Back to Archived Journals » Research and Reports in Endocrine Disorders » Volume 6
Prader–Willi syndrome: clinical problems in transition from pediatric to adult care
Authors Crinò A, Fintini D, Bocchini S, Carducci C, Grugni G ![]()
Received 29 February 2016
Accepted for publication 12 May 2016
Published 18 July 2016 Volume 2016:6 Pages 49—57
DOI https://doi.org/10.2147/RRED.S56401
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Mingzhao Xing
Antonino Crinò,1 Danilo Fintini,1 Sarah Bocchini,1 Chiara Carducci,1 Graziano Grugni,2
1Autoimmune Endocrine Diseases Unit, Bambino Gesù Children’s Hospital, Research Institute, Palidoro, Rome, 2Division of Auxology, Italian Auxological Institute, Research Institute, Piancavallo, Verbania, Italy
Abstract: Prader–Willi syndrome (PWS) represents the most common form of genetic obesity. Thanks to the advances in medical care and technology, many persons with PWS live longer and survive to adulthood. Currently, because of the many physical and behavioral manifestations, transitional health care is not easy for these patients and is considered a very important issue. Moreover, very few studies have examined these transitional problems in young adults with PWS. In recent years, there has been great interest in improving transition planning and support for young people with PWS reaching adulthood. In this article, we underline the main clinical problems in transition and give some advice to make this period less difficult and easier for adolescents with PWS. Special attention should be paid to obesity, diabetes mellitus, hypertension, osteoporosis, and sleep apnea during the period of transition. In PWS, for an effective transition from childhood to adulthood, a multidisciplinary team is needed, and should maintain the same approach to food, environment, and psychiatric issues. For comprehensive care, it is necessary to involve adult endocrinologists and other medical specialists in conjunction with the pediatric team. Parental involvement is, however, a great help for supervising adolescents with PWS during this particular period.
Keywords: Prader–Willi, transition period, intellectual disabilities
Introduction
Prader–Willi syndrome (PWS) is a complex multisystem genetic disorder that occurs in ∼1:10,000–1:30,000 live births and is considered the most frequent known genetic cause of obesity.1 PWS results from the loss of expression of paternally derived genes in the PWS critical region on chromosome 15q11-q13.2 Approximately 65%–70% of the cases are due to deletion of this region, 20%–30% are due to maternal uniparental disomy (UPD) of chromosome 15, and most of the remaining 2%–5% are due to an imprinting center defect.2,3 More recently, a significant change in the distribution of molecular mechanisms over time has been observed, with a larger proportion of UPD.4 The syndrome is typically characterized by neonatal hypotonia, behavioral problems, cognitive impairment, dysmorphic features, hypogonadism, short stature, early development of hyperphagia with food-seeking behavior, and progressive development of severe obesity unless eating is not promptly restricted.5 Obesity in these patients can be present early in childhood. A complex hypothalamic–pituitary dysregulation is currently thought to be partly responsible for the PWS phenotype, including the lack of satiety, short stature, and various endocrine abnormalities (ie, hypogonadism, growth hormone [GH] deficiency, central hypothyroidism, central adrenal insufficiency [CAI]). It is interesting to note that the clinical features of PWS differ significantly during the life span, becoming more evident in adulthood.3
Accumulation of body fat in subjects with PWS shows distinct phenotypic and metabolic characteristics that are not common to severe obesity itself. PWS harbors a higher fat mass than simple obesity, under the same degree of weight excess, both in children and in adults. Unlike in obesity, visceral fat deposits are reduced, and insulin sensitivity is higher.6,7
In spite of these metabolic benefits, PWS patients often die at an early age because of complications usually linked to obesity, which include respiratory problems, heart disease, and type 2 diabetes mellitus (T2DM).8,9 Choking (mainly during bouts of heavy eating) and stomach rupture or necrosis of tissue have also been described.10 However, by increasing the surveillance regarding weight and prevention of obesity, today PWS patients can hopefully live longer.9 In this context, the precocious use of GH gives an excellent advantage in reducing morbidity and modifies the profile of the disease in childhood, in teenage years, and in adulthood.5
The diagnosis of PWS can be confirmed very early during the neonatal period thanks to development of genetic testing. All three of the genetic abnormalities (deletion, UPD15, and imprinting center defect) may be detected by methylation analysis. However, high-resolution karyotyping is always necessary at all times in order to recognize the cases with translocations or other rare rearrangements that involve chromosome 15.3,11
The transition period
The transition term defines the management of chronic patients who pass the pediatric age and become adults. In other words, the transition represents the passage from pediatric health care, where the child is totally dependent on the parents, to that of adulthood, in which the subject should become independent.11
Therefore, the transition period is the span of time in which people complete their somatic and psychological development after reaching their full stature (also known as late teenage years, post adolescence, or young adulthood). It starts in late puberty and ends at ∼25 years of age, thus lasting from 3 years to 10 years. It includes hormonal and many lifestyle changes that lead to a different perception of the self and one’s capacity to relate to others.12
The purpose of transition is to offer patients a continuity of care, promote mental and physical health, and optimize the diagnostic and therapeutic protocols through the exchange of information among health care workers in order to better their skills and improve their quality of life.12
Clinical and endocrine problems during transition periods in PWS
The clinical features of PWS during the transition period depend on the treatment carried out during childhood and adolescence and therefore is characterized by great variability (Table 1).11,13
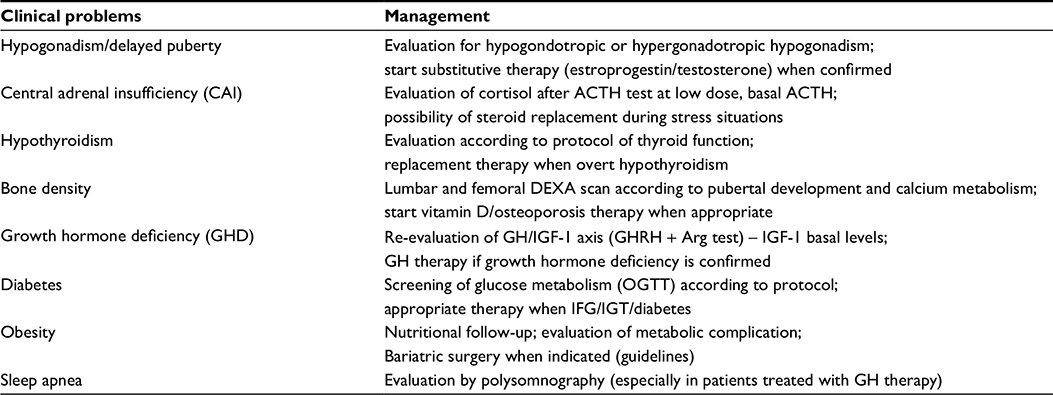 | Table 1 Clinical and endocrine problems during transition period in Prader–Willi Syndrome Abbreviations: ACTH, adrenocorticotropic hormone; Arg, arginine; DEXA, dual-energy X-ray absorptiometry; IFG, impaired fasting glucose; IGF-1, insulin-like growth factor-1; GH, growth hormone; GHRH, growth hormone-releasing hormone; OGTT, oral glucose tolerance test; IGT, impaired glucose tolerance. |
This period is usually characterized by ferocious bouts of hunger with compulsive eating, which leads to morbid obesity if caloric intake is uncontrolled. T2DM and impaired glucose tolerance frequently appear during or soon after puberty, affecting ∼10%–25% of these individuals.14 PWS patients with T2DM have a higher body weight and a greater probability of positive family history of diabetes. Hypertension is also quite common.
By adolescence, behavior and learning problems characteristically would have evolved as major issues for patients and their families. Adolescents with PWS have been described as stubborn, instinctive, manipulative, irritable, mood-labile, angry, persistent, egocentric, demanding, and prone to rage episodes when frustrated. Behavior and learning difficulties become more evident, and in particular temper tantrums and obsessive/compulsive behavior can negatively affect both the school and home environment. Psychiatric problems, such as anxiety, aggression, and perseveration, frequently revolve around food issues. Especially in persons with UPD, psychosis is frequent.3,15
Scoliosis is often more frequent at this age, and in some cases needs surgical treatment. A variable prevalence of scoliosis has been reported in PWS, ranging from 30% to 80% depending on age.16,17 Approximately 15% of patients with scoliosis develop severe curves, requiring bracing or surgery. Clinical detection can be challenging, because these patients are obese and have less vertebral rotation than seen in other patients with scoliosis curves of a similar size, leading to diagnostic delays. Marked generalized muscle hypotonia might influence the development and the progression of scoliosis. The role of GH therapy in development and progression of scoliosis has not been demonstrated yet.16
Sleep disorders (obstructive and/or central or mixed apnea), hypoventilation, significant oxygen desaturation, and frequent waking up during the night are particularly common but are not always related to obesity. Excessive daytime sleepiness is a very frequent problem in PWS patients, and almost 70%–100% of adults with PWS suffer from this problem. Young adult with PWS often fall asleep when they are inactive. Many of these patients have polisomnographic features of sleep-disordered breathing including the sleep apnea syndrome. Moreover, some of them need noninvasive ventilation.5
Skin-picking, in combination with reduced pain sensitivity and decreased gastrointestinal motility, is more frequently observed with increasing age.11
Tooth decay is frequent and can be related to the viscous saliva.3,11
Particularly in the transition period, PWS is associated with several endocrine disturbances, which should be carefully monitored and treated.
Hypogonadism with incomplete sexual development and delayed puberty is usually present in PWS. Especially in males, hypogonadism is multiform, ranging from severe gonadotropin deficiency to primary gonadal failure. A combined form of hypogonadism (of testicular and central origin) can be also present in these patients.18 The degree of hypogonadism is variable and is less severe in females than in males.19 Males only occasionally show secondary characteristics and voice change, male body habitus, or significant facial and body hair. Testes are usually very small with a volume of <6–7 mL, and pubertal stage rarely progresses over the Tanner genital stages 2–3. In females, usually menarche can occur spontaneously between the ages of 10 years and 38 years, although primary amenorrhea is reported in the majority of the patients older than 18 years, and oligomenorrhea is present in the rest of the patients. Regular menses are very rare. Fertility has not been reported in PWS men, while a few spontaneous pregnancies in females have been described. In spite of their hypogonadism, adolescents and adults with PWS show a strong interest in sexual and romantic thoughts and activities. Furthermore, in both sexes sex-steroid treatment might be beneficial.20
Available data indicate that some degree of CAI may be part of the PWS phenotype in the transitional period, although clinically relevant adrenal failure in PWS subjects appears to be rare.21 CAI has recently been hypothesized to be responsible of increased risk of sudden death in PWS, particularly during episodes of stress and infections. There are a number of features that support the view that CAI may form part of the PWS spectrum: the well-known hypothalamic dysfunction with multiple hormone deficiencies in PWS consistent with hypothalamic–hypophyseal brain imaging and histological anomalies; the observation of an altered adrenocorticotropic hormone (ACTH) adrenal response in some PWS children; the autopsy finding of hypoplastic adrenal glands in PWS patients dying suddenly; and the description of a previous death case with a proven poor cortisol response after an ACTH test.21,22
In any case, hydrocortisone treatment should be considered when clinically indicated and confirmed by hormonal tests.
In our clinical practice, we evaluate in all PWS patients the adrenal function by ACTH test at low dose (1 μg iv) and the families are advised of the possible adrenal insufficiency when patients are under stress. Therefore, the patients are given stress doses of hydrocortisone to be kept at home and to be taken when necessary, and we recommend the administration of a perioperative stress dose of steroids during surgery.21
Hypothyroidism is not common in PWS, but thyroid-stimulating hormone and thyroid hormones should be measured regularly. Similar to other endocrinopathies in PWS, the etiology of hypothyroidism is believed to be central in origin. Studies in PWS adult patients have shown that the frequency of thyroid diseases is ∼2%, which is almost similar to that of the general population.23 Studies investigating the thyroid gland disorders in the transition period of PWS are also needed.24
Osteoporosis is quite frequent and related to sex-steroid and GH deficiency, as well as to low muscular activity or deficient intake of calcium and vitamin D. To our knowledge, no guideline is currently available for diagnosis and follow-up of bone density in PWS adult subjects, especially in the transitional period.25,26 Therefore, osteoporosis seems to be underestimated and underdiagnosed, and a high risk of fractures is described in PWS adults. Consequently, regular assessment of hip and lumbar spine bone mineral density by dual-energy X-ray absorptiometry is recommended, particularly in the transition phase. However, the major difficulty in the management of these patients from pediatric to adult care is to establish in whom and when a clinical intervention has to be made to improve bone mineral density.27 A supplement with calcium and vitamin D is often necessary especially in patients on a restrictive diet and in the absence of substitution therapy with sex-steroid hormones. Finally, in order to safeguard bone health in PWS subjects, it seems useful to establish the appropriate timing of dual-energy X-ray absorptiometry evaluation in the transition period, as well as to perform a long-term follow-up study on fracture risk in these patients.
Most of the children with PWS have GH deficiency.22,28 In addition, previous reports support the hypothesis of an age-dependent derangement of the hypothalamus–pituitary axis occurring in PWS subjects, also regarding GH secretion. In fact, there is evidence of GH insufficiency after reaching final height, although study findings differ largely on the prevalence of this clinical feature in the adult.
Impaired GH secretion in PWS adults, however, is revealed not only by deficits in secretion amplitude parameters but also by abnormalities in the shape of the secretory response, as demonstrated by deconvolution analysis.29
GH replacement therapy in adult age improves body composition, increases bone mineral density, improves endurance in physical exercise and quality of life, and has an encouraging impact on development and behavior. GH is effective in keeping body proportions and metabolic balance during the transition period.30
Data about GH secretion through the transitional period into adulthood in PWS, however, are scarce, and controlled studies of continuous treatment in this period are not available.31 In this context, it has been reported that GH replacement therapy in PWS adults, including patients in the transitional period, causes a decrease in body adiposity and an increase in lean body mass, without changes in the body mass index.32 Similarly, long-term GH treatment was able to increase muscle strength and exercise tolerance.33 On the other hand, cessation of GH therapy in young PWS patients worsened the body mass index.34 Improvement in cognitive and motor performance tests during GH therapy, using a battery of neuropsychological tests, was found in 19 PWS patients with a median age of 25 years.35
Altogether, these nongrowth-promoting effects of GH should be considered essential for body homeostasis, and therefore PWS patients need lifelong GH therapy. Thus, a re-evaluation of the GH–insulin-like growth factor-1 (IGF-1) axis may be useful in all PWS adolescents reaching the transitional period. As such, reports from dynamic testing in adults suggest that GH deficiency (GHD) is not the rule as previously reported. Therefore, in many countries it is necessary to carry out tests before treating adult patients with GHD because of the reimbursement costs involved in health insurance for GH therapy. To our knowledge, criteria for determining GHD in PWS during the transitional phase are present only in Italy, and they are defined with one of the following: 1) three or more pituitary hormone deficiencies, or 2) a peak GH level after growth hormone-releasing hormone-arginine test of <4.1 μg/L after a period of rest for at least 1 month before retesting.
Data regarding the optimal dose in GH replacement for PWS during the period of transition is not available. Accordingly, with the indications for PWS adults, it seems reasonable to suggest a starting dose of 0.1–0.2 mg/d according to age, presence of edema, and concomitant oral estrogen.31 In the same way, a subsequent titration of the doses should be based on clinical response, age, and sex IGF-1 levels in the normal range.
Transition in intellectual disabilities
Transition in intellectual disabilities or other particular diseases, like genetic and metabolic syndromes, is rarely described in the literature.
The progress made in medical health care now allows many children affected by this disease to grow, survive, and reach adulthood.
In general, the principal elements to promote during the transition are high quality and well-coordinated health care without interruptions, and concentrated solely on the patient according to his/her age.12
During childhood, the medical care given is often multidisciplinary, and the patient is always in the care of their pediatrician. Having reached the adulthood, these children and their families do not find the same type of assistance, so they usually continue to have to visit pediatric centers into adult life, even if the health problems involved are different from those of pediatric age.36 Pediatricians should transfer their young patients from the pediatric center to general practitioners (GPs), who often are not familiar with the problems involved. So, in these cases the GPs advise their patients to visit specific specialists.37
This could involve endless hospital visits with various physicians and segmented health care, with no real coordination and scarcity of care management.38
In the past, people with disabilities lived mostly in segregated residential institutions, but today most of them live within their local community with their families or in community-integrated residential facilities supported by professionals. Thanks to these facilities, patients are given coordinated health care by GPs.37 It is important to establish that all patients with special health care needs should obtain medical assistance for transition.
A general plan of transition should include the following:
- A close cooperation between the pediatric case manager and the adult case manager.
- The drawing-up of a plan to help in the transfer of health care between pediatric and adult health care professionals.
- The drafting of a brief and clear medical history to facilitate the passing of the information.
- The drawing-up of a written transition plan, which should be made up and carried out with the help of both the patients and their families.39
Transition in PWS
An integrated multidisciplinary approach starting from the neonatal period is mandatory to ensure optimal management to improve quality of life of PWS patients throughout their lifetime. For a successful transition from childhood to adulthood in these patients, a multidisciplinary team needs to share clinical information, and should keep the same policy about food, environmental, and psychiatric issues.
However, controlled studies on the transition period in PWS are not available.13
The main aspect to promote the transition is collaboration between all the people involved in the health care of the patient with PWS.
The principal aspects of transition should have the following components:40
- Patient-centered approach
- High-quality health care
- Uninterrupted health care
- Coordinated health care between different specialists
- Development of an appropriate approach.
In this context, we have to underline that adolescents and young adults with PWS can show fairly good cognitive skills, while, on the other hand, some of them have maladaptive behaviors that prevent successful independence.
The difficulty to control food intake and hyperphagia has direct influence on the adult life. In fact, this aspect can be one of the biggest obstacles for an independent life to enjoy social events and for many types of work or tasks.
Consequently, specific syndrome-related problems are to be taken into consideration.
For PWS patients, the transition process must be carefully prepared in a specific period.
- there is a case manager for the transition;
- all the most important points of transition are clearly written down;
- pediatric and adult medical doctors are able to cooperate; and
- there are written guidelines of transition (disease-specific).
In a PWS transition process, the presence of obesity, diabetes, hypertension, heart diseases, osteoporosis, sleep apnea, difficult behavior, and psychiatric problems should always be taken into account.11
Weight reduction during the transitional phase, even if it is very difficult to achieve, remains one of the key issues in preventing morbidity and reducing mortality at a later age.2,8
During the transition phase, it is mandatory to underline the importance of some independence of the patients within a safe or supported environment.
Through the intervention of the national health care service, Parental Associations, and voluntary services, it is possible to carry out a project for the independence or semi-independence of adults with good cognitive abilities.13 However, we must bear in mind that the greater part of younger PWS patients are unable to live independently and frequently fail in small work projects.
Current practice during transition
In practice, the collaboration between two vocational figures is always necessary: pediatric or pediatric endocrinologist (pediatric team) and an adult specialist case manager. Moreover, a close cooperation between Patients’ Association and the GP is appropriate. Pediatricians have very important tasks: they have a deep knowledge of the PWS, and they know the history of the patient very well. For many years, the pediatrician has been responsible for the patient’s health by interacting with other professionals (Figure 1). The adult specialist takes the patient’s responsibility in the post-adolescent period (follow-up, interaction with other specialists). He or she must maintain the information network regarding the disease on a territorial basis and interact with the Patients’ Association. He or she must work together with the pediatrician to know the patient’s previous history. There are no pre-built and standard forms in the transition of various diseases, particularly those regarding PWS in different logistical, structural, and cultural situations.13,37,41
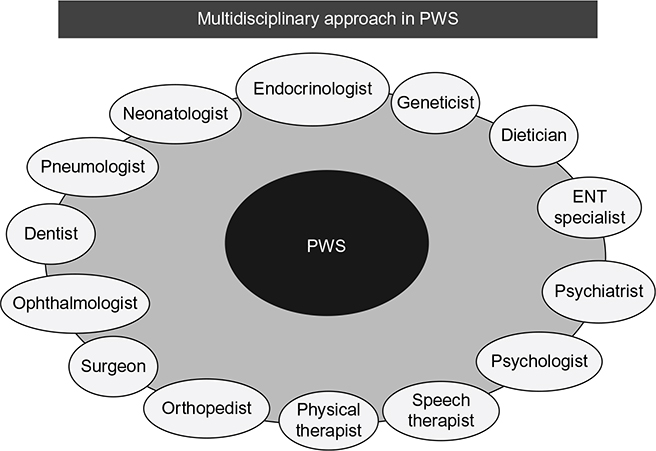 | Figure 1 Ideal multidisciplinary team for Prader–Willi syndrome (PWS) during transition. Abbreviation: ENT, ear-nose-throat. |
A practical model of transition provides a collegial visit together with the pediatrician, the medical team for adults, the patient, and the parents. The aim is to introduce the new medical staff who will follow the patient in adult life. At the same time, there should be the presentation of clinical problems and the setting of the future welfare-therapeutic programs. Planning should begin well before the patient reaches the period of transition and should take medical problems and learning disabilities into account (Figure 2).
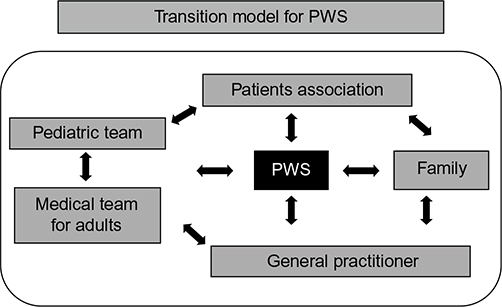 | Figure 2 A practical model of transition for Prader–Willi syndrome (PWS). |
The coordination of the transition will be mostly delegated to the pediatric team that had followed the patient, but at the same time it must work closely with the adult structure.
Unfortunately, transition care varies considerably between the different regions and the various medical facilities available. Therefore, patients and their families are often unable to find a uniform assistance, and this limitation can lead to different outcomes in the clinical development of the PWS patients.40,41
Potential barriers for a successful transition to adult care in PWS
To better analyze the process of transition, it is vital to underline the main barriers of this important process.
The main obstacles in all chronic diseases, including PWS, are the emotional ties not only from the patient’s point of view, but also from the pediatrician’s point of view.
PWS patients and their families very often have a long relationship with their pediatric endocrinologist, so young adults and their parents might be unwilling to leave their pediatrician and they may even refuse to do so. The pediatricians may also find it hard to let their patients and their families go.12
Generally, pediatricians see themselves as experts of childhood-onset diseases and know the multiple disciplines and different aspects involved during the health care process, which also includes the social aspects.42–44
Other barriers include the following:
- lack of institutional support
- lack or specific structures
- lack of specific professionals
- inappropriate planning
- geographic isolation.
The winning strategies that allow better addressing of the barriers of transition are sharing information, consulting meetings, and concurrence between pediatricians and adult caregivers.
All patients who go from pediatric care to adult care must be well prepared for this change.
The transition can take place only at a slow pace, and it is necessary to draw up a written plan in order for this change to come about both for the patients and their families. It is necessary to identify a senior liable person for this transition. However, it appears necessary to give the patient and the family the possibility to discuss the written plan of transition.13
Time allocation for outpatient visits and continuity of professional care between hospital visits may vary between pediatric and the adult units. Young persons with PWS and their families must be informed of these differences and made to acquire the skill to negotiate the transfer and ongoing adult care.
A visit to the adult unit and a key liaison member of staff are useful in this case. Problems of institutional support (eg, the inability to find an adult primary care provider) are significant limiting steps in developing an organized transition plan. Finally, in many countries the barrier of inadequate financing can exist.
The characteristic behavioral problems, more evident after adolescence, make the transition period more difficult with the new professional figures.13,15,38
To summarize, the main barriers to transitional healthcare are as follows:
- Emotional bond between patients, the family, and the pediatric team. Many difficulties can be faced in building up new relationships and in establishing trust in the adult sector.
- Adult PWS patients can present child-like and immature behavior and cognitive impairment. They may still have “pediatric” medical problems (such as pubertal delay and growth retardation) and still need a pediatric approach, the management of which may be unfamiliar to an adult team.
- The pediatric team may also not feel confident in the adult team in the management of the numerous problems of the syndrome.
- The adult team may not be experienced in problems of these patients. They may be less proficient in multiple specialities and less specialized than the pediatric team.
The team might not be interdisciplinary and less in harmony than the pediatric team. Both the conduct and psychiatric problems and the perennial search for food, which is typical of PWS patients, make all these problems very complex. It is hoped that, in the future, the prevalence of morbid and life-threatening obesity in adolescence will continue to decrease as seen in the past.
These benefits, if continued into adulthood, will need extension of comprehensive care and therefore will involve adult endocrinologists together with the aid of pediatricians, psychiatrists, and specialists for those who have intellectual disabilities.42
- There could be difficulties of some patients in entering the hospital’s organizational mechanism of adult patients.
- Psychiatric diseases become more prevalent in adult life, and behavioral problems can create obstacles both for the care and treatment. Therefore, a close collaboration with the psychiatric unit is important.41–43
In adult care, new health care settings become central:
- Autonomy
- Changes due to sexual development
- Psychological evolution
- Social issues
- Work possibilities
- Financial aspects
It is important to underline that the pediatric approach to patients with PWS requires the presence of a case manager, who organizes the overall care of the person. The patient and his family are always the center of their care and needs (Figure 2). The pediatric endocrinologist requires the support and expertise of various specialists, collects their indications, discusses them with the family, and supports the patient in the various stages of the care and growth. The person with PWS is a fragile subject because of the complexity of the medical conditions, disability, intellectual impairment, and psychological and psychiatric issues that emerge later in life. The pediatric approach seems to take this fragility into account. On the contrary, the transition seems to be a risk for the continuation of the care.41
Conclusion
The transitional period is a delicate dynamic process that should be centered on the patient, and should guarantee continuity, coordination, flexibility, and sensitive care according to the guidelines that were established previously. It has to be planned carefully, starting from the pediatric team up to the adult case manager.
Currently, because of many physical and behavioral problems, the transition from childhood into adulthood in PWS is not easy. Frequently, PWS patients are unable to live independently and are inadequate in competitive jobs. Most patients require special care and help throughout their lives. It is possible to find a young PWS adult who possesses good cognitive skill, while, on the other hand, the individual has maladaptive behaviors that are an obstacle for a successful independent existence.
Based on this evidence and the complex clinical picture of these patients, it is clear that a multidisciplinary approach is mandatory for patients with PWS. A multidisciplinary team with experience in the management of PWS can provide the best practice.11 Children with PWS and their families should have the opportunity to have caregivers at hand, and health specialists who can talk to them and make suggestions about the transitional period to adult medical care. We hope that a close collaboration between pediatricians, adolescents, and young adult health care professionals and patients with PWS will be able to provide answers to these problems in the future, and therefore improve the clinical manifestations during the transitional period. It would be a good idea to create a day-care clinic where the pediatric team works together with the medical adult doctors for a better exchange of patient data.
We suggest the implementation of common diagnostic and therapeutic protocols aimed at improving patient care. The creation of specific centers of reference for the diagnosis, follow-up, and treatment of PWS is desirable.
Some of these patients live in special communities that give them the possibilities to eat properly, live in safety, and work and socialize happily. New strategies that accompany PWS patients from childhood to adolescence and to adulthood should be taken into consideration such as the services and organizations like the Prader–Willi Association.
Furthermore, many public and private organizations, as well as many individuals, make it easier for the young adult with PWS to become an almost independent individual.
Finally, more attention should be paid to the development of organized model transition programs for patients with PWS.
Disclosure
The authors declare no conflicts of interest in this work.
References
Lionti T, Reid SM, White SM, Rowell MM. A population-based profile of 160 Australians with Prader-Willi syndrome: trends in diagnosis, birth prevalence and birth characteristics. Am J Med Genet A. 2015;167A(2):371–378. | ||
Butler MG, Manzardo AM, Forster JL. Prader-Willi syndrome: clinical genetics and diagnostic aspects with treatment approaches. Curr Pediatr Rev. 2016;12(2):136–166. | ||
Cassidy SB, Schwartz S, Miller JL, Driscoll DJ. Prader-Willi syndrome. Genet Med. 2012;14(1):10–26. | ||
Cho SY, Ki CS, Sohn YB, et al. The proportion of uniparental disomy is increased in Prader-Willi syndrome due to an advanced maternal childbearing age in Korea. J Hum Genet. 2013;58(3):150–154. | ||
Angulo MA, Butler MG, Cataletto ME. Prader-Willi syndrome: a review of clinical, genetic, and endocrine findings. J Endocrinol Invest. 2015;38(12):1249–1263. | ||
Brambilla P, Crinò A, Bedogni G, et al; Genetic Obesity Study Group of the Italian Society of Pediatric Endocrinology and Diabetology (ISPED). Metabolic syndrome in children with Prader-Willi syndrome: the effect of obesity. Nutr Metab Cardiovasc Dis. 2011;21(4):269–276. | ||
Grugni G, Crinò A, Bedogni G, et al. Metabolic syndrome in adult patients with Prader-Willi syndrome. Nutr Metab Cardiovasc Dis. 2013;23(11):1134–1140. | ||
Einfeld SL, Kavanagh SJ, Smith A, Evans EJ, Tonge BJ, Taffe J. Mortality in Prader-Willi syndrome. Am J Ment Retard. 2006;111(3):193–198. | ||
Grugni G, Crinò A, Bosio L, et al; Genetic Obesity Study Group of Italian Society of Pediatric Endocrinology and Diabetology (ISPED). The Italian National Survey for Prader-Willi syndrome: an epidemiologic study. Am J Med Genet A. 2008;146(7):861–872. | ||
Stevenson DA, Heinemann J, Angulo M, et al. Gastric rupture and necrosis in Prader-Willi syndrome. J Pediatr Gastroenterol Nutr. 2007;45(2):272–274. | ||
Goldstone AP, Holland AJ, Hauffa BP, Hokken-Koelega AC, Tauber M; speakers contributors at the Second Expert Meeting of the Comprehensive Care of Patients with PWS. Recommendations for the diagnosis and management of Prader-Willi syndrome. J Clin Endocrinol Metab. 2008;93(11):4183–4197. | ||
McDonagh JE, Kelly DA. Transitioning care of the pediatric recipient to adult caregivers. Pediatr Clin North Am. 2003;50(6):1561–1583. | ||
Kazemi E, Hodapp RM. Transition from adolescence to young adulthood: the special case of Prader-Willi syndrome. In: Butler MG, Lee ODK, Whitman Y, editors. Management of Prader-Willi syndrome. Berlin: Springer; 2006:356–369. | ||
Sinnema M, Maaskant MA, van Schrojenstein Lantman-de Valk HMJ, et al. Physical health problems in adults with Prader-Willi syndrome. Am J Med Genet A. 2011;155(9):2112–2124. | ||
Bonnot O, Cohen D, Thuilleaux D, Consoli A, Cabal S, Tauber M. Psychotropic treatments in Prader-Willi syndrome: a critical review of published literature. Eur J Pediatr. 2016;175(1):9–18. | ||
Odent T, Accadbled F, Koureas G, et al. Scoliosis in patients with Prader-Willi syndrome. Pediatrics. 2008;122(2):e499–e503. | ||
Nakamura Y, Murakami N, Iida T, et al. The characteristics of scoliosis in Prader-Willi syndrome (PWS): analysis of 58 scoliosis patients with PWS. J Orthop Sci. 2015;20(1):17–22. | ||
Radicioni A, Di Giorgio G, Grugni G, et al. Multiple forms of hypogonadism of central, peripheral or combined origin in males with Prader-Willi syndrome. Clin Endocrinol (Oxf). 2012;76(1):72–77. | ||
Gross-Tsur V, Hirsch HJ, Benarroch F, Eldar-Geva T. The FSH-inhibin axis in Prader-Willi syndrome: heterogeneity of gonadal dysfunction. Reprod Biol Endocrinol. 2012;10:39. | ||
Crinò A, Schiaffini R, Ciampalini P, et al. Genetic Obesity Study Group of Italian Society of Pediatric Endocrinology and Diabetology (SIEDP) Hypogonadism and pubertal development in Prader-Willi syndome. Eur J Pediatr. 2003;162(5):327–333. | ||
Emerick JE, Vogt KS. Endocrine manifestations and management of Prader-Willi syndrome. Int J Pediatr Endocrinol. 2013;2013(1):14. | ||
Burman P, Ritzén EM, Lindgren AC. Endocrine dysfunction in Prader-Willi syndrome: a review with special reference to GH. Endocr Rev. 2001;22(6):787–799. | ||
Butler MG, Theodoro M, Skouse JD. Thyroid function studies in Prader-Willi syndrome. Am J Med Genet A. 2007;143(5):488–492. | ||
Sharkia M, Michaud S, Berthier MT, et al. Thyroid function from birth to adolescence in Prader-Willi syndrome. J Pediatr. 2013;163(3):800–805. | ||
Schneider DL, Barrett-Connor EL, Morton DJ. Timing of postmenopausal estrogen for optimal bone mineral density. The Rancho Bernardo Study. JAMA. 1997;277(7):543–547. | ||
Butler MG, Haber L, Mernaugh R, Carlson MG, Price R, Feurer ID. Decreased bone mineral density in Prader-Willi syndrome: comparison with obese subjects. Am J Med Genet. 2001;103(3):216–222. | ||
Bianchi ML, Leonard MB, Bechtold S, et al. Bone health in children and adolescents with chronic diseases that may affect the skeleton: the 2013 ISCD Pediatric Official Positions. J Clin Densitom. 2014;17(2):281–294. | ||
Corrias A, Bellone J, Beccaria L, et al. GH/IGF-I axis in Prader-Willi syndrome: evaluation of IGF-I levels and of the somatotroph responsiveness to various provocative stimuli. Genetic Obesity Study Group of Italian Society of Pediatric Endocrinology and Diabetology. J Endocrinol Invest. 2000;23(2):84–89. | ||
Grugni G, Marostica E, Crinò A, Marzullo P, De Nicolao G, Sartorio A. Deconvolution-based assessment of pituitary GH secretion stimulated with GHRH+arginine in Prader-Willi adults and obese controls. Clin Endocrinol. 2013;79(2):224–231. | ||
Inzaghi E, Cianfarani S. The challenge of growth hormone deficiency diagnosis and treatment during the transition from puberty into adulthood. Front Endocrinol. 2013;4:34. | ||
Deal CL, Tony M, Höybye C, et al; 2011 Growth Hormone in Prader-Willi Syndrome Clinical Care Guidelines Workshop Participants. Growth Hormone Research Society workshop summary: consensus guidelines for recombinant human growth hormone therapy in Prader-Willi syndrome. J Clin Endocrinol Metab. 2013;98(6):E1072–E1087. | ||
Sanchez-Ortiga R, Klibanski A, Tritos NA. Effects of recombinant human growth hormone therapy in adults with Prader-Willi syndrome: a meta-analysis. Clin Endocrinol. 2012;77(1):86–93. | ||
Lafortuna CL, Minocci A, Capodaglio P, et al. Skeletal muscle characteristics and motor performance after 2-year growth hormone treatment in adults with Prader-Willi syndrome. J Clin Endocrinol Metab. 2014;99(5):1816–1824. | ||
Oto Y, Tanaka Y, Abe Y, et al. Exacerbation of BMI after cessation of growth hormone therapy in patients with Prader-Willi syndrome. Am J Med Genet A. 2014;164A(3):671–675. | ||
Hoybye C, Thoren M, Bohm B. Cognitive, emotional, physical and social effects of growth hormone treatment in adults with Prader-Willi syndrome. J Intellect Disabil Res. 2005;49(pt 4):245–252. | ||
Donckerwolcke RA, van Zeben-van der Aa DM. Transfer of the care for adolescents with chronic illnesses: from pediatrics to specialists for adults. Ned Tijdschr Geneeskd. 2002;146(14):675–678. | ||
Tracy J, Henderson D. Children and adolescents with developmental disabilities. The GP’s role. Aust Fam Physician. 2004;33(8):591–597. | ||
Rosen DS, Blum RW, Britto M, Sawyer SM, Siegel DM. Society for adolescent medicine-transition to adult health care for adolescents and young adults with chronic conditions: position paper of the Society for Adolescent Medicine. J Adolesc Health. 2003;33(4):309–311. | ||
Ciccarelli MR, Brown MW, Gladstone EB, Woodward JF, Swigonski NL. Implementation and sustainability of statewide transition support services for youth with intellectual and physical disabilities. J Pediatr Rehabil Med. 2014;7(1):93–104. | ||
Schrander-Stumpel CT, Sinnema M, van den Hout L, et al. Healthcare transition in persons with intellectual disabilities: general issues, the Maastricht model, and Prader-Willi syndrome. Am J Med Genet C Semin Med Genet. 2007;145C(3):241–247. | ||
Viner R. Transition from paediatric to adult care. Bridging the gaps or passing the buck? Arch Dis Child. 1999;81(3):271–275. | ||
Snow V, Beck D, Budnitz T, et al. Transitions of care consensus policy statement: American college of physicians, society of general internal medicine, society of hospital medicine, American geriatrics society, American college of emergency physicians, and society for academic emergency medicine. J Hosp Med. 2009;4(6):364–370. | ||
Bailey S, O’Connell B, Pearce J. The transition from paediatric to adult health care services for young adults with a disability: an ethical perspective. Aust Health Rev. 2003;26(1):64–69. | ||
Kripke CC. Primary care for adolescents with developmental disabilities. Prim Care. 2014;41(3):507–518. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.