Back to Journals » International Journal of Nanomedicine » Volume 11
Preclinical systemic toxicity evaluation of chitosan-solid–lipid nanoparticle-encapsulated aspirin and curcumin in combination with free sulforaphane in BALB/c mice
Authors Thakkar A, Chenreddy S, Thio A, Khamas W, Wang J, Prabhu S
Received 19 February 2016
Accepted for publication 11 May 2016
Published 20 July 2016 Volume 2016:11 Pages 3265—3276
DOI https://doi.org/10.2147/IJN.S106736
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Thomas Webster
Arvind Thakkar,1 Sushma Chenreddy,1 Astrid Thio,1 Wael Khamas,2 Jeffrey Wang,1 Sunil Prabhu1
1Department of Pharmaceutical Sciences, College of Pharmacy, 2College of Veterinary Medicine, Western University of Health Sciences, Pomona, CA, USA
Abstract: Our previous studies have established the efficacy of chemopreventive regimens of aspirin and curcumin (CUR) encapsulated within solid lipid nanoparticles (SLNs) in combination with free sulforaphane (ACS combination) to prevent or delay the initiation and progression of pancreatic cancer, classified as one of the deadliest diseases with very low chances of survival upon diagnosis. Although toxicity of individual drugs and SLN has been studied previously, there are no studies in current literature that evaluate the potential toxicity of a combined regimen of ACS, especially when encapsulated within chitosan-SLNs (c-SLNs). Hence, objective of the current study was to investigate the potential toxic effects of ACS c-SLN combined chemopreventive regimens following acute (3 days), subacute (28 days), and subchronic (90 days) administrations by oral gavage in BALB/c mice. Mice were administered the following regimens: saline, blank c-SLN, low-dose ACS c-SLN (2+4.5+0.16 mg/kg), medium-dose ACS c-SLN (20+45+1.6 mg/kg), and high-dose ACS c-SLN (60+135+4.8 mg/kg). The potential toxicity was evaluated based on animal survival, body weight, hematology, blood chemistry, and organ histopathology. During 3-day, 28-day, and 90-day study periods, no animal deaths were observed. Treatment with ACS c-SLNs did not cause alteration in complete blood counts and blood chemistry data. Histopathological examination of various organ sections (pancreas, heart, liver, kidney, and brain) appeared normal. Based on the results of this study, no signs of toxicity in acute, subacute, and subchronic studies following oral administration of ACS c-SLNs were found indicating that the oral dosing regimens were safe at the levels tested for long-term administration to prevent the onset of pancreatic cancer.
Keywords: pancreatic cancer, toxicity, chitosan-solid lipid nanoparticle, aspirin, curcumin, sulforaphane
Introduction
Adverse drug events affect millions of patients each year and are responsible for up to 5% of hospital admissions, resulting in a significant increase in the cost of health care.1,2 Drug–drug interactions (DDIs) are defined as an increase or decrease in the clinical effect of a given drug due to interference by another drug and could be a significant cause of morbidity and mortality worldwide.3 DDIs are characterized by adverse drug events; they are often predicted from published reports, toxicity studies, and pharmacological interactions. Some DDI events have fatal life-threatening concerns due to toxicity and may prompt the removal of approved medications from the marketplace.4–8 The reasons for toxicity vary depending on the mixture of drugs, for example, when digoxin and clarithromycin are used simultaneously, toxicity can develop in patients due to the inhibition of P-glycoprotein by clarithromycin,9 which in turn inhibits the renal clearance of digoxin.10,11
Our group recently published the results of a proof-of-concept 24-week chemopreventive study with the oral administration of a combination of aspirin (ASP) and curcumin (CUR) encapsulated within solid lipid nanoparticles (SLNs) and free sulforaphane (SFN), on N-nitrosobis(2-oxopropyl) amine-treated Syrian golden hamster model to suppress the progression of pancreatic intraepithelial neoplasms.12 Although previous reports have demonstrated ASP, CUR, and SFN (ACS) alone to be effective against different cancers, significantly higher drug concentrations have been used, which may be toxic and not feasible for long-term chemopreventive administration. In recent decades, chitosan-SLNs (c-SLNs) have gained wide attention due to their stability and scope of surface modifications.13–15 Recently, Arya et al16 demonstrated a targeted chitosan-complexed nanoparticle formulation for the delivery of gemcitabine drug to the pancreatic tumor site when administered to animals via tail vein injections. In the present study, we designed an oral delivery system for ASP and CUR encapsulated in c-SLN with free SFN. Chitosan is a nontoxic and biocompatible polysaccharide derived from the shells of crustaceans, with proven in vivo safety profiles.17,18 The addition of chitosan provided steric stabilization of nanoparticles by reducing their uptake by the reticuloendothelial system in the body.19,20 Chitosan-based nanoparticles exhibit a mucoadhesive feature because of their positive charge, thereby capable of prolonging their residence time in the negatively charged small intestine,21 thus increasing the drug concentration at the site of absorption. Moreover, chitosan is known to mediate the opening of tight junctions between epithelial cells reversibly, facilitating the paracellular transport of drug molecules ultimately leading to improved bioavailability of the drugs.22 Drug-loaded c-SLNs were coated with Eudragit L 100-55 polymer to avoid drug release in the stomach, instead to dissolve in the small intestine exposing c-SLNs in the duodenal region which was subsequently taken into the lymphatic circulation.23 The programmed release of ACS from c-SLNs ensured a sustained presence of these chemopreventive agents to allow a synergistic chemopreventive effect.
Individual components of the c-SLN formulation such as stearic acid (lipid), poloxamer 188 (surfactant), and chitosan have been reported to be noncytotoxic.24,25 However, there are no studies in the current literature that evaluate the potential toxicity of a combined regimen of ACS, especially when encapsulated within c-SLNs. While the incorporation of ACS chemopreventive regimens into lipid nanoparticles leads to improved efficacy, whether this incorporation will have any effects on the toxicity profile still remains an open question. Chemopreventive therapy requires long-term, frequent administration of the ACS nanoparticle regimen, thus making it very important to evaluate toxicity profiles of this novel regimen.
Low-dose ASP, commonly defined as 81 mg daily, is one of the most widely used drugs in the world and the mainstay of therapy for cardiovascular disease prevention.26 One survey suggested that over one-third of the US adult population and 80% of those with known cardiovascular disease use low-dose ASP regularly.27 At the high dose, ASP predominantly inhibits the cyclooxygenase (COX-1) isoform in the gastrointestinal mucosa leading to ulcerogenic effects,28 thus have been largely abandoned in clinical practice. The UGLA survey29 showed that 15.4% of ASP users demonstrated upper gastrointestinal symptoms and 70% gastroesophageal reflux, which resulted in a negative impact on treatment compliance in 12% of patients.
CUR, the principal curcuminoid of the popular spice turmeric, is obtained from the ground rhizomes of Curcuma longa. Published reports indicate that the majority effects of the CUR are associated with its ability to suppress inflammation.30 Clinical studies of CUR in healthy human subjects as well as cancer patients have demonstrated low systemic bioavailability even at the dose of 12 g/d following oral administration.31,32 However, no toxicity was evident even at this dose.
Finally, SFN has been shown to protect against acetaminophen-induced hepatotoxicity,33 transplacental toxicity,34 and alveolar epithelial cells exposed to cigarette smoke extract.35 However, there are no systemic subacute and subchronic toxicity studies of SFN reported in the literature.
The objective of the present study was to examine the potential toxicological aspects of the ACS combination within a nanoparticle oral delivery system in BALB/c mice. A toxicological profile is essential to design future clinical efficacy studies of these formulated nanoparticles. The current investigation addressed these issues by conducting acute (3 days), subacute (28 days), and subchronic (90 days) toxicity studies in BALB/c mice.
Material and methods
Chemicals and reagents
ASP and CUR were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Stearic acid, poloxamer 188, and chitosan were obtained from Spectrum Chemicals (Gardena, CA, USA). Ethanol and glacial acetic acid were obtained from Thermo Fisher Scientific (Waltham, MA, USA). Potassium chloride, potassium phosphate monobasic, and Tris base were purchased from BDH Chemicals (Radnor, PA, USA). Sodium Phosphate Dibasic was purchased from EMD Millipore (Billerica, MA, USA). Sodium chloride, Tween-20, and SFN were purchased from Santa Cruz Biotechnology Inc. (Dallas, TX, USA).
Chitosan-coated SLN preparation
The following formulations were prepared: (F1) ASP c-SLN (ASP) only, (F2) CUR c-SLN (CUR) only, (F3) free unmodified SFN only, and (F4) mixtures of ASP and CUR c-SLNs with free SFN. F1 and F2 formulations were prepared in bulk amounts, whereas F4 was prepared by blending known quantities of nanoparticles from F1, F2, and free-SFN. Blank nanoparticle formulations were also prepared using stearic acid without any drugs, which served as the vehicle control. Oil/water emulsion was prepared using the modified hot homogenization technique.24 Briefly, 1 g of stearic acid was melted by heating at 70°C. The drug (250 mg) was suspended in 5 mL of ethanol. The suspended drug solution was then added to the melted stearic acid. The 50 mL of 2% poloxamer solution (water phase) was heated to the same temperature as that of the oil phase. The oil phase was then added to the water phase dropwise under continuous high-speed (25,000 rpm) homogenization (Tissue Master 125; Omni International, Kennesaw, GA, USA). The pre-emulsion was further sonicated for 30 seconds using an ultrasonicator (Branson Sonifier 450; Emerson Electric, St Louis, MO, USA). The emulsion so formed was passed through a high-pressure homogenizer (Nano DeBEE; BEE International, South Easton, MA, USA) for five cycles and kept on a stirrer for 2 hours for solvent evaporation. The emulsion was then mixed with an equal volume of 0.1% chitosan dissolved in 0.1% glacial acetic acid. The mixture of SLNs with chitosan was stirred for 2 hours. The resulted c-SLNs were ultracentrifuged, and the pellet was collected. For the present study, mannitol was added as cryoprotectant in the ratio of 1:1 (nanoparticles:cryoprotectant). Five milliliter aqueous solution of mannitol was added to the pellet. This solution was mixed well and frozen in a deep freezer at -80°C. The samples were then lyophilized in a freeze dryer (FreeZone 4.5 Plus; Labconco, Kansas City, MO, USA) and coated with 30% Eudragit L 100-55 dispersion.
Determination of mean particle size, zeta potential, and polydispersity index
The mean particle size (z-average) and polydispersity index (PDI) were determined by photon correlation spectroscopy using Zetasizer (Nano ZS90; Malvern Instruments, Malvern, UK) at 25°C and 90° scattering angle. The Zetasizer works on dynamic light scattering technology. The results are represented in mean value from the percent intensity distribution (Z-average), and the PDI describes the distribution width. Briefly, 5 mg of the c-SLN formulation was suspended in 10 mL of phosphate-buffered saline (PBS, pH 7.4), and the suspension was sonicated for 30 seconds. Particle size was measured using 1 mL of the suspension. The surface charge was assessed by measuring zeta potential of c-SLNs based on the Smoluchowski Equation, using the same equipment at 25°C with an electric field strength of 23 V/cm.36
Chromatographic analysis of ASP and CUR
ASP and CUR were analyzed using a Shimadzu LC-20 binary (Shimadzu Corp., Kyoto, Japan) high-performance liquid chromatography (HPLC) system (Columbia, MD). The system consists of a Restek Ultra II C-18 column (4.6×150 mm, 5 μm) with mobile phase composed of acetonitrile and 0.1% acetic acid (60:40, v/v). The flow rate was 0.5 mL/min, and the detection wavelength was set at 223 nm for ASP and 423 nm for CUR using a photodiode array detector. The retention time of ASP and CUR was 4.2 minutes and 8.5 minutes, respectively.
Encapsulation efficiency determination
Encapsulation efficiency (EE%) was determined by incorporating 10 mg of the c-SLN formulation in 5 mL ethanol, which dissolved the stearic acid and released the drug entrapped within the lipid. Ethanol was evaporated under a current of inert air for 1 hour leaving behind a residue of the drug and lipid. The drug was separated from the lipid by dissolving it in 5 mL of acetonitrile. The drug was allowed to dissolve freely for ~30 minutes in a bath sonicator after which it was filtered through a 0.45 μm filter. The resulting solution was further diluted to 20 mL with acetonitrile and analyzed using Shimadzu LC-20 binary HPLC. Caffeine was used as an internal standard. EE was calculated using the following equation:
|
|

In vitro drug release kinetics
The cumulative release kinetics of ASP and CUR from c-SLN was determined in PBS, pH 7.4. Five milligram of the c-SLNs were suspended in 50 mL of PBS and placed in an incubator at 37°C with a shaking speed of 100 rpm. Drug release from c-SLNs was compared to PBS with blank c-SLNs. At predetermined time intervals (0 hour, 0.5 hour, 1 hour, 2 hours, 4 hours, 6 hours, 12 hours, 24 hours, 48 hours, and 72 hours), 1 mL of the buffer was withdrawn and replaced with equivalent volume of fresh buffer. All samples are centrifuged at 5,000 rpm for 10 minutes. The amount of drug released in the supernatant was analyzed using HPLC. The analysis was carried out in triplicate.
Experimental design and treatments of in vivo toxicity study
BALB/c mice
The study was conducted on male and female BALB/c mice, 6 weeks old with an average body weight of ~20 g. BALB/c mice were obtained from Taconic Biosciences (Hudson, NY, USA) and housed in a controlled environment (temperature 23°C±1°C, humidity 60.5%, and 12-hour light/12-hour dark cycle) in the animal facility of Western University of Health Sciences. The animals had access to commercially available feed pellets and water ad libitum as per the Western University of Health’s Guide for the Care and Use of Laboratory Animals. They were acclimatized for 1 week prior to experimentation. The animal protocol was approved by the Institutional Animal Care and Use Committee (IACUC) at Western University of Health Sciences. According to Organization for Economic Co-operation and Development (OECD) guidelines on short-term repeat dose toxicity study, the 28-day oral toxicity study (OECD 407, EC B.7) and 90-day oral toxicity tests (OECD 408, EC B.26) in rodents are the most commonly recommended.37,38 Hence, we performed a 3-day acute, 28-day subacute, and 90-day subchronic toxicity studies of c-SLNs in the BALB/c mice. Two male and two female BALB/c mice per group per time point were used. For the acute toxicity study, mice were orally gavaged for 3 days with 100 μL ACS c-SLNs at three doses: low-dose ACS (2+4.5+0.16 mg/kg; 1/30 of the high dose), medium-dose ACS (20+45+1.6 mg/kg; 1/3 of the high dose), and high-dose ACS (60+135+4.8 mg/kg). The mice were orally gavaged daily with 100 μL ACS c-SLNs from low to high dose. The control and vehicle control groups received 100 μL PBS buffer and blank c-SLN, respectively. The same experimental protocol was followed for 28-day subacute and 90-day subchronic toxicity studies. A total of 60 mice were used in this study (Table 1). Assessment of any evidence of toxicity included clinical observation, body weight, hematology, blood clinical chemistry, and histopathological evaluations.
 | Table 1 Treatment groups and dose levels of ACS c-SLNs |
LSL K-rasG12D/+/Pdx-1Cre/+ transgenic mice
The 6-week-old LSL-KrasG12D/+/pdx-1Cre/+ transgenic mice were randomly divided into five groups so that average body weights are equal in all groups (n=6/group). The treatment regimen and experimental conditions remained same as BALB/c mice study except this study was performed for 120 days. A total of 30 mice were used (Table 1).
In vivo observation and examination
Clinical signs
During the study, mice were monitored for signs of toxicity including ruffled fur, impeded movement, clear sign of abnormal constitution, aberrant behavior, obvious loss of weight (10%–15%), and diarrhea. Mice were weighed twice a week throughout the experimental period.
Hematology
Blood was collected from the heart after sacrificing the animal. One hundred microliter of blood was collected in a microtainer containing ethylenediaminetetraacetic acid (BD, Franklin Lakes, NJ, USA) and analyzed using Hemavet 950 (Drew Scientific, Dallas, TX, USA). The hematological parameters studied were as follows: white blood cell count, red blood cell (RBC) count, hemoglobin concentration, hematocrits, mean corpuscular volume, mean corpuscular hemoglobin, mean corpuscular hemoglobin concentration, red cell distribution width, platelet (PLT) count, mean PLT volume, percentage of neutrophils, percentage of lymphocytes, percentage of monocytes, percentage of eosinophils, and percentage of basophils.
Blood biochemistry
Four hundred microliter of blood was collected in a BD microtainer plasma separator tube with lithium heparin (BD). The blood samples were submitted to Antech Laboratories (Irvine, CA, USA) within 2 hours of collection. The parameters analyzed were blood urea nitrogen (BUN) and creatinine (CRE), which can indicate renal toxicity; total bilirubin (TBIL), alanine transaminase (ALT), and alkaline phosphatase (ALP) levels as indicators of hepatic toxicity, while glucose (GLU) and total protein are measures of healthy growth.
Histopathology
All organs of the thoracic and abdominal cavities were carefully examined in situ macroscopically after euthanization. The internal organs pancreas, kidney, heart, liver, and brain were collected for the 28-day and 90-day study, fixed in 10% (v/v) neutral-buffered formalin and submitted to the University of Southern California (USC) Histology Services to process for hematoxylin and eosin (H&E) staining. The morphological changes were assessed by independent assessor blinded to the study.
Statistical analysis
GraphPad Software, Inc. (La Jolla, CA, USA) was used for statistical analysis and plotting. The body weight data were expressed as mean ± standard error of mean, and hematology and blood chemistry results were expressed as mean ± standard deviation and analyzed by one-way analysis of variance (ANOVA) and two-way ANOVA followed by Dunnett’s multiple comparisons test. A P-value of ≤0.05 was considered statistically significant.
Results and discussion
Particle size, encapsulation efficiency, zeta potential, and polydispersity index of drug encapsulated c-SLNs
The mean particle size was measured by using a dynamic light scattering technique. The mean particle size of ASP SLN and c-SLNs was 361 nm and 430 nm, respectively, and CUR SLN and c-SLNs was 400 nm and 440 nm, respectively (Table 2). The c-SLNs of ASP and CUR were within the nanometer range and showed optimal particle size with low variability. The difference in mean particle size between SLNs and c-SLNs was 40 nm to 70 nm, indicating additional layer of chitosan on SLNs. The EE% of ASP and CUR c-SLNs was 65% and 72%, respectively.
 | Table 2 Optimization of aspirin and curcumin encapsulated in SLNs and c-SLNs formulations |
Surface charge potential plays an important role in nanoparticle stability due to electrostatic repulsion. Since surfactant poloxamer 188 is a nonionic surfactant, lipid cores may be responsible for the negative surface charge of SLN formulations. The negative charge is likely caused by the slightly ionized fatty acids from glycerides. The coating of cationic-charged chitosan altered the surface charge of SLNs (-10.6 mV to -13.3 mV) to c-SLNs (16–17 mV), indicating coating with chitosan on the SLN surface. The low PDI was observed in both ASP and CUR SLNs ranging from 0.19 to 0.21, indicating the homogenous nature of the formulation. The particle size, zeta potential, and PDI remained same before and after lyophilization because mannitol was added as cryoprotectant.
In vitro drug release from the c-SLNs
The ability of nanoparticles to deliver drugs was examined by determining the drug release, as shown in Figure 1. The release of the drug from nanoparticles prepared using stearic acid was conducted over a period of 72 hours. The percentage of drug released from c-SLN was plotted as a function of time. The encapsulation of the drug in c-SLNs significantly prolonged the release. As shown in Figure 1, the release of ASP was faster compared to CUR. A cumulative drug release of ~98% of ASP was observed within 72 hours of the study showing faster release pattern in comparison to the CUR (94%). Both formulations exhibited slow sustained release of the drug.
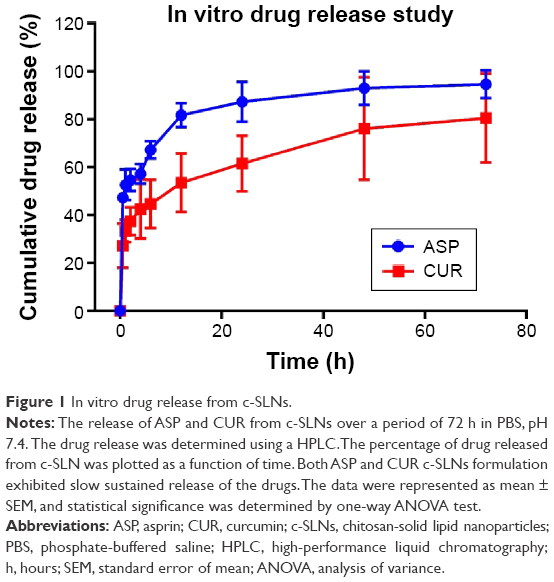 | Figure 1 In vitro drug release from c-SLNs. |
Drug-encapsulated c-SLN stability study
Stability studies of lyophilized c-SLN powder were performed under various storage conditions. In the present work, the effect of lyophilization and temperature storage of c-SLNs on their physical and chemical characteristics was evaluated. For stability study, all the formulations were stored in amber colored bottles at 4°C and 25°C. The accelerated stability study was conducted at 40°C temperature with 65% humidity. The formulations were analyzed for particle size, EE%, zeta potential, and PDI after being stored at specific temperatures for 5 days, 28 days, and 90 days. The effect of storage time and temperature is shown in Table 3.
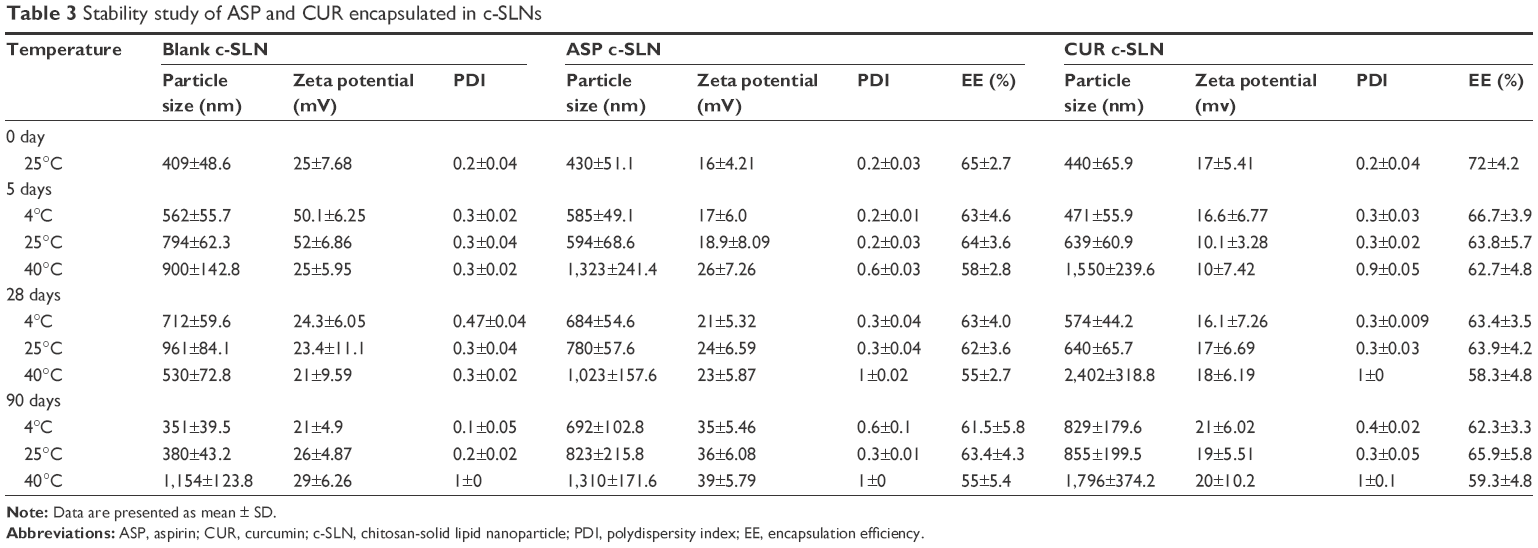 | Table 3 Stability study of ASP and CUR encapsulated in c-SLNs |
Particle size and polydispersity index
Mean particle size of ASP c-SLNs was 430 nm at the start of the study. After 3 months of storage at three different temperatures of 4°C, 25°C, and 40°C, the particle size was observed to be 692 nm, 823 nm, and 1,310 nm, respectively. Similarly, for CUR c-SLNs, the mean particle size was 440 nm at the start of the stability study but at the end of 3 months, the particle size increased to 829 nm, 855 nm, and 1,796 nm at 4°C, 25°C, and 40°C, respectively. Thus, both ASP and CUR c-SLNs exhibited significant increases in mean particle size but were <1,000 nm at lower temperatures (4°C and 25°C). However, some agglomeration was evident at accelerated stability temperature (40°C and 65% humidity), which demonstrated an increase in particle size >1,000 nm (Table 3). The higher temperature and humid conditions of the sample storage may be responsible for the aggregation.39 The PDI values all remained <0.4, which is generally considered the acceptable limit of monodispersity.40 However, increases were observed for accelerated storage conditions at 40°C temperature and 65% humidity.
Zeta potential
Surface potential plays an important role in nanoparticle stability due to electrostatic repulsion. Surface charge is an indicator of the stability of nanoparticles in suspensions. A higher charge on the surface of the c-SLNs will prevent aggregation in buffer solution because of the strong repellent forces among particles.41,42 As a rule of thumb, surface charge values >10 mV or <-10 mV provide good stability.43,44 For ASP c-SLN, the zeta potential was 16 mV at the start of the study. The surface charge values were between 17 mV and 26 mV for 5 days and 28 days of stability study, respectively. After 90 days of storage at 4°C, 25°C, and 40°C, surface charge was increased to 35 mV, 36 mV, and 39 mV, respectively. For CUR c-SLN, surface charge remained constant for 5 days, 28 days, and 90 days at various storage temperatures (Table 3).
Encapsulation efficiency (EE%)
After 3 months of storage at various temperatures, the EE% of ASP c-SLNs was found to be 61.5% (4°C), 63.4% (25°C), and 55% (40°C). This compared well to the EE% at the start of the study (65%), although at accelerated stability conditions (40°C and 65% humidity), there appeared to be a significant decrease as shown previously. For CUR c-SLNs, EE% was measured at 72% at the start of the study, but after 3 months storage at different temperatures, EE% values were decreased to 62.3% (4°C), 65.9% (25°C), and 59.3% (40°C; Table 3). In both formulations, encapsulation seemed to be adversely affected by storage conditions at 40°C temperature and 65% humidity.
Results of these studies demonstrated that storage at higher temperatures of 40°C and humidity of 65% could result in increase in particle size and reduction in encapsulation efficiency; however, at lower temperatures, encapsulation efficiencies were comparable to the original formulations, especially for ASP c-SLNs.
3-day and 28-day in vivo toxicity study
Acute toxicity tests study the probability of the test compounds to cause toxicity following short duration, whereas subacute toxicity tests evaluate the probability of drugs to generate collective hematological, biochemical, and physiological changes following multiple administrations. In toxicity studies, the hematological data are very important observations because it directly reflects possible tissue toxicity or injury. The ACS c-SLNs were orally administered to male and female BALB/c mice at the doses of low (2+4.5+0.16 mg/kg), medium (20+45+1.6 mg/kg), and high (60+135+4.8 mg/kg) for 3 days, 28 days, and 90 days. During the entire course of study, treatment with ACS c-SLNs did not cause any mortality concerns. In addition, there were no significant changes indicated by clinical observation, body weight, hematology, and blood biochemistry except for PLT count (Figures 2 and 3; Table 4; 3-day study data not shown). The PLT count was lower in low-dose and medium-dose groups compared to control group, but PLT count was not changed in the high-dose group. The changes noted in PLT count were not dose dependent, and hence considered incidental changes/biological variations, not as treatment-related effects. Based on the results of this study, the same three doses were used for the 90-day toxicity study (Table 1).
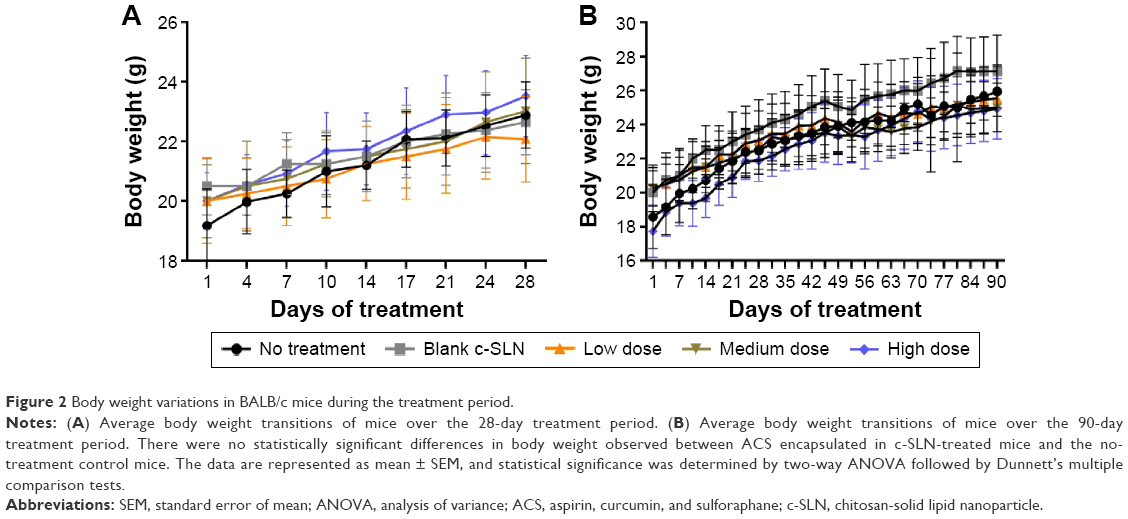 | Figure 2 Body weight variations in BALB/c mice during the treatment period. |
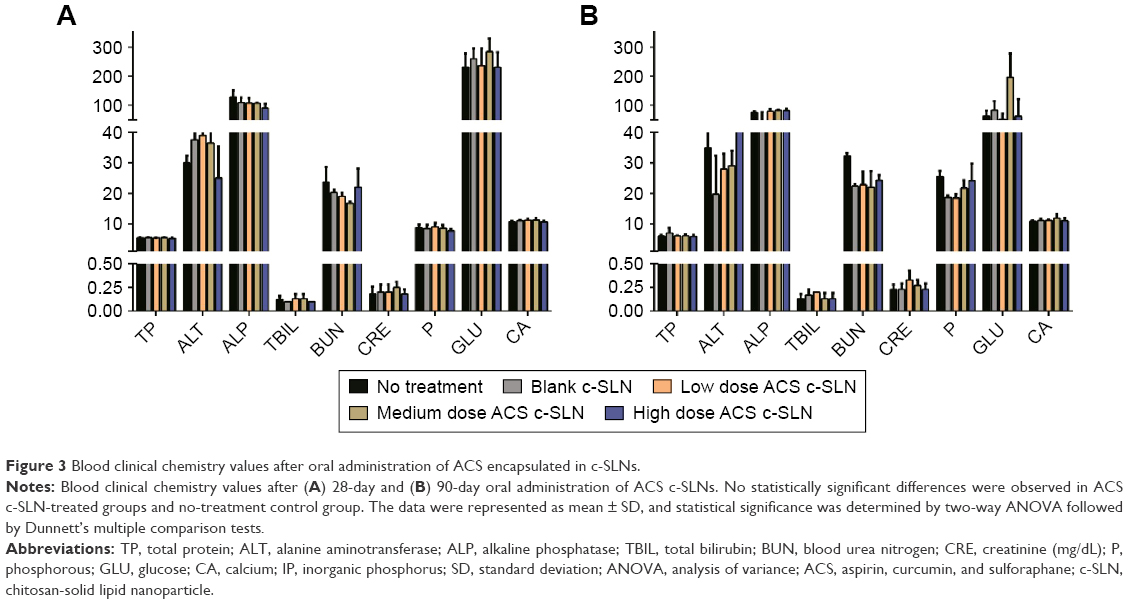 | Figure 3 Blood clinical chemistry values after oral administration of ACS encapsulated in c-SLNs. |
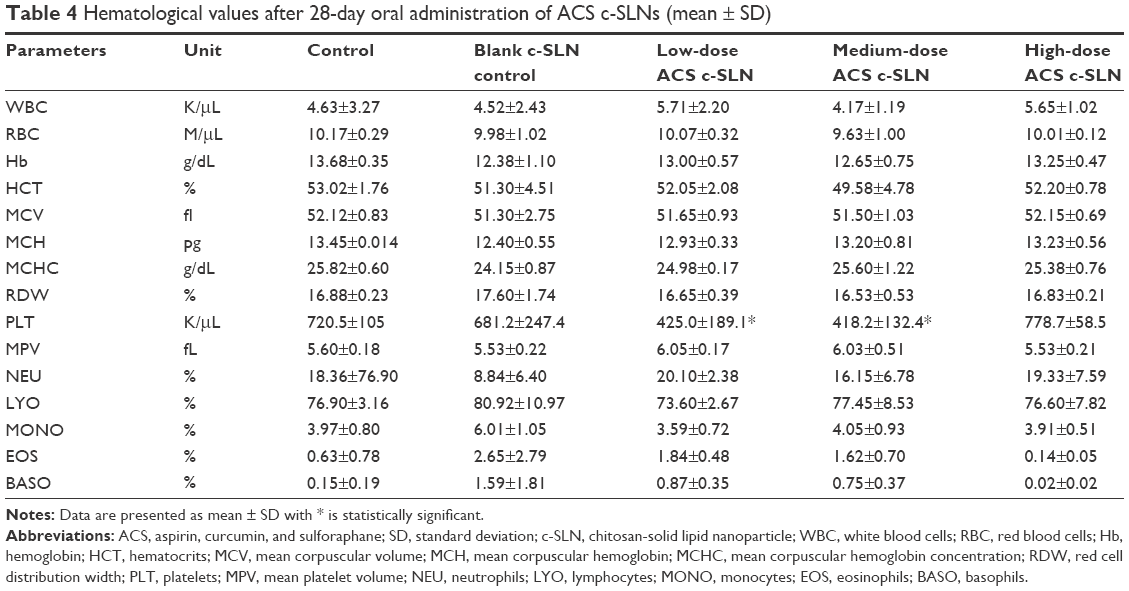 | Table 4 Hematological values after 28-day oral administration of ACS c-SLNs (mean ± SD) |
90-day in vivo toxicity study
Body weight measurement
Body weight gain or loss may be an important measure or indication of an indirect immunotoxic effect. Figure 2 shows body weight of mice treated with different doses of ACS c-SLNs. All mice were weighed twice per week over the study period to observe for signs of any weight loss. Overall, the body weight transition curves of all the mice groups showed a steady gain from start to the end of the study period. Treatment with ACS c-SLN for 90 days did not cause obvious adverse effects on growth because no statistically significant differences in body weight were observed between treatment and control groups. Furthermore, no abnormal clinical signs or behavior were detected in either group. Finally, necropsy at the end of the experiment did not show any macroscopic changes in the organs in any of the treatment groups.
Effect of ACS c-SLN on hematology studies
In this study, hematological observations of all treated mice were conducted to measure any changes in different blood parameters. Results exhibited normal levels of total and differential white blood cells, RBCs, hemoglobin, as well as packed cell volume as compared to untreated control mice (Table 5). Thrombocytopenia, leukopenia, pancytopenia, or anemia was not observed in ACS c-SLN-treated mice. There was an increase in PLT counts in low and medium ACS c-SLN-treated group compared to control group (P<0.01), but no change in PLT count in high-dose treatment group compared to control group was noted. Also, it is important to note that PLT count was elevated in the blank c-SLN group, thus strongly indicating that changes noted were not dose dependent among treatment groups and hence were considered incidental changes/biological variations and not due to ACS treatment-related effects.
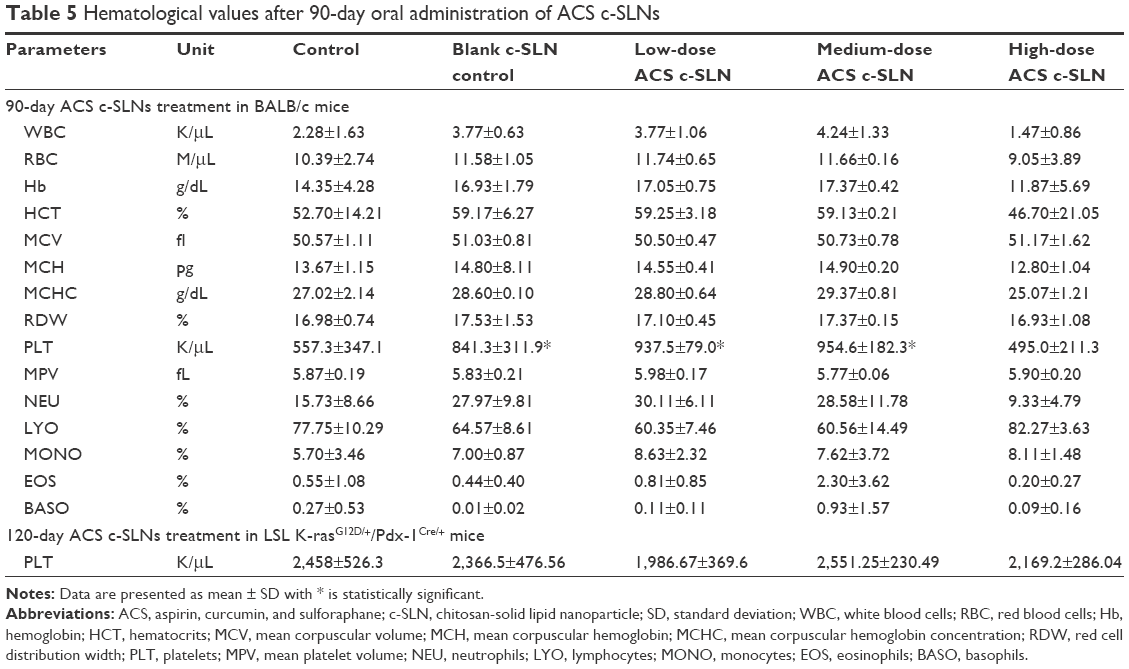 | Table 5 Hematological values after 90-day oral administration of ACS c-SLNs |
To support our hypothesis that changes noted in PLT count were incidental/biological variations, we included PLT count data from ACS c-SLN treatment for 120 days in LSL K-rasG12D/+/Pdx-1Cre/+ (C57BL/6 genetic background) pancreatic cancer transgenic mice. In this study, we did not observe any changes in PLT count in low and medium dose ACS c-SLNs compared to no-treatment control group (Table 5). We conclude that changes in PLT count observed in BALB/c mice were incidental, as we did not observe similar increase in PLT count in LSL K-rasG12D/+/Pdx-1Cre/+ pancreatic cancer transgenic mice.
Effect of ACS c-SLN on clinical chemistry
Enzymes such as ALT and ALP are mainly present in liver cells, and their concentrations increase in the blood with necrosis of liver cells. The amount of these enzymes in the blood corresponds well with the extent of liver damage and are commonly used as indicators of liver function.45–48 In the 90-day study, there were no statistically significant difference in the levels of ALT and ALP in any of the ACS c-SLN treatment group compared to control group, indicating nontoxicity to liver (Figure 3).
TBIL is an organic compound formed by the reticuloendothelial system during the normal and abnormal destruction of RBCs.45 It is a chemical normally present in the blood in small amounts and used by the liver to produce bile. There were no changes in TBIL level after 90 days of ACS c-SLN treatment (Figure 3).
BUN and creatinine (CRE) are important indicators of kidney function. BUN is a product of cellular metabolism, while CRE is a specific product of muscle breakdown. A high level of BUN indicates acute renal dysfunction, and a high level of CRE indicates chronic renal dysfunction. When the serum CRE is significantly higher than normal, kidney function is seriously damaged.49,50 For the ACS c-SLN treatment regimen, there was no significant difference in the levels of BUN and CRE, indicating nontoxicity effects to kidneys (Figure 3).
A significant increase in GLU level was observed in medium-dose ACS c-SLN compared to no treatment control group, but low- and high-dose ACS c-SLNs were within normal range and hence considered as incidental changes/biological variations and not as treatment-related effects. There were no other statistically significant differences between control and treatment groups when compared by two-way ANOVA.
Histopathological examination
Histopathological evaluation was conducted on five major organs, namely pancreas, kidney, heart, liver, and brain for 28-day and 90-day study for blank c-SLN, low-, medium-, and high-dose ACS c-SLNs. Figure 4 represents the H&E-stained images for 90-day treatment regimen. All of the organs evaluated appeared normal in the ACS c-SLN-treated groups (Figure 4). Pancreatic tissue of control and ACS c-SLN groups did not indicate the destruction of pancreatic acinar cells and development of pancreatic fibrosis, an indication of chronic pancreatitis (Figure 4). The toxicity to kidney was evaluated based on the following characteristics: the overall cellularity of the glomerulus, the symmetry of the glomerulus, and the thickness of the capillary walls. The H&E-stained kidney did not show any morphological changes in ACS c-SLN-treated groups compared to control group. The heart sections of ACS c-SLN-treated groups showed no signs of infiltration with leukocytes, fibrosis, hypertrophied, and early signs of necrosis, ie, loss of nuclei and eosinophilia. Toxic change in the liver is characterized by fatty change, pigment accumulation, and proliferation of sinusoidal lining cells. The histopathological assessment of H&E-stained liver sections showed no sign of hepatocyte degeneration, fatty change, oval cell hyperplasia, and pigment deposition, indicating no toxicity to liver (Figure 4). No histopathological, neuronal vacuolation, and necrosis changes were observed in brain sections of low, medium, and high doses of ACS c-SLN (Figure 4). Thus, the microscopic evaluations of the selected organs did not reveal morphological abnormalities that could be attributed to the oral administration of ACS c-SLN to the mice.
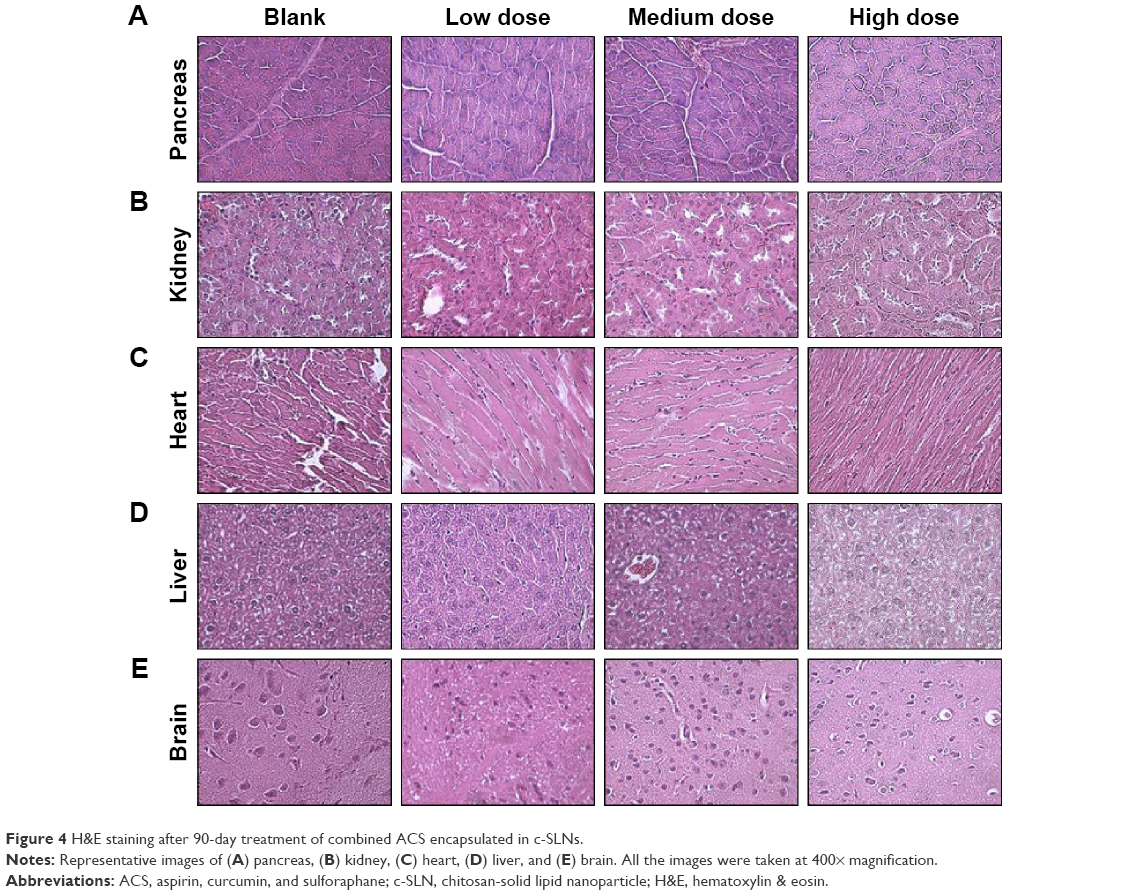 | Figure 4 H&E staining after 90-day treatment of combined ACS encapsulated in c-SLNs. |
In the present study, statistically significant changes were noted in some of the blood parameters in the ACS c-SLN-treated groups. For example, statistically significant increase in PLT counts was noted in the low- and medium-dose treatment groups, but no change in the high-dose group. This dose-independent increase in PLT counts is difficult to explain. In the absence of changes in other blood parameters, or blood clinical chemistry observations, increase in PLT counts is generally not considered a cause of concern. There was significant increase in GLU level in medium-dose group, but no change in low- and high-dose groups. The previously described and other statistically significant changes noted in hematology and serum chemistry parameters were considered as incidental and not related to treatment, as they lacked dose response and were not supported by any other changes in related clinical parameters or histopathological observations. No such changes are observed in our current long-term chemoprevention efficacy study.
Conclusion
Previous and ongoing studies have established the chemopreventive potential of the ACS c-SLN combined regimens, but no studies have been conducted to evaluate the toxicity of this novel regimen. Thus, the objective of this study was to establish toxicity profiles, if any, associated with the combined administration of ACS c-SLNs in mice. Overall, the animals exhibited signs of good health and well-being throughout the study. There was no significant change in the body weight in treatment groups compared to the control group. During the 3-day, 28-day, and 90-day study periods, treatment with ACS c-SLNs did not cause an alteration in blood count. There were no statistically significant changes in blood biochemistry data related to administration of ACS c-SLN. A histopathology assessment of the pancreas, kidney, heart, liver, and brain were correlated with the hematological count and plasma levels of the biochemical markers. The results indicate the oral safety of the developed formulation for short-term and prolonged administration. Thus, a safe toxicological profile of the nanoparticle regimen supports their potential evaluation in in vivo efficacy models and further in human trials to establish their clinical benefits as an effective prevention therapy against pancreatic cancer.
Acknowledgments
This work was supported by a R15 grant from the National Institute of Health, USA (1 R15 CA182834-01A1) to SP. The abstract of this paper was presented at the AACR 106th Annual Meeting 2015 as a poster presentation with interim findings. The poster’s abstract was published in “Supplements section” in Cancer Research Journal: Cancer Res August 1, 2015 75;2621; http://cancerres.aacrjournals.org/content/75/15_Supplement/2621.abstract.
Disclosure
The authors report no conflicts of interests in this work.
References
Lazarou J, Pomeranz BH, Corey PN. Incidence of adverse drug reactions in hospitalized patients: a meta-analysis of prospective studies. JAMA. 1998;279(15):1200–1205. | ||
Einarson TR. Drug-related hospital admissions. Ann Pharmacother. 1993;27(7–8):832–840. | ||
Scripture CD, Figg WD. Drug interactions in cancer therapy. Nat Rev Cancer. 2006;6(7):546–558. | ||
Wynn RL. Erythromycin and ketoconazole (Nizoral) associated with terfenadine (Seldane)-induced ventricular arrhythmias. Gen Dent. 1993;41(1):27–29. | ||
Wysowski DK, Bacsanyi J. Cisapride and fatal arrhythmia. N Engl J Med. 1996;335(4):290–291. | ||
Michalets EL, Williams CR. Drug interactions with cisapride: clinical implications. Clin Pharmacokinet. 2000;39(1):49–75. | ||
Wysowski DK, Corken A, Gallo-Torres H, Talarico L, Rodriguez EM. Postmarketing reports of QT prolongation and ventricular arrhythmia in association with cisapride and food and drug administration regulatory actions. Am J Gastroenterol. 2001;96(6):1698–1703. | ||
Juurlink DN, Mamdani M, Kopp A, Laupacis A, Redelmeier DA. Drug-drug interactions among elderly patients hospitalized for drug toxicity. JAMA. 2003;289(13):1652–1658. | ||
Wakasugi H, Yano I, Ito T, et al. Effect of clarithromycin on renal excretion of digoxin: interaction with P-glycoprotein. Clin Pharmacol Ther. 1998;64(1):123–128. | ||
Brown BA, Wallace RJ Jr, Griffith DE, Warden R. Clarithromycin-associated digoxin toxicity in the elderly. Clin Infect Dis. 1997;24(1):92–93. | ||
Guerriero SE, Ehrenpreis E, Gallagher KL. Two cases of clarithromycin-induced digoxin toxicity. Pharmacotherapy. 1997;17(5):1035–1037. | ||
Grandhi BK, Thakkar A, Wang J, Prabhu S. A novel combinatorial nanotechnology-based oral chemopreventive regimen demonstrates significant suppression of pancreatic cancer neoplastic lesions. Cancer Prev Res. 2013;6(10):1015–1025. | ||
Vauthier C, Dubernet C, Chauvierre C, Brigger I, Couvreur P. Drug delivery to resistant tumors: the potential of poly(alkyl cyanoacrylate) nanoparticles. J Control Release. 2003;93(2):151–160. | ||
Ramalingam P, Ko YT. Enhanced oral delivery of curcumin from N-trimethyl chitosan surface-modified solid lipid nanoparticles: pharmacokinetic and brain distribution evaluations. Pharm Res. 2015;32(2):389–402. | ||
Luo Y, Teng Z, Li Y, Wang Q. Solid lipid nanoparticles for oral drug delivery: chitosan coating improves stability, controlled delivery, mucoadhesion and cellular uptake. Carbohydr Polym. 2015;122:221–229. | ||
Arya G, Vandana M, Acharya S, Sahoo SK. Enhanced antiproliferative activity of Herceptin (HER2)-conjugated gemcitabine-loaded chitosan nanoparticle in pancreatic cancer therapy. Nanomedicine. 2011;7(6):859–870. | ||
Roy K, Mao HQ, Huang SK, Leong KW. Oral gene delivery with chitosan – DNA nanoparticles generates immunologic protection in a murine model of peanut allergy. Nat Med. 1999;5(4):387–391. | ||
Maezaki Y, Tsuji K, Nakagawa Y, et al. Hypocholesterolemic effect of chitosan in adult males. Biosci Biotechnol Biochem. 1993;57(9):1439–1444. | ||
Chen MC, Mi FL, Liao ZX, et al. Recent advances in chitosan-based nanoparticles for oral delivery of macromolecules. Adv Drug Deliv Rev. 2013;65(6):865–879. | ||
Yuan H, Jiang SP, Du YZ, Miao J, Zhang XG, Hu FQ. Strategic approaches for improving entrapment of hydrophilic peptide drugs by lipid nanoparticles. Colloids Surf B Biointerfaces. 2009;70(2):248–253. | ||
Sakloetsakun D, Perera G, Hombach J, Millotti G, Bernkop-Schnürch A. The impact of vehicles on the mucoadhesive properties of orally administrated nanoparticles: a case study with chitosan-4-thiobutylamidine conjugate. AAPS PharmSciTech. 2010;11(3):1185–1192. | ||
Fonte P, Andrade F, Araújo F, Andrade C, Neves J, Sarmento B. Chitosan-coated solid lipid nanoparticles for insulin delivery. Methods Enzymol. 2012;508:295–314. | ||
Cole ET, Scott RA, Connor AL, et al. Enteric coated HPMC capsules designed to achieve intestinal targeting. Int J Pharm. 2002;231(1):83–95. | ||
Sutaria D, Grandhi BK, Thakkar A, Wang J, Prabhu S. Chemoprevention of pancreatic cancer using solid lipid nanoparticulate delivery of a novel aspirin, curcumin and sulforaphane drug combination regimen. Int J Oncol. 2012;41(6):2260–2268. | ||
Thakkar A, Sutaria D, Grandhi BK, Wang J, Prabhu S. The molecular mechanism of action of aspirin, curcumin and sulforaphane combinations in the chemoprevention of pancreatic cancer. Oncol Rep. 2013;29(4):1671–1677. | ||
Antithrombotic Trialists’ (ATT) Collaboration, Baigent C, Blackwell L, et al. Aspirin in the primary and secondary prevention of vascular disease: collaborative meta-analysis of individual participant data from randomised trials. Lancet. 2009;373(9678):1849–1860. | ||
Ajani UA, Ford ES, Greenland KJ, Giles WH, Mokdad AH. Aspirin use among U.S. adults: behavioral risk factor surveillance system. Am J Prev Med. 2006;30(1):74–77. | ||
Lanas A. Gastrointestinal bleeding associated with low-dose aspirin use: relevance and management in clinical practice. Expert Opin Drug Saf. 2011;10(1):45–54. | ||
Cayla G, Collet JP, Silvain J, Thiefin G, Woimant F, Montalescot G. Prevalence and clinical impact of upper gastrointestinal symptoms in subjects treated with low dose aspirin: the UGLA survey. Int J Cardiol. 2012;156(1):69–75. | ||
Sikora E, Scapagnini G, Barbagallo M. Curcumin, inflammation, ageing and age-related diseases. Immun Ageing. 2010;7(1):1. | ||
Cheng AL, Hsu CH, Lin JK, et al. Phase I clinical trial of curcumin, a chemopreventive agent, in patients with high-risk or pre-malignant lesions. Anticancer Res. 2001;21(4B):2895–2900. | ||
Dhillon N, Aggarwal BB, Newman RA, et al. Phase II trial of curcumin in patients with advanced pancreatic cancer. Clin Cancer Res. 2008;14(14):4491–4499. | ||
Noh JR, Kim YH, Hwang JH, et al. Sulforaphane protects against acetaminophen-induced hepatotoxicity. Food Chem Toxicol. 2015;80:193–200. | ||
Philbrook NA, Winn LM. Sub-chronic sulforaphane exposure in CD-1 pregnant mice enhances maternal NADPH quinone oxidoreductase 1 (NQO1) activity and mRNA expression of NQO1, glutathione S-transferase, and glutamate-cysteine ligase: potential implications for fetal protection against toxicant exposure. Reprod Toxicol. 2014;43:30–37. | ||
Jiao Z, Zhang Q, Chang J, et al. A protective role of sulforaphane on alveolar epithelial cells exposed to cigarette smoke extract. Exp Lung Res. 2013;39(9):379–386. | ||
Sze A, Erickson D, Ren L, Li D. Zeta-potential measurement using the Smoluchowski equation and the slope of the current-time relationship in electroosmotic flow. J Colloid Interface Sci. 2003;261(2):402–410. | ||
OECD. Test No 407: Repeated Dose 28-day Oral Toxicity Study in Rodents. OECD Publishing. Paris. | ||
OECD. Test No 408: Repeated Dose 90-Day Oral Toxicity Study in Rodents. OECD Publishing. Paris. | ||
de Castro MD, Izquierdo A. Lyophilization: a useful approach to the automation of analytical processes? J Automat Chem. 1990;12(6):267–279. | ||
Yen FL, Wu TH, Tzeng CW, Lin LT, Lin CC. Curcumin nanoparticles improve the physicochemical properties of curcumin and effectively enhance its antioxidant and antihepatoma activities. J Agric Food Chem. 2010;58(12):7376–7382. | ||
Sawant KK, Dodiya SS. Recent advances and patents on solid lipid nanoparticles. Recent Pat Drug Deliv Formul. 2008;2(2):120–135. | ||
Kedar U, Phutane P, Shidhaye S, Kadam V. Advances in polymeric micelles for drug delivery and tumor targeting. Nanomedicine. 2010;6(6):714–729. | ||
Wissing SA, Kayser O, Müller RH. Solid lipid nanoparticles for parenteral drug delivery. Adv Drug Deliv Rev. 2004;56(9):1257–1272. | ||
Jacobs C, Kayser O, Müller RH. Nanosuspensions as a new approach for the formulation for the poorly soluble drug tarazepide. Int J Pharm. 2000;196(2):161–164. | ||
Dufour DR, Lott JA, Nolte FS, Gretch DR, Koff RS, Seeff LB. Diagnosis and monitoring of hepatic injury. II. Recommendations for use of laboratory tests in screening, diagnosis, and monitoring. Clin Chem. 2000;46(12):2050–2068. | ||
Amacher DE. A toxicologist’s guide to biomarkers of hepatic response. Hum Exp Toxicol. 2002;21(5):253–262. | ||
Ozer J, Ratner M, Shaw M, Bailey W, Schomaker S. The current state of serum biomarkers of hepatotoxicity. Toxicology. 2008;245(3):194–205. | ||
Zhang XD, Wu D, Shen X, et al. Size-dependent in vivo toxicity of PEG-coated gold nanoparticles. Int J Nanomedicine. 2011;6:2071–2081. | ||
Miller WG, Myers GL, Ashwood ER, et al. Creatinine measurement: state of the art in accuracy and interlaboratory harmonization. Arch Pathol Lab Med. 2005;129(3):297–304. | ||
Rosner MH, Bolton WK. Renal function testing. Am J Kidney Dis. 2006;47(1):174–183. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.