Back to Journals » International Medical Case Reports Journal » Volume 19
Prenatal Diagnosis and Genotype-Phenotype Correlation in 8q21.11 Microdeletion Syndrome: A Case Report
Authors Libotte F, Margiotti K ![]() , Fabiani M, Mesoraca A, Giorlandino C
, Fabiani M, Mesoraca A, Giorlandino C
Received 18 July 2025
Accepted for publication 24 December 2025
Published 17 February 2026 Volume 2026:19 542044
DOI https://doi.org/10.2147/IMCRJ.S542044
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Thomas E Hutson
Francesco Libotte,* Katia Margiotti,* Marco Fabiani, Alvaro Mesoraca, Claudio Giorlandino
Human Genetics Lab, Altamedica Main Centre, Rome, 00198, Italy
*These authors contributed equally to this work
Correspondence: Katia Margiotti, Human Genetics Lab, Altamedica Main Centre, Viale Liegi 45, Rome, 00198, Italy, Tel +39 06 8505805, Fax +39 068505815, Email [email protected]
Abstract: 8q21.11 microdeletion syndrome is a rare chromosomal disorder characterized by a highly variable phenotype, including mild to moderate intellectual disability, distinctive facial dysmorphisms, and congenital anomalies such as ocular defects, cardiac malformations, and limb abnormalities. The deletion size in 8q21.11 microdeletion syndrome ranges from 0.12 to 13.15 Mb, with a critical small region of overlap (SRO) of 539.77 Kb. The ZFHX4 gene in this region is implicated in neurodevelopmental disorders and ocular anomalies. Other genes, including PEX2 and PMP2, contribute to the complex clinical presentation by affecting metabolic and immune functions. Here, we present a prenatal diagnosis of 8q21.11 microdeletion syndrome in a fetus with increased nuchal translucency detected via ultrasound. This case underscores the importance of high-resolution genomic testing and genetic counseling in the management of 8q21.11 microdeletion syndrome, providing valuable insights into prenatal assessment of this rare condition.
Keywords: 8q21.11 microdeletion syndrome, prenatal diagnosis, nuchal translucency, ZFHX4
Introduction
8q21.11 microdeletion syndrome is a rare chromosomal disorder resulting from a heterozygous deletion in the 8q21.11 region. First described by Palomares et al,1 the syndrome is characterized by a highly variable phenotype that includes mild to moderate intellectual disability, distinctive facial dysmorphisms, and a range of congenital anomalies such as ocular defects, cardiac malformations, and limb abnormalities.1,2 The phenotypic variability and lack of specific prenatal markers make early diagnosis challenging, often leading to postnatal confirmation.2 However, the advent of high-resolution genomic technologies like array comparative genomic hybridization (aCGH) has enhanced the detection of submicroscopic deletions, including those in the 8q21.11 region, during prenatal assessments. This advancement allows for more accurate diagnoses and informed genetic counseling.2 The size of the deletion in 8q21.11 microdeletion syndrome can range from approximately 0.12 to 13.15 Mb and may encompass multiple genes. Using a high-density targeted oligonucleotide array, Palomares et al1 defined a small region of overlap (SRO) of 539.77 Kb. Interestingly, there is no clear correlation between the size of the deletion and the severity of the phenotype, which complicates genotype-phenotype interpretations.2 This underscores the importance of analysing the specific gene content within each deletion to better understand the clinical implications. One critical gene located within the SRO is ZFHX4 (zinc finger homeobox 4) (OMIM#606940). ZFHX4 encodes a transcription factor with four homeodomains and 22 zinc finger motifs, playing a significant role in regulating gene expression during neuronal and mesenchymal cell differentiation.3 Haploinsufficiency of ZFHX4 has been associated with congenital ocular abnormalities—such as ptosis, sclerocornea, and microphthalmia—as well as neurodevelopmental delays.4 Recently, a crucial role for ZFHX4 in the neurodevelopmental disorders (NDDs) was observed among 42 cases, reinforcing its relevance in 8q21.11 deletion syndromes.5 Other genes within the deleted region, including PEX2, PMP2, and IL7, are implicated in metabolic processes and immune function, contributing to the complexity of the clinical presentation.2 Previous reports have highlighted the broad spectrum of anomalies associated with deletions in the 8q21.11 region. For example, Donahue et al6 reported a prenatal case involving severe congenital heart defects linked to an 8q21.11 deletion, emphasizing the potential for serious cardiovascular involvement. Additionally, studies have suggested that deletions encompassing the HEY1 gene may contribute to developmental anomalies, although its exact role remains less defined compared to ZFHX4.7 HEY1 is also located within the smallest region of overlap (SRO) described for 8q21.11 microdeletion syndrome, suggesting that both genes may act synergistically in determining the neurodevelopmental phenotype. In this case report, we present a prenatal diagnosis of 8q21.11 microdeletion syndrome in a fetus identified through increased nuchal translucency on ultrasound examination. The deletion was confirmed via G-banding karyotype and aCGH analyses. We discuss the phenotypic implications in the context of existing literature, aiming to enhance the understanding of this rare syndrome and to underscore the importance of comprehensive genetic counselling in similar cases.
Materials and Methods
Chorionic villus sampling (CVS) was performed at 11 weeks and 5 days of gestation due to abnormal ultrasound findings, specifically an increased nuchal translucency (NT) measurement of 5 mm. Villus cells were cultured according to standard protocols for karyotyping. Chromosomal analysis was conducted using G-banding techniques at a resolution of 400 bands per haploid set, following the International System for Human Cytogenomic Nomenclature (ISCN 2020) guidelines. To determine whether the chromosomal abnormality was inherited, peripheral blood samples from both parents were collected and analysed using the same cytogenetic methods.8 Genomic DNA was extracted from uncultured chorionic villi using the QIAamp DNA Mini Kit (Qiagen), following the manufacturer’s instructions. Array comparative genomic hybridization (aCGH) was performed using the Agilent CytoSure ISCA v2 Microarray 4x44K platform, which offers high-resolution genome-wide coverage. DNA labeling, hybridization, and scanning were carried out according to the Agilent Technologies protocol with genomic coordinates aligned to the GRCh37/hg19 genome build. Decipher platform was used (https://www.deciphergenomics.org/) to interpret genomic variations.
Case Report
A 25-year-old gravida 3, para 0 woman was referred to our prenatal diagnostic center at 11 weeks and 5 days of gestation due to an abnormal ultrasound finding of increased nuchal translucency measuring 5 mm (Figure 1). Both the woman and her husband were healthy, non-consanguineous, and had no significant family history of genetic disorders or congenital malformations. The patient had previously experienced two spontaneous abortions at 16 and 18 weeks of gestation, attributed to placental abruption and toxoplasmosis infection, respectively. Given the ultrasound findings, chorionic villus sampling (CVS) was performed to investigate potential chromosomal anomalies. Cytogenetic analysis using G-banding at a resolution of 400 bands revealed an abnormal karyotype in the fetus: 46,XY,del(8)(q21.11q21.13). The deletion was observed in all 20 metaphases examined. Both parents had normal karyotypes, confirming de novo origin of deletion (Figure 2). To further characterize the chromosomal aberration, array comparative genomic hybridization (aCGH) was conducted. The aCGH analysis identified a 5.07 Mb deletion on chromosome 8q21.11–q21.13, spanning genomic coordinates chr8:77,491,659–82,561,762 (GRCh37/hg19) (Figure 2). There are 12 OMIM genes within the deleted region, of which 5 are classified as disease-causing according to the DECIPHER database, ZFHX4, PMP2, PEX2, IL7, and MRPS28 gene.9 The alignment of our case with all previously described overlapping deletions reported ZFHX4 gene as common denominator2 (Figure 3). The overlap region encompasses the ZFHX4 gene, resides within the previously established SRO associated with 8q21.11 microdeletion syndrome.1 Subsequent ultrasound examinations up to 14 weeks of gestation did not reveal any additional structural abnormalities beyond the increased nuchal translucency. There were no detectable cardiac defects, limb anomalies, or other organ malformations at this gestation stage. Comprehensive genetic counselling was provided to the couple, detailing the potential phenotypic outcomes associated with the 8q21.11 microdeletion syndrome. Discussions included the risks of intellectual disability, developmental delays, congenital ocular anomalies, and the variability in clinical expression due to incomplete penetrance and variable expressivity.2,5,6 The uncertainty in predicting the exact phenotype was emphasized, given the limited number of reported cases and the broad spectrum of possible manifestations. After careful consideration, the couple elected to terminate the pregnancy at 14 weeks of gestation, rising concerns over the high risk of severe developmental issues and the potential impact on quality of life. The termination was performed in accordance with institutional protocols and ethical guidelines. An autopsy was not conducted.
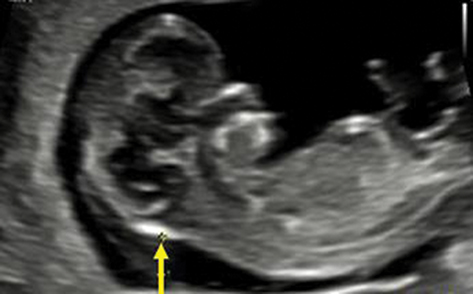 |
Figure 1 The yellow arrow indicates the presence of increased nuchal translucency. |
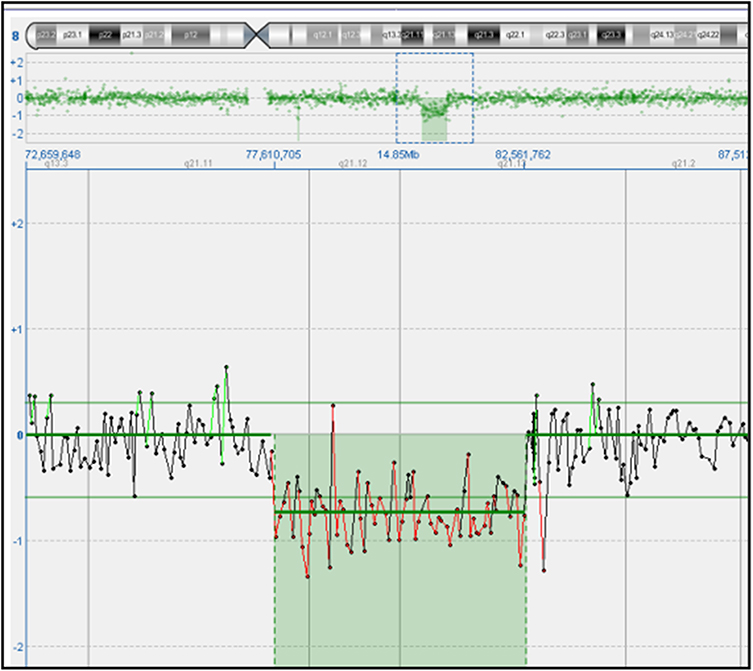 |
Figure 2 Array comparative genomic hybridization (aCGH) result of the fetus showing a 5.07 Mb deletion at 8q21.11–q21.13 (chr8:77,491,659–82,561,762, GRCh37/hg19) encompassing 12 OMIM genes. |
 |
Figure 3 Schematic representation of the 8q21.11 microdeletion region in the fetus, overlapping pathogenic or likely pathogenic deletions (red bars) reported in the DECIPHER database. The shared overlap involving the ZFHX4 gene (light blue vertical bar) is highlighted. |
Discussion and Conclusion
The prenatal diagnosis of an 8q21.11 microdeletion syndrome in this case highlights the complexities associated with interpreting chromosomal deletions that encompass multiple genes with variable clinical significance. The initial finding of increased NT served as a non-specific marker that warranted further genetic investigation. Although increased NT is associated with chromosomal abnormalities and genetic syndromes, it lacks specificity, necessitating advanced diagnostic tools for precise identification.10 Nonetheless, reporting cases in which increased NT is the only prenatal finding remains clinically relevant, as this feature can represent the earliest detectable sign of a submicroscopic chromosomal imbalance. In this context, the identification of a 5.07 Mb de novo deletion in the 8q21.11–q21.13 region via aCGH underscores the utility of high-resolution genomic technologies in prenatal diagnostics. ZFHX4 emerged as the most critical gene within the deleted region due to its established role in neurodevelopmental processes and ocular development. Haploinsufficiency of ZFHX4 has been consistently associated with intellectual disability and congenital ocular anomalies such as ptosis and microphthalmia.4,5 Therefore, its deletion is likely to contribute significantly to the neurodevelopmental risks associated with 8q21.11 microdeletion syndrome. However, while ZFHX4 is a plausible contributor to the phenotype, the large size of the deleted region encompassing multiple OMIM genes (including PEX2, PMP2, HEY1, and IL7) suggests that a multigenic mechanism may underlie the observed features. This broader interpretation better reflects the complexity of 8q21.11 microdeletion syndrome and aligns with the variability reported in previous studies. This highlights the necessity of detailed genetic counselling in prenatal settings where SRO-associated genes like ZFHX4 are implicated, as they carry significant prognostic implications for neurodevelopment and morphology. The deletion also encompassed other genes such as PEX2, PMP2, and HEY1, which may contribute to the phenotypic spectrum. PEX2 is involved in peroxisome biogenesis, and mutations can lead to peroxisomal disorders affecting metabolic functions.11 PMP2 plays a role in myelin sheath formation in peripheral nerves, and its alteration could potentially impact neural conductivity.12 HEY1 is a transcriptional repressor involved in neural and cardiac development, although its exact clinical significance in humans remains less defined.2 Given the size and gene content of the deleted segment, it is plausible that the observed phenotype results from a combined haploinsufficiency of multiple genes rather than the loss of a single driver gene. Moreover, epigenetic mechanisms or the influence of modifier genes could modulate the expressivity of the deletion, contributing to the broad variability described across reported cases. This multigenic and multifactorial interpretation may better explain the non-specific features observed in the present case. Previous studies have reported significant phenotypic variability associated with 8q21.11 microdeletions. For instance, Ben Ayed et al described,2 patients with intellectual disability, facial dysmorphisms, and limb anomalies, but with varying degrees of severity. Donahue et al reported,6, a prenatal case with severe congenital heart defects, emphasizing the potential for critical cardiovascular involvement. The absence of additional clinical signs besides the increased NT in our case highlights the unpredictability of phenotypic expression and the challenges in prenatal counselling. Moreover, it is important to report cases like ours, where there is no clear genotype-phenotype correlation. In fact, as seen in our case at 18 weeks of gestation, this rare microdeletion syndrome only showed an increase in NT, whereas other cases with similar microdeletions have presented more definitive clinical signs, such as cardiac defects or intellectual disability.1,2 The limited number of studies on this microdeletion necessitates reporting even cases like ours, which may not be completely clear or well defined, to promote and stimulate scientific debate. Given the scarcity of well documented cases in the literature, it is essential to bring such cases to the scientific community’s attention. This helps increase awareness of a microdeletion that is still not well understood, fostering further research and a deeper understanding of its genetic and clinical implications. A limitation of the current case is the lack of an autopsy, which restricts comprehensive phenotypic correlation and represents an important consideration for clinical interpretation. Future studies should aim to integrate detailed postmortem findings where available to strengthen genotype-phenotype correlations. The decision to terminate the pregnancy was made after comprehensive counselling and reflects the parents’ weighing of potential quality-of-life issues for the child and family. This emphasizes the need for sensitive and supportive counselling that respects the values and decisions of the family.
In conclusion, high-resolution genomic analysis remains vital in prenatal diagnostics of syndromic microdeletions, and detailed genetic counselling must accompany prenatal diagnosis to adequately prepare families for variable clinical outcomes.
Institutional Review Board Statement
The study and the publication of the case details were approved by the Ethics Committee of Artemisia SPA (Rome, Italy) on 21/08/2024 (reference number: 2024_57399).
Informed Consent Statement
Written informed consent has been obtained from the patient to publish this paper.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research received no external funding.
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Palomares M, Delicado A, Mansilla E, et al. 8q21.11-q21.3 submicroscopic deletions: evidence for a recognizable phenotype including mild intellectual disability, facial dysmorphisms, and limb anomalies. Am J Hum Genet. 2011;89(2):228–6. doi:10.1016/j.ajhg.2011.06.012
2. Ben Ayed I, Bouzid A, Kammoun F, et al. 8q21.11 microdeletion syndrome: Delineation of HEY1 as a candidate gene in neurodevelopmental and cardiac defects. Mol Genetics Genomic Med. 2021;9(11):e1811. doi:10.1002/mgg3.1811
3. Shannon M, Hamilton AT, Gordon L, Branscomb E, Stubbs L Differential expansion of zinc-finger transcription factor loci in homologous human and mouse gene clusters. Genome Res. 2003;13(6A). doi:10.1101/gr.963903
4. Hofmann K, et al. ZFHX4 mutations are associated with congenital ptosis, microphthalmia, and mental retardation. Human Mutation. 2011;32(10):1045–1049. doi:10.1002/humu.21541
5. Del Rocio P. B. M. Loss-of-function of the Zinc Finger Homeobox 4 (ZFHX4) gene underlies a neurodevelopmental disorder. Am J Hum Genet. 2025;112(6):1388–1414. doi:10.1016/j.ajhg.2025.04.008
6. Donahue ML, Ryan RM. Interstitial deletion of 8q21—>22 associated with minor anomalies, congenital heart defect, and Dandy-Walker variant. Am. J. Med. Genet. 1995;56:97–100. doi:10.1002/ajmg.1320560122
7. Kuroda Y, Ohashi I, Saito T, et al. Refinement of the deletion in 8q22.2-q22.3: the minimum deletion size at 8q22.3 related to intellectual disability and epilepsy. Am J Med Genet A. 2014;164A(8):2104–8. doi:10.1002/ajmg.a.36604
8. Libotte F, Fabiani M, Margiotti K, et al. De novo 3q13.13q21.2 interstitial deletion and paternal 12p13.3 microdeletion in a fetus with dysplasia of the corpus callosum and ventriculomegaly: A case report. Exp Ther Med. 2023;25(2):100. doi:10.3892/etm.2023.11799
9. Firth HV, Richards SM, Bevan AP, et al. DECIPHER (Database of Chromosomal Imbalance and Phenotype in Humans using Ensembl Resources). AmJ Hum Genet. 2009;84:524–533. doi:10/1016/j.ajhg.2009.03.010
10. Braverman NE, Raymond GV, Rizzo WB, et al. Peroxisome biogenesis disorders in the Zellweger spectrum: An overview of current diagnosis, clinical manifestations, and treatment guidelines. Mol Genet Metab. 2016;117(3):313–21. doi:10.1016/j.ymgme.2015.12.009
11. Nicolaides K. H. Increased fetal nuchal translucency at 11–14 weeks. Prenatal Diagnosis. 2002;22(4):308–315. doi:10.1002/pd.308
12. Uusitalo M, Klenow MB, Laulumaa S, et al. Human myelin protein P2: from crystallography to time-lapse membrane imaging and neuropathy-associated variants. FEBS J. 2021;288(23):6716–6735. doi:10.1111/febs.16079
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.