Back to Journals » Drug Design, Development and Therapy » Volume 20
Preliminary Exploration of Fluvastatin Inhibiting Proliferation, Migration and Invasion of Lung Cancer Cells and Reversing Paclitaxel Resistance: Mechanism Exploration Based on Multi-Omics Analysis
Authors Xu H, Du Z, Shui J, Li X, Tang J, Li G
Received 20 November 2025
Accepted for publication 27 January 2026
Published 16 February 2026 Volume 2026:20 579427
DOI https://doi.org/10.2147/DDDT.S579427
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Hongyu Xu,1 Zedong Du,2 Jia Shui,1 Xueting Li,1 Juan Tang,1 Guangquan Li3
1Department of Oncology, 363 Hospital, Chengdu, Sichuan, 610041, People’s Republic of China; 2Department of Oncology, Sichuan Second Hospital of T.C.M, Chengdu, Sichuan, People’s Republic of China; 3Clinical Laboratory, 363 Hospital, Chengdu, Sichuan, 610041, People’s Republic of China
Correspondence: Guangquan Li, Clinical Laboratory, 363 Hospital, No. 108, Daosangshu Street, Wuhou District, Chengdu, Sichuan, 610041, People’s Republic of China, Tel +86-18980798528, Email [email protected]
Background: Lung cancer is one of the leading causes of cancer-related deaths, among which NSCLC accounts for approximately 80– 85% of all lung cancer cases. Paclitaxel (TAX) is a commonly used chemotherapeutic drug, but it is easy to cause drug resistance. Fluvastatin has anti-cancer potential, but the mechanism of its reversal of drug resistance is unclear.
Methods: The study was divided into four groups: the A549 control group, the A549/Tax control group, the A549 Fluvastatin-treated group, and the A549/Tax Fluvastatin-treated group. CCK-8, Transwell, and flow cytometry assays were used to detect fluvastatin’s effects on cell proliferation, migration, invasion, and apoptosis. Transcriptomics, proteomics, and acetylomics were combined to explore the potential molecular mechanisms.
Results: The preliminary results showed that fluvastatin inhibited the proliferation, migration and invasion of A549 and A549/Tax cells and promoted their apoptosis. Multi-omics analysis revealed that a large number of differentially expressed molecules were detected in both the A549-Fluvastatin vs A549-NC group and the A549/Tax-Fluvastatin vs A549/Tax-NC group, and these molecules were significantly enriched in multiple biological processes and signaling pathways. This suggests that fluvastatin may exert its effects through the synergistic regulation of multiple molecules and pathways. Integrated multi-omics analysis identified several key molecules (for example, HMGCR, RDH11, HSPB1) and acetylated protein-target gene pairs (for example, P09874-BCL2, P42224-PTGS2, P04150-CCND3), which may mediate the antitumor mechanism of fluvastatin.
Conclusion: This study indicates that fluvastatin has the potential to reverse TAX resistance in lung cancer, and the results of multi-omics analysis provide a theoretical basis for the exploration of potential therapeutic targets in the future.
Keywords: lung cancer, paclitaxel, fluvastatin, resistance, multi-omics
Introduction
Globally, the number of lung cancer cases and deaths is rising, which is the leading cause of cancer death.1 Lung cancer is classified into small cell lung cancer and non-small cell lung cancer (NSCLC), and approximately 80–85% of patients are diagnosed with NSCLC via pathological examination.2–4 The treatment strategies for lung cancer include surgical, chemotherapy, radiotherapy, immunotherapy, targeted therapy or combination therapy, depending on the type and stage of the disease.5,6 To date, chemotherapy is the mainstream treatment of the disease.7 However, treatment resistance frequently results in therapeutic failure, thereby imposing limitations on the application of chemotherapy. As a commonly known chemotherapy drug, paclitaxel (TAX) is utilized for treating a variety of cancers, including lung cancer.2 Long-term use of TAX in treatment can lead to the development of drug resistance.8 Therefore, continuous exploration of effective strategies to alleviate or even overcome TAX resistance is of crucial significance for the clinical treatment of lung cancer.
As one of the HMG-CoA reductase inhibitors, fluvastatin is widely applied in the treatment of dyslipidemia and coronary artery disease, and additionally has anticancer activity.9 Research has shown that fluvastatin can down-regulate SATB1 via the Wnt/β-Catenin pathway, significantly inhibiting the in vitro tumor progression of H292 cells.10 Fluvastatin can also prevent bone metastasis of lung adenocarcinoma by inducing autophagy in cancer cells.11 Moreover, fluvastatin can also be involved in the treatment of ferroptosis in lung adenocarcinoma.12 Additionally, studies have also shown that fluvastatin can alleviate EGFR TKI resistance and cisplatin resistance in NSCLC,13,14 suggesting its broad potential in overcoming cancer drug resistance. Fluvastatin can also enhance the antitumor activity of TAX in bladder cancer cells.15 However, to date, the therapeutic mechanism of fluvastatin in lung cancer has not been fully elucidated, and its role in lung cancer drug resistance, especially in TAX resistance, remains to be further explored.
Recent advances in omics technologies have enabled large-scale molecular analysis of biological systems. However, studies focusing solely on a single omics type fail to clearly reveal the dependencies between biological features and the relationships across different molecular layers.16,17 The information spanning multiple omics layers can provide richer evidence for biological mechanisms at different molecular layers, helping to gain a deeper understanding of the complex mechanisms underlying biological processes and complex phenotypes. Transcriptome reflects the dynamic changes of gene expression at the transcriptional level.18 Proteomics is employed to detect significantly altered proteins that represent the contents of cells, tissues, organisms, or biofluids. Presently, LC-MS is a widely utilized method in proteomic investigations.19 Acetylome targets protein acetylation, a conserved and widespread post-translational modification that plays a crucial role in regulating protein function and various cellular processes.20 One study reported that TAX-resistant H460 lung cancer cells, generated by a stepwise increase in TAX, exhibited markedly increased tubulin acetylation and consequently acquired TAX resistance.21 Moreover, a renal cancer study has shown that fluvastatin affects histone acetylation.9 These evidences highlight the importance of studying acetylation in cancer biology and therapy.
In this study, A549 cells and A549/Tax cells were used as research objects, and four groups were set up: A549 control group (A549-NC), A549/Tax control group (A549/Tax-NC), A549 fluvastatin-treated group (A549-Fluvastatin), and A549/Tax fluvastatin-treated group (A549/Tax-Fluvastatin). First, the CCK-8 assay, Transwell assay, and flow cytometry were used to detect the effects of fluvastatin on the proliferation, migration, invasion, and apoptosis of the two cell lines. Then, the potential molecular mechanism of fluvastatin in regulating non-resistant and TAX-resistant A549 cells was explored by combining transcriptomics, proteomics and acetylomics sequencing analysis. This study aims to provide a new theoretical basis and potential targets for the clinical reversal of TAX resistance in lung cancer.
Materials and Methods
Cell Source and Grouping
A549 cells (Cat. No.: CL-0016) and A549/Tax cells (Cat. No.: CL-0585) were purchased from Pricella Biotechnology Co., Ltd. A549 and A549/Tax cells were cultured in an incubator at 37°C with 5% CO2 and 95% relative humidity, using cell-specific culture medium for subculture. Preliminary pre-experiments showed that the half-maximal inhibitory concentration (IC50) of fluvastatin on A549 cells at 72 h was 62.41 μM, while its IC50 on A549/Tax cells at 72 h was 136 μM. Therefore, concentrations of 62.41 μM and 136 μM were selected in this study to treat A549 cells and A549/Tax cells, respectively, with a treatment duration of 72 hours. The study was divided into four groups: the A549-NC group, the A549/Tax-NC group, the A549-Fluvastatin group, and the A549/Tax-Fluvastatin group. All cell experiments were performed with three independent biological replicates.
CCK-8 Assay
The CCK-8 assay was used to analyze the effect of fluvastatin on cell proliferation. The cell suspension (concentration was 1.5*104 cells/100μL) was prepared, inoculated in the 96-well plate (100 μL per well) and cultured in 5% carbon dioxide incubator at 37°C. The corresponding concentration of fluvastatin was added to each well and cultured in 5% carbon dioxide incubator at 37°C for 24 h, 48 h and 72 h, respectively. Add 10 μL of CCK-8 (APEXBIO, K1018-5mL) solution to 96-well cell culture plate and incubate for 4h, and measure the absorbance at 450 nm with a microplate reader.
Detection of Invasion and Migration by Transwell
Matrigel was taken from the −20°C refrigerator, and the required Transwell chambers were placed in new 24-well plate, allowing them to equilibrate to room temperature in a sterile laminar flow hood (invasion assay). Subsequently, 500 μL of serum-free medium was added to both the upper and lower chambers, followed by incubation at 37°C for 2 h to rehydrate the Matrigel matrix layer (invasion assay). Meanwhile, for the migration assay, Transwell chambers were directly placed in new 24-well plate, with 100 μL of serum-free medium added to the upper chambers and incubated at 37°C for 1 h. Both assays required the preparation of serum-free cell suspensions, which were counted (typically 1×105 cells per well in 24-well plates). After the matrix layer rehydration was complete, the chambers were transferred to new plates (only for the invasion assay). The medium in the upper chambers was discarded, and 200 μL of cell suspension was added to the upper chambers for the invasion assay, while 100 μL was added for the migration assay. Then, 500 μL of medium containing 10% FBS was added to the lower chambers in both cases, followed by incubation at 37°C. Subsequently, the chambers were inverted on absorbent paper to remove medium, and non-migrated cells in the upper chambers were gently wiped off with cotton swabs, followed by 3 washes with PBS. The chambers were fixed with 4% paraformaldehyde for 30 min, then inverted on a 24-well plate lid and air-dried on the lower surface in a fume hood for approximately 30min. Another 24-well plate (without chambers) was prepared with 500 μL of 0.1% crystal violet, and the chambers were placed in it to immerse the membranes for staining at 37°C for 30 min. After 3 washes with PBS, images were captured under a microscope: 3 random 100× magnification fields were photographed per chamber.
Detection of Cell Apoptosis by Flow Cytometry
The cells were digested with trypsin free of EDTA, then cell culture solution was added and cells were gently dispersed. The cell suspension was transferred to a centrifuge tube and centrifuged at 1000 rpm for 5 min. The supernatant was carefully aspirated, leaving approximately 50 μL medium to avoid cell loss, followed by resuspension in 1 mL pre-cooled PBS and another centrifugation to precipitate cells. Subsequently, binding buffer was diluted 1:9 with deionized water (2 mL 10× Binding Buffer + 18 mL deionized water), and cells were resuspended in binding buffer to a concentration of 1–5×106 cells/mL. Next, 100 μL of the cell suspension was transferred to a 5 mL flow cytometry tube, mixed with 5 μL Annexin V/FITC, and incubated at room temperature in the dark for 5 min. Finally, 5 μL propidium iodide (PI) solution and 400 μL PBS were added, and flow cytometry analysis was performed immediately.
mRNA Library Construction, Sequencing and Raw Data Processing
Total RNA was extracted from cell samples using the Trizol kit, and mRNA was obtained from the total RNA using oligodT. Then, mRNA fragmentation, cDNA synthesis, terminal repair and connector linking were performed. PCR reaction and product recovery were performed to complete the preparation of the library. The DNBSEQ platform was used to sequence cDNA library. Fastp (v0.23.42) software was used for quality control of raw sequencing data to obtain high-quality clean data. Hisat2 (version 2.2.1) was used to align the clean data with the human reference genome (reference genome from the Ensemble database, genome version GRCh38). The samtools (v1.19) was used to convert the sam file into bam file, and StringTie (v2.2.1) was used to quantitatively analyze the RNA-seq data.
Protein Extraction and Enrichment of Acetylated Peptides
Samples were processed using a proteomic sample preparation kit (Cat. No.: PF-226089). Specifically, 300 μL of lysis buffer was added to the samples, which were then disrupted by sonication using a cell ultrasonic disruptor. After incubation on ice for 30 min, the samples were centrifuged at 13,000 rpm for 15 min at 4°C. The supernatant was carefully aspirated, aliquoted, and stored at −80°C. Protein concentration was determined using a BCA kit, and absorbance at 562 nm was measured with a microplate reader. Subsequently, enzymatic digestion was performed, and the resulting peptides were desalted according to the instructions for C18 ZipTips (for proteomic analysis) and HLB (for acetylome analysis), followed by vacuum drying. The PTMScan® Acetyl-Lysine Motif [Ac-K] Kit (Cell Signaling technology, 13416) was used to enrich acetylated peptides from desalted peptide samples according to the manufacturer’s instructions.
Sequencing and Analysis of Proteomics and Acetylation Modification
For LC-MS/MS analysis of acetylation and proteome, peptide fractions were first redissolved in 8 μL and 10 μL of 0.1% formic acid (FA) solution, respectively. Subsequently, separation was performed using a U3000 high-performance liquid chromatography (HPLC) system coupled with a nanoViper C18 column (75 μm × 250 mm, 2 μm), with mobile phase A being 0.1% FA in water and mobile phase B being 80% acetonitrile containing 0.1% FA. The gradient setting was as follows: 0–5 min (A: 100%→99%, B: 0→1%, flow rate: 5→1.2 μL/min), 5–23 min (A: 99%→85%, B: 1%→15%, flow rate: 1.2→0.6 μL/min), 23–55.30 min (A: 85%→43%, B: 15%→57%, flow rate: 0.6 μL/min), 55.30–59 min (A: 43%→10%, B: 57%→90%, flow rate: 0.6→1.2 μL/min), 59–66 min (A: 10%→60%→0%, B: 90%→40%→0%, flow rate: 1.2→0 μL/min). Then, both were analyzed by an Orbitrap Exploris 480 mass spectrometer in high-sensitivity mode. The instrument parameters were set as follows: each full scan was a high-speed signal-dependent scan with a duration of 66 min; the first-level full-scan resolution was 120,000, the scan range was 350–1200 m/z, the automatic gain control (AGC) target was 3e6, and the maximum injection time was 50 ms; the second-level scan resolution was 30,000, collision energies were 25%, 30%, and 35%, the AGC target was 2e5, the maximum injection time was 50 ms, and 80 data-independent acquisition (DIA) scan windows were set. The obtained mass spectrometry data were searched using Spectronaut software (version 17.2), and the database used was uniprotkb_proteome_UP000005640_SP_human_2024_04_18.fasta.
Identification of Differentially Expressed Genes (DEGs), Differentially Expressed Proteins (DEPs) and Differentially Acetylated Sites (DASs)
Differential expression analysis of genes between the two groups was performed using DESeq2. The screening criteria for DEGs were P < 0.05 and |log2fold change| > 1. T-test was performed on the protein data of the two groups, and DEPs and DASs were screened according to P < 0.05 and |log2Ratio| > 1. Motif analysis was carried out on the relatively highly abundant acetylation sites in cells using the Position weight matrix (PWM) scores algorithm. The DAVID database (https://david.ncifcrf.gov/tools.jsp) was used to perform Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) functional enrichment analyses on DEGs, DEPs, and differentially acetylated modified proteins to explore their involved molecular mechanisms. P < 0.05 was set as the cut-off value.
Integrated Analysis of Transcriptome, Proteome, and Acetylome
Molecules with consistent up/down-regulated in transcriptome and proteome were obtained via intersection analysis. Meanwhile, proteins with consistent up/down-regulated in proteome and acetylated sites were obtained via intersection analysis. In addition, target genes of differentially acetylated modified proteins were predicted using the TRRUST database (https://www.grnpedia.org/trrust/). Subsequently, these target genes were intersected with DEGs to obtain differential target genes. The three groups of obtained molecules were integrated to screen key potential molecular mechanisms by which fluvastatin participates in drug resistance therapy through the combination of transcriptome, proteome, and acetylome.
Results
Effects of Fluvastatin on Proliferation, Migration, Invasion and Apoptosis of A549 and A549/Tax Cells
Compared with the A549-NC group, the cell proliferation rate of the A549-Fluvastatin group was reduced, the cell migration and invasion ability was down-regulated, and the apoptosis rate was increased (Figure 1). In addition, compared with the A549/Tax-NC group, the cells in the A549/Tax-Fluvastatin group also showed proliferation inhibition, decreased migration and invasion, and increased apoptosis (Figure 1). The preliminary results indicate that fluvastatin not only has an inhibitory effect on conventional lung cancer cells, but also can effectively reverse the drug resistance phenotype of Tax-resistant cells, suggesting that it may have potential application value in clinical lung cancer treatment.
 |
Figure 1 Effects of fluvastatin on proliferation, migration, invasion and apoptosis of A549 and A549/Tax cells. (A) Effects of fluvastatin on proliferation of A549 and A549/Tax cells were detected by CCK-8 assay; (B) Effects of fluvastatin on migration and invasion of A549 cells were detected by Transwell assay; (C) Effects of fluvastatin on migration and invasion of A549/Tax cells were detected by Transwell assay; (D) Effects of fluvastatin on apoptosis of A549 and A549/Tax cells were detected by flow cytometry. |
Identification and Functional Annotation of DEGs
Compared with the A549-NC group, a total of 1798 DEGs were identified in the A549-Fluvastatin group, among which 829 DEGs were up-regulated and 969 DEGs were down-regulated (Figure 2A and B). In GO-BP term, DEGs were mainly involved in cholesterol biosynthetic process, cell adhesion, signal transduction, inflammatory response, and cell-cell adhesion (Figure 2C). In GO-CC term, DEGs were mainly distributed in plasma membrane, extracellular space, collagen-containing extracellular matrix, extracellular region, and apical plasma membrane (Figure 2D). In GO-MF term, DEGs were mainly associated with extracellular matrix structural constituent, integrin binding, oxidoreductase activity, calcium ion binding, and signaling receptor binding (Figure 2E). KEGG analysis showed that DEGs were significantly enriched in steroid biosynthesis, cytoskeleton in muscle cells, complement and coagulation cascades, retinol metabolism, and steroid hormone biosynthesis (Figure 2F).
 |
Figure 2 Identification and functional annotation of DEGs in A549-Fluvastatin vs A549-NC group. (A) Volcanic map of DEGs in A549-Fluvastatin vs A549-NC group; (B) Heat map of DEGs in A549-Fluvastatin vs A549-NC group; (C) The BP term of GO functional annotation of DEGs in A549-Fluvastatin vs A549-NC group; (D) The CC term of GO functional annotation of DEGs in A549-Fluvastatin vs A549-NC group; (E) The MF term of GO functional annotation of DEGs in A549-Fluvastatin vs A549-NC group; (F) KEGG functional annotation of DEGs in A549-Fluvastatin vs A549-NC group. |
Compared with the A549/Tax-NC group, a total of 2089 DEGs were identified in the A549/Tax-Fluvastatin, among which 933 DEGs were up-regulated and 718 DEGs were down-regulated (Figure 3A and B). Subsequently, functional annotations were performed on 2089 DEGs. GO analysis revealed that the enrichment results of DEGs in the A549/Tax-Fluvastatin vs A549/Tax-NC group largely overlapped with those of the A549-Fluvastatin vs A549-NC group (Figure 3C–E). KEGG functional enrichment analysis indicated that the DEGs in the A549/Tax-Fluvastatin vs A549/Tax-NC group were specifically enriched in the PPAR signaling pathway and the cytokine-cytokine receptor interaction pathway (Figure 3F).
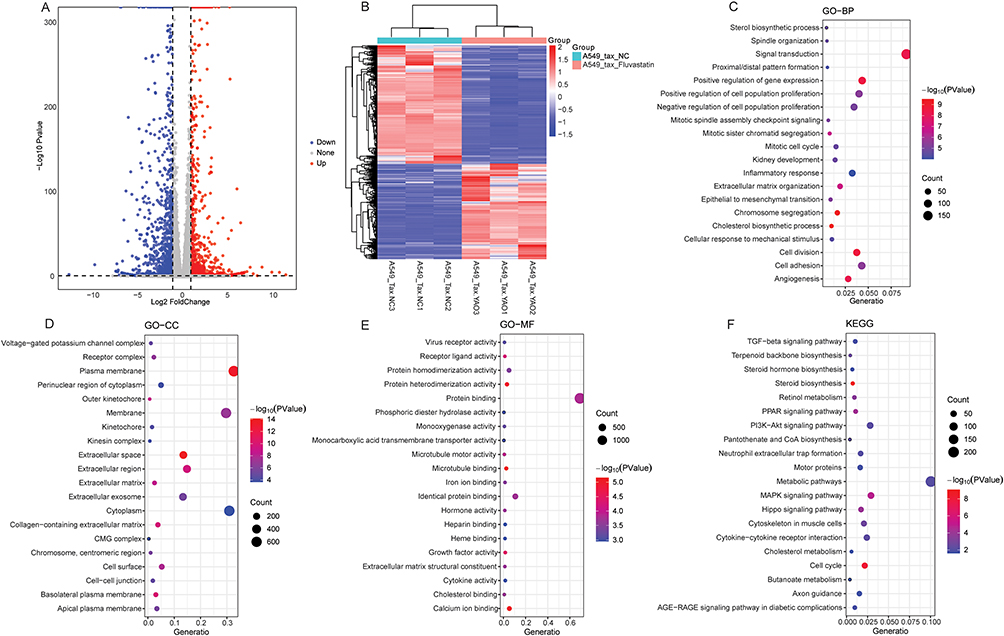 |
Figure 3 Identification and functional annotation of DEGs in A549/Tax-Fluvastatin vs A549/Tax-NC group. (A) Volcanic map of DEGs in A549/Tax-Fluvastatin vs A549/Tax-NC group; (B) Heat map of DEGs in A549/Tax-Fluvastatin vs A549/Tax-NC group; (C) The BP term of GO functional annotation of DEGs in A549/Tax-Fluvastatin vs A549/Tax-NC group; (D) The CC term of GO functional annotation of DEGs in A549/Tax-Fluvastatin vs A549/Tax-NC group; (E) The MF term of GO functional annotation of DEGs in A549/Tax-Fluvastatin vs A549/Tax-NC group; (F) KEGG functional annotation of DEGs in A549/Tax-Fluvastatin vs A549/Tax-NC group. |
Identification and Functional Annotation of DEPs
Compared with the A549-NC group, a total of 1112 DEPs were identified in the A549-Fluvastatin group, among which 740 DEPs were up-regulated and 372 DEPs were down-regulated (Figure 4A and B). In GO-BP term, DEPs were related to mitochondrial respiratory chain assembly, cholesterol metabolism, and cellular respiration (Figure 4C). In GO-CC term, DEPs were associated with structures such as the mitochondria, endoplasmic reticulum, and Golgi apparatus (Figure 4D). In GO-MF term, DEPs were associated with enzyme activity, protein binding, and virus receptor activity (Figure 4E). KEGG analysis showed that DEPs were significantly enriched in metabolic pathways, oxidative phosphorylation, and neurodegenerative disease-related pathways, etc (Figure 4F).
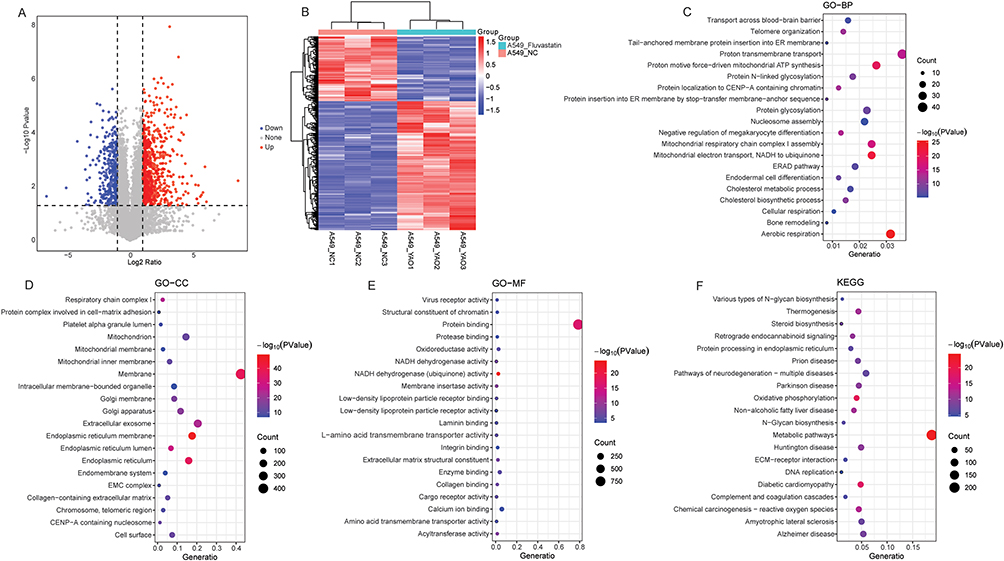 |
Figure 4 Identification and functional annotation of DEPs in A549-Fluvastatin vs A549-NC group. (A) Volcanic map of DEPs in A549-Fluvastatin vs A549-NC group; (B) Heat map of DEPs in A549-Fluvastatin vs A549-NC group; (C) The BP term of GO functional annotation of DEPs in A549-Fluvastatin vs A549-NC group; (D) The CC term of GO functional annotation of DEPs in A549-Fluvastatin vs A549-NC group; (E) The MF term of GO functional annotation of DEPs in A549-Fluvastatin vs A549-NC group; (F) KEGG functional annotation of DEPs in A549-Fluvastatin vs A549-NC group. |
Compared with the A549/Tax-NC group, a total of 1118 DEPs were identified in the A549/Tax-Fluvastatin, among which 400 DEPs were up-regulated and 718 DEPs were down-regulated (Figure 5A and B). In GO-BP term, DEPs were mainly involved in DNA replication and repair, chromosome separation, and cholesterol biosynthetic process (Figure 5C). In GO-CC term, DEPs were associated with mitochondrion, cytosol, and nucleus (Figure 5D). In GO-MF term, DEPs were associated with molecular binding, enzyme activity, chromatin/DNA-related functions, and protein modification and regulatory functions (Figure 5E). KEGG analysis showed that DEPs were significantly enriched in amino acid degradation, genetic information maintenance, p53 signaling pathway, etc (Figure 5F).
 |
Figure 5 Identification and functional annotation of DEPs in A549/Tax-Fluvastatin vs A549/Tax-NC group. (A) Volcanic map of DEPs in A549/Tax-Fluvastatin vs A549/Tax-NC group; (B) Heat map of DEPs in A549/Tax-Fluvastatin vs A549/Tax-NC group; (C) The BP term of GO functional annotation of DEPs in A549/Tax-Fluvastatin vs A549/Tax-NC group; (D) The CC term of GO functional annotation of DEPs in A549/Tax-Fluvastatin vs A549/Tax-NC group; (E) The MF term of GO functional annotation of DEPs in A549/Tax-Fluvastatin vs A549/Tax-NC group; (F) KEGG functional annotation of DEPs in A549/Tax-Fluvastatin vs A549/Tax-NC group. |
Identification of DASs and Functional Annotation of Differentially Acetylated Modified Proteins
Compared with the A549-NC group, a total of 368 DASs (corresponding to 301 proteins) were identified in the A549-Fluvastatin group, among which 196 DASs were up-regulated and 172 DASs were down-regulated (Figure 6A). Subsequently, GO and KEGG functional annotations were performed for 301 differentially acetylated modified proteins. In GO-BP term, differentially acetylated modified proteins were related to tricarboxylic acid cycle, translation and transcription processes, immune response, and regulation of apoptosis (Figure 6B). In GO-CC term, differentially acetylated modified proteins were associated with extracellular exosome, mitochondrion, cytosol, and nucleus (Figure 6C). In GO-MF term, differentially acetylated modified proteins were associated with molecular binding, oxidoreductase activity, structural constituent of ribosome, and ATP-dependent protein folding chaperone (Figure 6D). KEGG analysis showed that differentially acetylated modified proteins were significantly enriched in multiple metabolic pathways and Ferroptosis (Figure 6E). The results of motif analysis revealed a significant amino acid sequence preference around the modification site centered on lysine (K), with marked enrichment of amino acids such as glutamic acid (E) at position −1 and leucine (L) at position +1 (Figure 6F).
 |
Figure 6 Identification of DASs and functional annotation of differentially acetylated modified proteins in A549-Fluvastatin vs A549-NC group. (A) Volcanic map of DASs in A549-Fluvastatin vs A549-NC group; (B) The BP term of GO functional annotation of differentially acetylated modified proteins in A549-Fluvastatin vs A549-NC group; (C) The CC term of GO functional annotation of differentially acetylated modified proteins in A549-Fluvastatin vs A549-NC group; (D) The MF term of GO functional annotation of differentially acetylated modified proteins in A549-Fluvastatin vs A549-NC group; (E) KEGG functional annotation of differentially acetylated modified proteins in A549-Fluvastatin vs A549-NC group; (F) The motif analysis results of DASs in A549-Fluvastatin vs A549-NC group. |
Compared with the A549/Tax-NC group, a total of 135 DASs (corresponding to 114 proteins) were identified in the A549/Tax-Fluvastatin, among which 67 DASs were up-regulated and 68 DEPs were down-regulated (Figure 7A). Subsequently, GO and KEGG functional annotations were performed for 114 differentially acetylated modified proteins. In GO-BP term, differentially acetylated modified proteins were mainly involved in cardiac development and electrophysiological regulation, calcium signaling pathway, glucose metabolism, and mRNA alternative splicing (Figure 7B). In GO-CC term, differentially acetylated modified proteins were associated with extracellular exosome, mitochondrion, cytosol, and nucleus (Figure 7C). In GO-MF term, differentially acetylated modified proteins were mainly associated with molecular binding, ATP-dependent protein folding chaperone, transcription coactivator activity, and calcium channel inhibitor activity (Figure 7D). KEGG functional enrichment analysis indicated that the differentially acetylated modified proteins in the A549/Tax-Fluvastatin vs A549/Tax-NC group were specifically enriched in the estrogen signaling pathway and the HIF-1 signaling pathway (Figure 7E). The results of motif analysis revealed a significant amino acid sequence preference around the modification site centered on lysine (K), with marked enrichment of amino acids such as aspartic acid (D) at position −1 and leucine (L) at position +1 (Figure 7F).
 |
Figure 7 Identification of DASs and functional annotation of differentially acetylated modified proteins in A549/Tax-Fluvastatin vs A549/Tax-NC group. (A) Volcanic map of DASs in A549/Tax-Fluvastatin vs A549/Tax-NC group; (B) The BP term of GO functional annotation of differentially acetylated modified proteins in A549/Tax-Fluvastatin vs A549/Tax-NC group; (C) The CC term of GO functional annotation of differentially acetylated modified proteins in A549/Tax-Fluvastatin vs A549/Tax-NC group; (D) The MF term of GO functional annotation of differentially acetylated modified proteins in A549/Tax-Fluvastatin vs A549/Tax-NC group; (E) KEGG functional annotation of differentially acetylated modified proteins in A549/Tax-Fluvastatin vs A549/Tax-NC group; (F) The motif analysis results of DASs in A549/Tax-Fluvastatin vs A549/Tax-NC group. |
Integrated Analysis of Transcriptome, Proteome, and Acetylome in A549-Fluvastatin vs A549-NC Group
Intersection analysis of DEGs and DEPs in the A549-Fluvastatin vs A549-NC group identified 88 molecules with up-regulated at both transcriptional and protein levels, and 54 molecules with down-regulated at both levels (Figure 8A). Similarly, intersection analysis of DEPs and differentially acetylated modified proteins in the A549-Fluvastatin vs A549-NC group revealed 47 molecules with up-regulated at both protein and acetylation levels, and 11 molecules with down-regulated at both levels (Figure 8B). Target genes corresponding to differentially acetylated modified proteins in the A549-Fluvastatin vs A549-NC group were downloaded from the TRRUST database. Intersection analysis of these target genes with DEGs identified 67 target regulatory pairs, among which 27 pairs showed exhibited consistent regulatory trends.
 |
Figure 8 Integrated analysis of transcriptome, proteome, and acetylome in A549-Fluvastatin vs A549-NC group. (A) Venn diagram of DEGs and DEPs in A549-Fluvastatin vs A549-NC group; (B) Venn diagram of DEPs and differentially acetylated modified proteins in A549-Fluvastatin vs A549-NC group; (C) Molecules with consistent expression trends in transcriptome, proteome and acetylome; (D) Identification of the same direction changes of acetylated proteins and their downstream target genes and target gene-encoded proteins; (E) Identification of the same direction changes of DEPs and their acetylated modification status and downstream target genes. DEAc-down represents proteins with down-regulated acetylation level, and DEAc-up represents proteins with up-regulated acetylation level. V-shaped represents acetylation sites, ellipse represents gene, and rectangle represents protein. Blue indicates down-regulated and red indicates up-regulated. |
Subsequently, the obtained three groups of molecules were integrated. The results showed that in the A549-Fluvastatin vs A549-NC group, 9 genes and their encoded protein products exhibited consistent directional changes at the transcriptome (RDH11, FDFT1, HSPB1, HMGCR, FDPS, ECH1, EBP, FASN, and CTSB), proteome (Q8TC12, P37268, P04792, P04035, P14324, Q13011, Q15125, P49327, and P07858) and acetylome levels (Figure 8C). In addition, it was found that the up-regulated of acetylated protein Q02447 (at site Q02447_K593_M1) may regulate the up-regulated of downstream target genes ITGAV, PLAUR, and TYMP, thereby increasing the expression of proteins encoded by these target genes (Figure 8D). Most notably, we also observed that the down-regulated of P09874 and its corresponding acetylation sites (P09874_K108_M1 and P09874_K621_M1) may lead to decreased expression of its downstream target genes BCL2 and SERPINF1 (Figure 8E).
Integrated Analysis of Transcriptome, Proteome, and Acetylome in A549/Tax-Fluvastatin vs A549/Tax-NC Group
Intersection analysis of DEGs and DEPs in the A549/Tax-Fluvastatin vs A549/Tax-NC group identified 67 molecules with up-regulated at both transcriptional and protein levels, and 109 molecules with down-regulated at both levels (Figure 9A). Similarly, intersection analysis of DEPs and differentially acetylated modified proteins in the A549/Tax-Fluvastatin vs A549/Tax-NC group revealed 8 molecules with up-regulated at both protein and acetylation levels, and 5 molecules with down-regulated at both levels (Figure 9B). Target genes corresponding to differentially acetylated modified proteins in the A549/Tax-Fluvastatin vs A549/Tax-NC group were downloaded from the TRRUST database. Intersection analysis of these target genes with DEGs identified 29 target regulatory pairs, among which 12 pairs showed exhibited consistent regulatory trends.
 |
Figure 9 Integrated analysis of transcriptome, proteome, and acetylome in A549/Tax-Fluvastatin vs A549/Tax-NC group. (A) Venn diagram of DEGs and DEPs in A549/Tax-Fluvastatin vs A549/Tax-NC group; (B) Venn diagram of DEPs and differentially acetylated modified proteins in A549/Tax-Fluvastatin vs A549/Tax-NC group; (C) Molecules with consistent expression trends in transcriptome, proteome and acetylome; (D) Identification of the same direction changes of acetylated proteins and their downstream target genes and target gene-encoded proteins; (E) Identification of the same direction changes of DEPs and their acetylated modification status and downstream target genes. DEAc-down represents proteins with down-regulated acetylation level, and DEAc-up represents proteins with up-regulated acetylation level. V-shaped represents acetylation sites, ellipse represents gene, and rectangle represents protein. Blue indicates down-regulated and red indicates up-regulated. |
Then, the obtained three groups of molecules were integrated. The results showed that in the A549/Tax-Fluvastatin vs A549/Tax-NC group, 1 gene and its encoded protein product exhibited consistent directional changes at the transcriptome (HMGCR), proteome (P04035) and acetylome levels (Figure 9C). Furthermore, it was found that the up-regulated of acetylated protein P42224 (at site P42224_K152_M1) may regulate the up-regulated of downstream target genes PTGS2, thereby increasing the expression of proteins encoded by these target genes (Figure 9D). Moreover, it was also found that the down-regulated of acetylated protein P04150 (at site P04150_K154_M1) may regulate the down-regulated of downstream target genes CCND3, thereby increasing the expression of proteins encoded by these target genes (Figure 9D). Notably, we also observed that the down-regulated of P04150 and its corresponding acetylation site (P04150_K154_M1) may lead to decreased expression of its downstream target genes BRCA1, CCND1, CCND3, NR1I2, and RLN2 (Figure 9E).
Discussion
TAX resistance is one of the main causes of chemotherapy failure in lung cancer. Our in vitro preliminary experimental results showed that fluvastatin not only inhibits the proliferation, migration, and invasion of A549 cells while promoting their apoptosis, but also effectively suppresses the proliferation, migration, and invasion of A549/Tax cells and accelerates the apoptosis of these cells. These findings indicate that fluvastatin exhibits antitumor activity against conventional lung cancer cells and can reverse TAX resistance in A549/Tax cells.
In the comparative analysis of A549-Fluvastatin vs A549-NC group, a total of 1798 DEGs, 1112 DEPs, and 301 differentially acetylated modified proteins were identified. Functional annotation of these differential molecules showed that they were significantly enriched in various key biological processes/pathways, including steroid metabolism, respiration, immune regulation, inflammatory response, apoptosis, and ferroptosis. Steroid metabolism disorders play a significant role in the occurrence and progression of lung cancer.22,23 The tricarboxylic acid cycle and oxidative phosphorylation are two key stages of aerobic respiration. Genetic variations in the key enzyme genes of the tricarboxylic acid cycle are associated with the prognosis of lung cancer,24 while oxidative phosphorylation is closely linked to the progression of lung cancer.25 Immune regulation plays a key role in the occurrence, development and treatment of lung cancer. It may inhibit tumor by enhancing immune function or promote tumor progression due to immune disorders. Currently, immunotherapy has become one of the important treatment methods for lung cancer.26 Inflammatory cells and mediators are important components of the tumor microenvironment and play a decisive role in the initiation, proliferation, survival, promotion, invasion or metastasis of lung cancer.27 Apoptosis refers to the spontaneous and orderly death of cells controlled by genes, which helps maintain the stability of the internal environment. The dysregulation of the apoptotic mechanism is associated with the occurrence and drug resistance of lung cancer.28 Ferroptosis is mainly caused by the accumulation of iron and lipid peroxides within cells. Studies have shown that ferroptosis plays a crucial role in the progression of tumors, especially lung cancer. Specifically, the induction of ferroptosis within cells can inhibit the growth of tumor cells, which in turn reverses the process of tumor formation.29 Collectively, the differential molecules identified in the A549-Fluvastatin vs A549-NC group may exert regulatory effects on the therapeutic effects of fluvastatin through the aforementioned key biological processes/pathways, which provides an important research direction for further exploration of the mechanism of fluvastatin in the treatment of lung cancer.
In the comparative analysis of A549/Tax-Fluvastatin vs A549/Tax-NC group, a total of 2089 DEGs, 1118 DEPs, and 114 differentially acetylated modified proteins were identified. Functional annotation results revealed that the biological processes/pathways enriched by these differential molecules not only shared substantial overlap with those enriched by the differential molecules in the A549-Fluvastatin vs A549-NC group, but also included unique biological processes/pathways, including PPAR signaling pathway, cytokine-cytokine receptor interaction pathway, genetic information maintenance, p53 signaling pathway, and HIF-1 signaling pathway. Studies have shown that PPAR signaling pathway is related to the regulation of lung cancer metastasis, and it is also related to the treatment of lung cancer.30,31 Cytokine-cytokine receptor interaction pathway is closely related to the prognosis of lung cancer and apoptosis of lung cancer cells.32,33 Genetic information maintenance is a core process for cells to ensure genomic integrity through a series of mechanisms such as DNA replication fidelity, damage repair, and chromosome stability regulation. The abnormality of this process is a key driving factor for the occurrence, development and drug resistance of lung cancer.34,35 The p53 signaling pathway is a key tumor suppressor signaling pathway in cells, and it plays a central role in regulating biological processes such as apoptosis and autophagy of lung cancer cells.36,37 Moreover, it is also involved in inhibiting cisplatin resistance.38 Furthermore, the deficiency of p53 can also trigger the activation mechanism of HIF-1-dependent hypoxic signaling. The abnormal activation of hypoxia-inducible transcription factor HIF-1 and the dysfunction of tumor suppressor p53 can induce the malignant phenotype and therapeutic resistance of cancer.39 Notably, in the A549/Tax-Fluvastatin vs A549/Tax-NC group, the biological processes/pathways enriched by differential molecules not only showed substantial overlap with those enriched by differential molecules in the A549-Fluvastatin vs A549-NC group, but also additionally revealed unique pathways specific to the former. This finding not only uncovers the potential specific mechanism of action of fluvastatin in TAX resistant A549 cells, but also provides critical basis for exploring the potential therapeutic targets of fluvastatin in reversing lung cancer chemoresistance.
The data of transcriptome, proteome and acetylation group from the A549-Fluvastatin vs A549-NC group were integrated and analyzed. The results showed that the expression changes of 9 molecules in transcriptome, proteome and acetylation group were consistent. Among them, RDH11,40 FDFT1,41 HSPB1,42 HMGCR,43 ECH1,44 FASN,45 and CTSB46 have been reported to be associated with lung cancer. Furthermore, HMGCR47 is also related to drug sensitivity. However, the specific mechanisms of these molecules in lung cancer development and lung cancer treatment have not been fully elucidated. This study found that these molecules are enriched in biological processes such as retinol metabolism, steroid metabolism, apoptosis, or inflammation, suggesting that they may exert a regulatory role in fluvastatin-mediated lung cancer treatment by regulating these processes. Additionally, we identified key regulatory pairs between acetylated proteins and downstream target genes. The up-regulated of acetylated protein Q02447 (at site Q02447_K593_M1) may regulate the up-regulated of downstream target genes ITGAV, PLAUR, and TYMP, thereby increasing the expression of proteins encoded by these target genes. Previous studies have shown that ITGAV and PLAUR are involved in regulating lung cancer progression,48,49 while TYMP is associated with reduced lung cancer risk.50 Notably, we also observed that the down-regulated of P09874 and its corresponding acetylation sites (P09874_K108_M1 and P09874_K621_M1) may lead to decreased expression of its downstream target genes BCL2 and SERPINF1. BCL2 is an anti-apoptotic protein that participates in regulating ferroptosis and apoptosis in lung cancer.51,52 SERPINF1, also known as PEDF, can participate in regulating the migration and invasion of lung cancer cells.53 The identified acetylated protein-target gene pairs may mediate the effects of fluvastatin on lung cancer progression, apoptosis, ferroptosis, migration, and invasion. This finding provides new mechanistic insights into how fluvastatin regulates lung cancer-related biological processes. However, the specific mechanisms of action still require further in-depth investigation.
In the integrated analysis of the A549/Tax-Fluvastatin vs A549/Tax-NC group, it was also found that the expression trend of HMGCR in transcriptome, proteome and acetylation group was consistent. This suggests that HMGCR not only exerts effects when fluvastatin treats normal A549 cells, but also plays a functional role in TAX-resistant A549 cells, thereby further highlighting the potential research and application value of HMGCR. In addition, some acetylated protein-target gene pairs were identified, such as P42224 (at site P42224_K152_M1)-PTGS2, P04150 (at site P04150_K154_M1)-CCND3 and P04150 (at site P04150_K154_M1)-BRCA1. PTGS2 is overexpressed in lung cancer, which promotes tumor proliferation, invasion, angiogenesis, and resistance to apoptosis. In addition, this molecule is also a key therapeutic target for lung cancer treatment with the drug HQi-sRNA-2.54 The downregulation of CCND3 in lung cancer is involved in tumor suppression.55 BRCA1 is overexpressed in NSCLC drug-resistant cells and may regulate the occurrence and development of lung cancer through the PI3K/AKT signaling pathway.56 Overall, these findings from the integrated analysis provide some initial clues about HMGCR and the acetylated protein-target gene pairs in TAX-resistant A549 cells under fluvastatin treatment. However, the specific roles of these in regulating drug resistance and the activity of lung cancer cells remain unclear and require more experimental data for validation.
This study initially investigated fluvastatin’s effects and potential molecular mechanisms on A549 and A549/Tax cells. However, this study also has certain limitations. Firstly, it only completed preliminary in vitro experimental verification and lacks animal models to verify the in vivo antitumor effect and drug resistance reversal effect of fluvastatin. Secondly, the study lacks clinical relevance, as it did not include clinical lung cancer samples, thus failing to verify the expression patterns of the differential molecules identified in vitro in clinical samples. Thirdly, the specific mechanisms of the molecules and related biological processes/pathways screened by multi-omics in the antitumor and drug resistance reversal effects of fluvastatin remain unclear. Therefore, more in vivo and in vitro studies are needed in the future to improve the relevant conclusions. Moreover, advanced technologies such as PROTAC probe technology57 and cutting-edge proteomics technology58 will also be used to verify the identified key molecules and pathways to deepen the exploration of potential mechanisms. Fourthly, the study has limitations regarding sample size. Each experimental group comprises n=3 (three independent repetitions). Although three independent repetitions in cell experiments can reflect preliminary intergroup trends, the small sample size leads to insufficient statistical power, which may make the observed experimental trends susceptible to random factors. In subsequent studies, we will expand the sample size and further validate the stability and reproducibility of the current preliminary results. In conclusion, this study indicates that fluvastatin has the potential to reverse TAX resistance in lung cancer, and the results of multi-omics analysis provide a theoretical basis for the subsequent clinical development of strategies to reverse chemotherapy resistance in lung cancer and the exploration of potential therapeutic targets.
Data Sharing Statement
All data generated or analyzed during this study are included in this published article. Further inquiries can be directed to the corresponding authors upon reasonable request.
Funding
This study was funded by the “Sichuan Provincial Medical Research Project (S22034)”, the “Chengdu Medical Research Project (2022485)”, the “Wu Jieping Medical Foundation (320.6750.2020-10-123)”, and the “Capacity Building and Continuing Education Center of the National Health Commission (GWJJMB202510010014).
Disclosure
The author(s) declare no competing interests.
References
1. Bade BC, Dela Cruz CS. Lung cancer 2020: epidemiology, etiology, and prevention. Clin Chest Med. 2020;41(1):1–16. doi:10.1016/j.ccm.2019.10.001
2. Chen F, Xiang W, Qiang G. Tanshinone IIA affects the proliferation of A549/Tax by affecting the expression of MMP7 through the PI3K-AKT-mTOR signaling pathway. Discov Oncol. 2025;16(1):369. doi:10.1007/s12672-025-02152-8
3. Bai Y, Xiong L, Zhu M, Yang Z, Zhao J, Tang H. Co-expression network analysis identified KIF2C in association with progression and prognosis in lung adenocarcinoma. Cancer Biomark. 2019;24(3):371–382. doi:10.3233/cbm-181512
4. Relli V, Trerotola M, Guerra E, Alberti S. Abandoning the notion of non-small cell lung cancer. Trends Mol Med. 2019;25(7):585–594. doi:10.1016/j.molmed.2019.04.012
5. Zappa C, Mousa SA. Non-small cell lung cancer: current treatment and future advances. Transl Lung Cancer Res. 2016;5(3):288–300. doi:10.21037/tlcr.2016.06.07
6. Maslyar DJ, Jahan TM, Jablons DM. Mechanisms of and potential treatment strategies for metastatic disease in non-small cell lung cancer. Semin Thorac Cardiovasc Surg. 2004;16(1):40–50. doi:10.1053/j.semtcvs.2003.12.003
7. Han ML, Zhao YF, Tan CH, et al. Cathepsin L upregulation-induced EMT phenotype is associated with the acquisition of cisplatin or paclitaxel resistance in A549 cells. Acta Pharmacol Sin. 2016;37(12):1606–1622. doi:10.1038/aps.2016.93
8. Cui H, Arnst K, Miller DD, Li W. Recent advances in elucidating paclitaxel resistance mechanisms in non-small cell lung cancer and strategies to overcome drug resistance. Curr Med Chem. 2020;27(39):6573–6595. doi:10.2174/0929867326666191016113631
9. Okubo K, Isono M, Miyai K, Asano T, Sato A. Fluvastatin potentiates anticancer activity of vorinostat in renal cancer cells. Cancer Science. 2020;111(1):112–126. doi:10.1111/cas.14225
10. Xu HY, Xue JX, Gao H, et al. Fluvastatin-mediated down-regulation of SATB1 affects aggressive phenotypes of human non-small-cell lung cancer cell line H292. Life Sci. 2019;222:212–220. doi:10.1016/j.lfs.2018.12.022
11. Yang Z, Su Z, DeWitt JP, et al. Fluvastatin prevents lung adenocarcinoma bone metastasis by triggering autophagy. EBioMedicine. 2017;19:49–59. doi:10.1016/j.ebiom.2017.04.017
12. Wang W, Fu F, Huang Z, et al. Inhalable biomimetic protein corona-mediated nanoreactor for self-amplified lung adenocarcinoma ferroptosis therapy. ACS Nano. 2022;16(5):8370–8387. doi:10.1021/acsnano.2c02634
13. Otahal A, Aydemir D. Delineation of cell death mechanisms induced by synergistic effects of statins and erlotinib in non-small cell lung cancer cell (NSCLC) lines. Scientific Reports. 2020;10(1):959. doi:10.1038/s41598-020-57707-2
14. Bi YY, Chen Q, Yang MY, Xing L, Jiang HL. Nanoparticles targeting mutant p53 overcome chemoresistance and tumor recurrence in non-small cell lung cancer. Nat Commun. 2024;15(1):2759. doi:10.1038/s41467-024-47080-3
15. Li S, De Souza P. Ras isoprenylation and pAkt inhibition by zoledronic acid and fluvastatin enhances paclitaxel activity in T24 bladder cancer cells. Cancers. 2011;3(1):662–674. doi:10.3390/cancers3010662
16. Agamah FE, Bayjanov JR, Niehues A, et al. Computational approaches for network-based integrative multi-omics analysis. Front Mol Biosci. 2022;9:967205. doi:10.3389/fmolb.2022.967205
17. Lu Z, Qian P, Chang J, et al. Multi-omics analysis explores the effect of chronic exercise on liver metabolic reprogramming in mice. Front Cell Dev Biol. 2023;11:1199902. doi:10.3389/fcell.2023.1199902
18. Jiang C, Li P, Ma Y, et al. Comprehensive gene profiling of the metabolic landscape of humanized livers in mice. J Hepatol. 2024;80(4):622–633. doi:10.1016/j.jhep.2023.11.020
19. Qian X, Zhang HY, Li QL, et al. Integrated microbiome, metabolome, and proteome analysis identifies a novel interplay among commensal bacteria, metabolites and candidate targets in non-small cell lung cancer. Clin Transl Med. 2022;12(6):e947. doi:10.1002/ctm2.947
20. Qian P, Ma F, Zhang W, et al. Chronic exercise remodels the lysine acetylome in the mouse hippocampus. Front Mol Neurosci. 2022;15:1023482. doi:10.3389/fnmol.2022.1023482
21. Wattanathamsan O, Thararattanobon R, Rodsiri R, Chanvorachote P, Vinayanuwattikun C, Pongrakhananon V. Tubulin acetylation enhances lung cancer resistance to paclitaxel-induced cell death through Mcl-1 stabilization. Cell Death Disc. 2021;7(1):67. doi:10.1038/s41420-021-00453-9
22. Xiu W, Liu X, Hu K, Zhang Q, Shi H. The role of cholesterol metabolism in lung cancer. Oncol Res. 2024;32(10):1613–1621. doi:10.32604/or.2024.047933
23. Asavasupreechar T, Chan MSM, Saito R, Miki Y, Boonyaratanakornkit V, Sasano H. Sex steroid metabolism and actions in non-small cell lung carcinoma. J Steroid Biochem Mol Biol. 2019;193:105440. doi:10.1016/j.jsbmb.2019.105440
24. Guo X, Li D, Wu Y, et al. Genetic variants in genes of tricarboxylic acid cycle key enzymes are associated with prognosis of patients with non-small cell lung cancer. Lung Cancer. 2015;87(2):162–168. doi:10.1016/j.lungcan.2014.12.005
25. Rao S, Mondragón L, Pranjic B, Hanada T, Stoll G. AIF-regulated oxidative phosphorylation supports lung cancer development. Cell Research. 2019;29(7):579–591. doi:10.1038/s41422-019-0181-4
26. Castillo-Peña A, Molina-Pinelo S. Landscape of tumor and immune system cells-derived exosomes in lung cancer: mediators of antitumor immunity regulation. Front Immunol. 2023;14:1279495. doi:10.3389/fimmu.2023.1279495
27. Shi L, Wang L, Hou J, et al. Targeting roles of inflammatory microenvironment in lung cancer and metastasis. Cancer Metastasis Rev. 2015;34(2):319–331. doi:10.1007/s10555-015-9570-4
28. Xu CX, Jin H, Cho MH. Apoptosis and apoptosis-based therapy in lung cancer. Anticancer Agents Med Chem. 2009;9(9):952–957. doi:10.2174/187152009789377682
29. Xiong R, He R. Ferroptosis: a new promising target for lung cancer therapy. Oxidative Med Cell Longevity. 2021;2021:8457521. doi:10.1155/2021/8457521
30. Chen J, Liu A, Lin Z, et al. Downregulation of the circadian rhythm regulator HLF promotes multiple-organ distant metastases in non-small cell lung cancer through PPAR/NF-κb signaling. Cancer Lett. 2020;482:56–71. doi:10.1016/j.canlet.2020.04.007
31. Reka AK, Goswami MT, Krishnapuram R, Standiford TJ, Keshamouni VG. Molecular cross-regulation between PPAR-γ and other signaling pathways: implications for lung cancer therapy. Lung Cancer. 2011;72(2):154–159. doi:10.1016/j.lungcan.2011.01.019
32. Yang F, Zhang S, Meng Q, et al. CXCR1 correlates to poor outcomes of EGFR-TKI against advanced non-small cell lung cancer by activating chemokine and JAK/STAT pathway. Pulm Pharmacol Ther. 2021;67:102001. doi:10.1016/j.pupt.2021.102001
33. Li B, Wang Z, Zhong Y, Lan J, Li X, Lin H. CCR9-CCL25 interaction suppresses apoptosis of lung cancer cells by activating the PI3K/Akt pathway. Med Oncol. 2015;32(3):66. doi:10.1007/s12032-015-0531-0
34. Morris BB, Wages NA, Grant PA, et al. MYBL2-Driven transcriptional programs link replication stress and error-prone DNA repair with genomic instability in lung adenocarcinoma. Front Oncol. 2020;10:585551. doi:10.3389/fonc.2020.585551
35. Venkatesan S, Angelova M. Induction of APOBEC3 exacerbates DNA replication stress and chromosomal instability in early breast and lung cancer evolution. Cancer Discovery. 2021;11(10):2456–2473. doi:10.1158/2159-8290.cd-20-0725
36. Zhong G, Chen X, Fang X, Wang D, Xie M, Chen Q. Fra-1 is upregulated in lung cancer tissues and inhibits the apoptosis of lung cancer cells by the P53 signaling pathway. Oncol Rep. 2016;35(1):447–453. doi:10.3892/or.2015.4395
37. Zhu J, Ao H, Liu M, Cao K, Ma J. UBE2T promotes autophagy via the p53/AMPK/mTOR signaling pathway in lung adenocarcinoma. J Transl Med. 2021;19(1):374. doi:10.1186/s12967-021-03056-1
38. Lin L, Zou Y, Zhang D. Silencing ribosome biogenesis regulator 1 homolog (RRS1) inhibits angiogenesis and cisplatin resistance of lung cancer cells by activating ferroptosis mediated by p53 pathway. Tissue Cell. 2025;94:102796. doi:10.1016/j.tice.2025.102796
39. Koyasu S, Horita S. ZBTB2 links p53 deficiency to HIF-1-mediated hypoxia signaling to promote cancer aggressiveness. EMBO Reports. 2023;24(1):e54042. doi:10.15252/embr.202154042
40. Kuznetsova ES, Zinovieva OL, Oparina NY, et al. Abnormal expression of genes that regulate retinoid metabolism and signaling in non-small-cell lung cancer. Mol Biol. 2016;50(2):255–265. doi:10.7868/s0026898416020130
41. Dehghani M, Samani Z, Abidi H, Manzouri L, Mahmoudi R, Hosseini Teshnizi S. Relationship of SNP rs2645429 in farnesyl-diphosphate farnesyltransferase 1 gene promoter with susceptibility to lung cancer. Cancer Cells.2018;2018:4863757. doi:10.1155/2018/4863757
42. Choi SH, Nam JK, Kim BY, et al. HSPB1 inhibits the Endothelial-to-Mesenchymal transition to suppress pulmonary fibrosis and lung tumorigenesis. Cancer Res. 2016;76(5):1019–1030. doi:10.1158/0008-5472.can-15-0952
43. Liu L, Chai L, Ran J, Yang Y, Zhang L. BAI1 acts as a tumor suppressor in lung cancer A549 cells by inducing metabolic reprogramming via the SCD1/HMGCR module. Carcinogenesis. 2020;41(12):1724–1734. doi:10.1093/carcin/bgaa036
44. Dai L, Li J, Tsay JJ, et al. Identification of autoantibodies to ECH1 and HNRNPA2B1 as potential biomarkers in the early detection of lung cancer. Oncoimmunology. 2017;6(5):e1310359. doi:10.1080/2162402x.2017.1310359
45. Zhan N, Li B, Xu X, Xu J, Hu S. Inhibition of FASN expression enhances radiosensitivity in human non-small cell lung cancer. Oncol Lett. 2018;15(4):4578–4584. doi:10.3892/ol.2018.7896
46. Zhang J, Zhou B, Jin S, et al. Mechanism of action of Panax notoginoside against lung cancer in mice based on response to CTSB gene. BMC Complement Med Ther. 2020;20(1):367. doi:10.1186/s12906-020-03159-0
47. Yuan H, Wu H, Cheng J, Xiong J. SIAH1 ubiquitination-modified HMGCR inhibits lung cancer progression and promotes drug sensitivity through cholesterol synthesis. Cancer Cell Int. 2023;23(1):71. doi:10.1186/s12935-023-02914-w
48. Taheri Baghmisheh S, Wu YY, Wu JE, Hsu KF, Chen YL, Hong TM. CASZ1 promotes migration, invasion, and metastasis of lung cancer cells by controlling expression of ITGAV. Am J Cancer Res. 2023;13(1):176–189.
49. Cheng Y, Bao S, Zhang H, et al. Identification of plasminogen activator urokinase receptor-related non-coding RNA and immune prognostic signature for non-small cell lung cancer. Clinics. 2025;80:100711. doi:10.1016/j.clinsp.2025.100711
50. Zhang W, Chen N, Li C, et al. Causal association between plasma proteins and lung adenocarcinoma: a two-sample mendelian randomization study. BMC Pulm Med. 2025;25(1):280. doi:10.1186/s12890-025-03751-5
51. Huang F, Pang J, Xu L, et al. Hedyotis diffusa injection induces ferroptosis via the Bax/Bcl2/VDAC2/3 axis in lung adenocarcinoma. Phytomedicine. 2022;104:154319. doi:10.1016/j.phymed.2022.154319
52. Alam S, Mohammad T, Padder RA, Hassan MI. Thymoquinone and quercetin induce enhanced apoptosis in non-small cell lung cancer in combination through the Bax/Bcl2 cascade. J Cell Biochem. 2022;123(2):259–274. doi:10.1002/jcb.30162
53. Huang WT, Chong IW, Chen HL, et al. Pigment epithelium-derived factor inhibits lung cancer migration and invasion by upregulating exosomal thrombospondin 1. Cancer Lett. 2019;442:287–298. doi:10.1016/j.canlet.2018.10.031
54. Lin Y, Sun N, Liu D, Yang X, Dong Y, Jiang C. COX-2/PTGS2-targeted herbal-derived oligonucleotide drug HQi-sRNA-2 was effective in spontaneous mouse lung cancer model. IUBMB Life. 2024;76(11):937–950. doi:10.1002/iub.2858
55. Xie C, Zhou X. ZNF652 exerts a tumor suppressor role in lung cancer by transcriptionally downregulating cyclin D3. Cell Death Disease. 2024;15(11):792. doi:10.1038/s41419-024-07197-1
56. Wang S, Liu F, Zhu J, et al. DNA repair genes ERCC1 and BRCA1 expression in non-small cell lung cancer chemotherapy drug resistance. Med Sci Monit. 2016;22:1999–2005. doi:10.12659/msm.896606
57. Yan S, Zhang G, Luo W, et al. PROTAC technology: from drug development to probe technology for target deconvolution. Eur J Med Chem. 2024;276:116725. doi:10.1016/j.ejmech.2024.116725
58. He SJ, Li J, Zhou JC, Yang ZY, Liu X, Ge YW. Chemical proteomics accelerates the target discovery of natural products. Biochem Pharmacol. 2024;230(Pt 3):116609. doi:10.1016/j.bcp.2024.116609
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.