity of anticancer drugs incorporated in solid lipid nanoparticles on HT-29 colorectal cancer cell line. Eur J Pharm Biopharm. 2004;58(3):673–680. | |
Zbinden G, Flury-Roversi M. Significance of the LD50-test for the toxicological evaluation of chemical substances. Arch Toxicol. 1981;47(2):77–99. | |
Watson WC, Gordon RS. Studies on the digestion, absorption and metabolism of castor oil. Biochem Pharmacol. 1962;11:229–236. | |
DeMerlis CC, Schoneker DR. Review of the oral toxicity of polyvinyl alcohol (PVA). Food Chem Toxicol. 2003;41(3):319–326. | |
Ghaffari S, Varshosaz J, Saadat A, Atyabi F. Stability and antimicrobial effect of amikacin-loaded solid lipid nanoparticles. Int J Nanomedicine. 2011;6:35–43. | |
Xie S, Pan B, Shi B, et al. Solid lipid nanoparticle suspension enhanced the therapeutic efficacy of praziquantel against tapeworm. Int J Nanomedicine. 2011;6:2367–2374. | |
Paliwal R, Rai S, Vaidya B, et al. Effect of lipid core material on characteristics of solid lipid nanoparticles designed for oral lymphatic delivery. Nanomedicine. 2009;5(2):184–191. | |
Shegokar R, Singh KK, Müller RH. Production and stability of stavudine solid lipid nanoparticles – from lab to industrial scale. Int J Pharm. 2011;416(2):461–470. | |
Venkateswarlu V, Manjunath K. Preparation, characterization and in vitro release kinetics of clozapine solid lipid nanoparticles. J Control Release. 2004;95(3):627–638. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.