Back to Journals » Drug Design, Development and Therapy » Volume 19
Population Pharmacokinetic and Pharmacodynamic Modeling of Enteric-Coated Aspirin Capsule and Tablet Formulations in Healthy Subjects
Authors Koh J ![]() , Khwarg J
, Khwarg J ![]() , Yu KS
, Yu KS ![]() , Lee S
, Lee S ![]() , Jang IJ
, Jang IJ ![]() , Lee S
, Lee S ![]()
Received 23 April 2025
Accepted for publication 1 August 2025
Published 9 September 2025 Volume 2025:19 Pages 7853—7863
DOI https://doi.org/10.2147/DDDT.S533428
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tuo Deng
JaeEun Koh,1 Juyoung Khwarg,1 Kyung-Sang Yu,1 SeungHwan Lee,1 In-Jin Jang,1 Soyoung Lee2
1Department of Clinical Pharmacology and Therapeutics, Seoul National University College of Medicine and Hospital, Seoul, Republic of Korea; 2College of Pharmacy, Chungnam National University, Daejeon, Republic of Korea
Correspondence: Soyoung Lee, College of Pharmacy, Chungnam National University, 99, Daehak-ro, Yuseong-gu, Daejeon, 34134, Republic of Korea, Tel +82428215915, Email [email protected]
Purpose: This study aimed to develop a population pharmacokinetic-pharmacodynamic (PK-PD) model to predict the PKs of acetylsalicylic acid (ASA) and salicylic acid (SA), and their effects on thromboxane B2 (TXB2) inhibition following oral administration of two enteric-coated aspirin formulations.
Patients and Methods: Data from two Phase I studies in healthy Korean subjects were used to develop the PK-PD model. A nonlinear mixed effect modeling approach was implemented using Monolix®, based on 669 plasma concentrations of ASA and SA and 83 serum TXB2 concentrations from 44 subjects. Simulx® was used for model-based simulation and external validation using published literature data. Differences in absorption profiles between two formulations were assessed as a covariate effect.
Results: The PK of aspirin was well described by a one-compartment model for ASA and a two-compartment model for SA, incorporating pre-systemic metabolism and dual absorption. A turnover model with an Emax function captured the TXB2 inhibition. The capsule formulation showed faster absorption (0.22 h− 1) than the tablet (0.053 h− 1), but this did not affect TXB2 inhibition. Body weight significantly influenced ASA-to-SA metabolism and SA clearance. External validation confirmed that the model adequately predicted PK and PD profiles at both 80 mg and 160 mg doses, with simulated TXB2 inhibition showing similar responses between formulations at steady state, exceeding 80%.
Conclusion: This model adequately described the PK and PD of enteric-coated aspirin and demonstrated comparable TXB2 inhibition between the capsule and tablet formulations, supporting their potential interchangeability in clinical practice.
Keywords: enteric-coated formulation, aspirin, salicylic acid, thromboxane B2, population pharmacokinetic and pharmacodynamic modeling, simulation
Introduction
Platelets circulate in the bloodstream and primarily regulate hemostasis and thrombosis.1 After activation, they play an important role in thrombus formation and stabilization following vascular injury or insult. However, excessive platelet activation can lead to thrombosis and atherosclerosis, potentially resulting in cardiovascular events and sudden death.2 A major pathway of platelet activation is mediated by the cyclooxygenase-1 (COX-1) enzyme, which converts arachidonic acid into prostaglandins (PGs) and thromboxane A2 (TXA2).3
Aspirin has been used as an antiplatelet agent for the prevention and treatment of atherothrombotic complications.4,5 The acetyl group of aspirin irreversibly binds to a serine residue of COX-1, subsequently inhibiting the synthesis of PG and TXA2.6 Aspirin can be absorbed through the stomach and small intestine; however, its systemic bioavailability is approximately 50% due to extensive biotransformation into the inactive metabolite salicylic acid (SA) by esterases distributed in the plasma, intestine, and liver.7 Although aspirin has a short plasma half-life of 20 minutes, its antiplatelet effects persist longer due to the irreversible inhibition of platelets derived from megakaryocytes in the bone marrow.6,7 To evaluate the pharmacodynamic (PD) effects of aspirin, thromboxane B2 (TXB2) has been used as a validated biomarker, as TXA2 is nonenzymatically hydrolyzed into the more stable TXB2.3,8,9
Enteric-coated aspirin is designed to dissolve in the small intestine rather than the stomach, resulting in delayed absorption.10 It was developed to minimize direct gastric mucosal irritation and to reduce the risk of gastrointestinal side effects such as ulcers and bleeding associated with aspirin use.11 A notable pharmacokinetic (PK) characteristic of enteric-coated aspirin is its variable bioavailability, which results from delayed and often incomplete absorption in the small intestine.12 In Korea, various enteric-coated aspirin formulations, including capsules and tablets, have been approved for clinical use, but evidence on their PK and PD differences remains limited.
While several PK models of aspirin have been developed, most have focused on immediate-release formulations. Earlier models employed one-compartment structures for the parent compound (ASA) and one or two compartments for the metabolite (SA), incorporating lag time, pre-systemic compartments, or first-pass effect to describe absorption and metabolism.13,14 However, to the best of our knowledge, no population PK/PD model has been reported specifically for enteric-coated aspirin formulations that simultaneously evaluate dual absorption processes and the downstream PD effects via TXB2 inhibition.
The aim of this study was to develop a population PK-PD model to characterize the PKs of acetylsalicylic acid (ASA) and its metabolite, as well as the PD effect of TXB2 inhibition following oral administration of two different enteric-coated aspirin formulations. Furthermore, the study aimed to explore whether formulation-related differences in PKs would affect PD responses.
Materials and Methods
Study Data and Population
Data from two phase I studies in healthy Korean subjects were used to develop the population PK-PD model of enteric-coated aspirin. In each study, two different enteric-coated aspirin formulation were used: an enteric-coated capsule (Astrix®, Boryungbio, Korea) and an enteric-coated tablet (Aspirin Protect®, Bayer, Germany). Both studies were designed as open-label, two-period, one-sequence crossover studies to evaluate the PK and PD interactions between rabeprazole and aspirin in healthy Korean subjects (Figure S1). Period 1 involved multiple administrations of aspirin 100 mg once daily for 5 days to assess the baseline PK and PD profiles of aspirin without the perpetrator. Period 2 included multiple administrations of rabeprazole at 5 mg/day followed by coadministration of aspirin at 100 mg/day. The plasma concentrations of ASA and SA were analyzed for PK assessment, and serum TXB2 concentrations were analyzed for PD assessment. To construct the PK-PD model of aspirin, we used pooled data from period 1 of the two studies.
The key inclusion and exclusion criteria were identical in both phase I studies. Eligible participants were healthy men or women aged 19 to 50 years with no childbearing potential, a body mass index (BMI) between 18.0 and 27.0 kg/m2, and a body weight between 50.0 and 90.0 kg. Individuals with a clinically significant medical history or known hypersensitivity to aspirin, salicylic acid, or other nonsteroidal anti-inflammatory drugs were excluded. Additional exclusion criteria included a platelet count < 150,000/µL, a prothrombin time (international normalized ratio) > 1.2, or an activated partial thromboplastin time > 40 seconds at screening.
These studies were conducted at Seoul National University Hospital, Republic of Korea, in accordance with the Declaration of Helsinki and the Korean Good Clinical Practice guidelines. Both studies were registered at ClinicalTirals.gov (NCT05481307 and NCT05699070). Both study protocols were approved by the Institutional Review Board of Seoul National University Hospital and the Ministry of Food and Drug Safety, and written informed consent was obtained from all subjects prior to study initiation.
Sample Collection and Analytical Methodology
Serial blood samples for aspirin PK were collected at 0 h (pre-dose), 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 5, 6, 7, 8, 12, and 24 h post-dose on day 1 and day 5. PK blood samples were collected into EDTA-K2 tubes at each sampling point and centrifuged at 3000 rpm for 10 minutes at 4 °C. Plasma samples were aliquoted and stored at −70°C until analysis. Plasma concentrations of ASA and SA were simultaneously determined by protein precipitation, followed by a validated ultra-performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS). UPLC-MS/MS analysis was performed using a Waters ACQUITY UPLCTM System (Waters, USA) coupled with a Waters XevoTM TQ MS (Waters, USA). MS/MS was operated in multiple reaction monitoring with negative electrospray ionization mode. The lower limit of quantification of ASA and SA was 10.0 and 200 ng/mL, respectively.
Blood samples for aspirin PD evaluation were collected at 0 h (pre-dose) on day 1 and 24 h post-dose on day 5. PD blood samples were collected in serum separator tubes at each sampling point and centrifuged at 3000 rpm for 10 minutes at 4 °C after 30 minutes of clotting time. Serum samples were aliquoted and stored at −70°C until analysis. Serum concentrations of TXB2 were determined using the R&D ELISA kit (KGE011) (R&D Systems, USA).
Population PK-PD Model Development
The population PK-PD model of enteric-coated aspirin was developed using a nonlinear mixed effect (NLME) modeling approach with Monolix software version 2021R1 (Lixoft, Antony, France). Parameters were estimated by maximum likelihood using the Stochastic Approximation Expectation Maximization (SAEM) algorithm of Monolix. Concentrations below the limit of quantification were ignored in the estimation of population parameters using the M1 method.15,16
In this study, several PK structural models were evaluated using a simultaneous modeling approach that estimates the plasma concentrations of ASA and its metabolite, SA, concurrently. Prior to model estimation, ASA and SA concentrations were converted to molar units. One- and two-compartment models with linear elimination were assessed as potential PK structural models. To best characterize the absorption profile of enteric-coated aspirin, various absorption models such as first-order, transit compartment, dual (first- and zero-order), enterohepatic recirculation, pre-systemic compartment, and first-pass effect models with or without lag time were investigated. Following the selection of the final PK model, a sequential PD model was developed to describe the inhibitory effect of ASA on TXB2 production. A turnover model was employed to capture the production and elimination of endogenous TXB2, and a sigmoid Emax model was assessed to explain the drug effect, as expressed by the following equation:
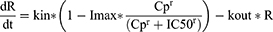
where R is the serum TXB2 concentration at time t; kin is the zero-order production rate of endogenous TXB2; kout is the first-order elimination rate constant of endogenous TXB2; Imax is the maximum TXB2 inhibition; IC50 is the concentration yielding half-maximal inhibition of TXB2; and Cp is the parent (ASA) plasma concentration. The baseline TXB2 concentration (R0) was estimated as the ratio of kin to kout.
The inter-individual variability (IIV) for each PK and PD parameter was described by the following exponential model and was assumed to follow a log-normal distribution, except for the fraction of dose absorbed by zero-order process (fr0).
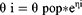
For the fr0, a logit-normal distribution between 0 and 1 was assumed.
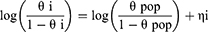
where θi is the individual parameter; θpop is the typical value of the population parameter; and ηi is the random variable that is normally distributed with a mean of 0 and variance of ω2. Furthermore, the additive, proportional, and combined (additive and proportional) error models were explored for the residual error. The final model code is provided in the Supplementary Materials (Appendix 1).
Covariate Analysis
Several demographic data, including age, height, weight, and BMI as well as enteric-coated formulations were evaluated as potential covariates. Correlations between continuous covariates and model parameters were initially explored using regression analysis. Candidate covariates showing plausible relationships were subsequently tested in the model using exponential functions normalized to their median values. The equations are as follows:
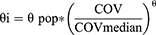
where COVmedian is the median value of the covariate; θ is the estimated exponent of the parameter as a power relationship for a typical subject. For categorical covariate, the effect of enteric-coated formulations was evaluated to account for potential variability in aspirin PKs. Covariates included in the final model were selected using a stepwise procedure consisting of forward selection (ΔOFV > - 3.84, p < 0.05) and backward elimination (ΔOFV > −6.63, p < 0.01).
Population PK-PD Model Evaluation and Simulation
Various models were evaluated using both numerical and visual diagnostics to optimize the structural model, residual error model, and covariate selection, as well as to verify the final model. Numerical criteria included parameter precision, relative standard errors (RSE) of parameter estimates, objective function value (OFV; calculated as −2 times the log-likelihood, - 2LL), Akaike Information Criteria (AIC), and Bayesian Information Criteria (BIC). Visual evaluation was based on goodness-of-fit (GOF) diagnostics and individual plots. The GOF plot was assessed using several diagnostic scatter plots, including observations vs population predicted (PRED) concentrations, observations vs individual predicted (IPRED) concentrations, population weighted residuals (PWRES) vs PRED, PWRES vs time, individual weighted residual (IWRES) vs PRED, and IWRES vs time. The predictive performance of the final model was further evaluated using a visual predictive check (VPC), in which the observed data were overlaid with the median and 90% prediction intervals (10th and 90th percentiles) of 1000 simulated datasets.
External model evaluation was performed using ASA, SA, and TXB2 data obtained from a previously published study. This external study was a randomized crossover trial involving 16 healthy volunteers who received enteric-coated aspirin at doses of 80 and 160 mg/day for 7 days.17 For the external evaluation, model-based simulations were conducted using Simulx version 2021R1 (Lixoft, Antony, France) based on the final model to predict the concentration-time profiles of ASA, SA, and TXB2 following single and multiple (7-day) oral administrations of 80 and 160 mg enteric-coated aspirin. Multiple dosing over 7 days was considered sufficient to achieve steady state based on the t1/2 of ASA and to allow accumulation of the PD effect through sustained inhibition of platelet COX-1.4,5
A total of 200 virtual subjects were simulated. The predicted PK and PD concentration-time profiles were overlaid with observed external data. R software (version 4.2.2) was used for post-processing and visualization. The percentage inhibition of TXB2 was calculated using the following equation and compared with the external data:
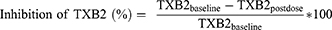
An inhibition level of 80–90% was considered adequate, as this range has been associated with the efficacy of aspirin in patients with essential thrombocythemia.18 In addition, the simulation results were used to explore potential differences in TXB2 inhibition between the enteric-coated tablet and capsule formulations.
Results
Demographics of Model Datasets
A total of 779 concentration measurements from 44 subjects were used to develop a population PK-PD model for enteric-coated aspirin. Specifically, 317 observations were ASA plasma concentrations, 379 observations were SA plasma concentrations, and 83 observations were TXB2 serum concentrations. The age of participants ranged from 20 to 49 years, height from 157.7 to 185.5 cm, weight from 55.6 to 89.5 kg, and BMI from 19.5 to 26.9 kg/m2. The demographic characteristics of all included subjects are summarized in Table S1.
Development of Structural Model
The PK of aspirin was described using a one-compartment model for ASA and a two-compartment model for SA, incorporating dual absorption and pre-systemic metabolism. The PK model was parameterized using the following variables: absorption rate constants for the capsule and tablet formulations (ka), zero-order absorption duration (Tk0), lag time (Lag0), fraction absorbed via zero-order process (fr), transfer rate constants (k23, k24, k34), metabolic clearance of SA (CLm/F), intercompartmental clearance (Q/F), and apparent volume of distribution (V3/F for ASA, V4/F for central SA, and V5/F for peripheral SA). The overall model structure is illustrated in Figure 1.
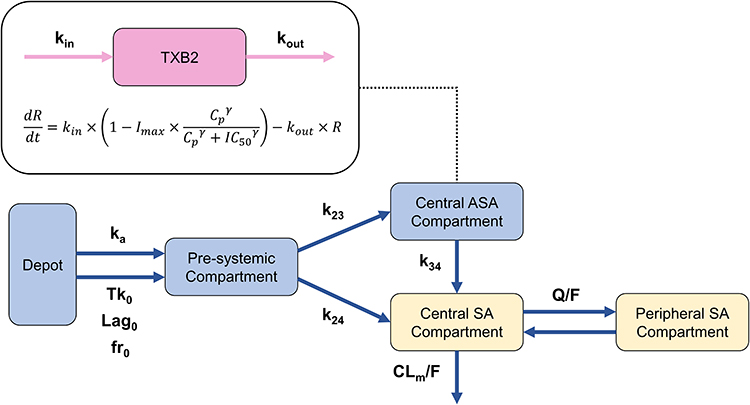 |
Figure 1 Structure of population pharmacokinetic-pharmacodynamic model describing orally administered enteric-coated aspirin. |
To characterize the absorption profile of enteric-coated formulations, a dual absorption model was implemented. In this model, a fraction of the dose (fr) was absorbed via the zero-order process, defined by Lag0 and Tk0, while the remaining fraction (1-fr) was absorbed through first-order kinetics (ka), generating the observed biphasic absorption pattern. A pre-systemic compartment was included to reflect the gastrointestinal metabolism of ASA to SA prior to systemic absorption (Figure 1). SA in the central compartment (V4/F) originated either directly from the pre-systemic compartment or through the metabolism of ASA in the central ASA compartment (V3/F). Central SA was further distributed to the peripheral compartment (V5/F) via intercompartmental flow (Q/F) or eliminated via CLm/F. To aid model convergence, k24, Q/F, and V5/F were fixed based on a previous study.14
The PD effect of ASA on TXB2 inhibition was described by a turnover model incorporating an Emax function. TXB2 production and elimination were described by rate constants kin and kout, respectively. The inhibitory effect of ASA on TXB2 synthesis was modeled using a Emax function mediated by parameters Imax, IC50, and gamma. Due to the limited number of TXB2 sampling time points, IC50 and gamma were fixed at 0.0036 mol/L and 1, respectively, based on literature value.19 The estimated baseline concentration of TXB2 (R0) was 26.4 μg/L.
IIV was included on fr, ka, Tk0, lag0, k34, CLm/F in the PK model, and on R0 in the PD model. Residual unexplained variability was modeled using a proportional residual error model for ASA and SA concentrations, and an additive error model for TXB2 concentrations.
Covariate Analysis
In the final model, covariate analysis was conducted to identify factors contributing to IIV in the PK and PD of enteric-coated aspirin. Among the absorption related parameters, ka best captured the differences between the enteric-coated capsule and tablet formulations, rather than Tk0, Lag0, or fr. Based on the constant estimation results, the ka of the tablet formulation was fixed at 0.053 during subsequent covariate analysis to stabilize the model. The inclusion of body weight as a covariate on the ASA-to-SA metabolism rate constant (k34) and the clearance of SA, modeled using a power function, significantly improved the model fit. Parameter estimates of the final population PK-PD model are summarized in Table 1.
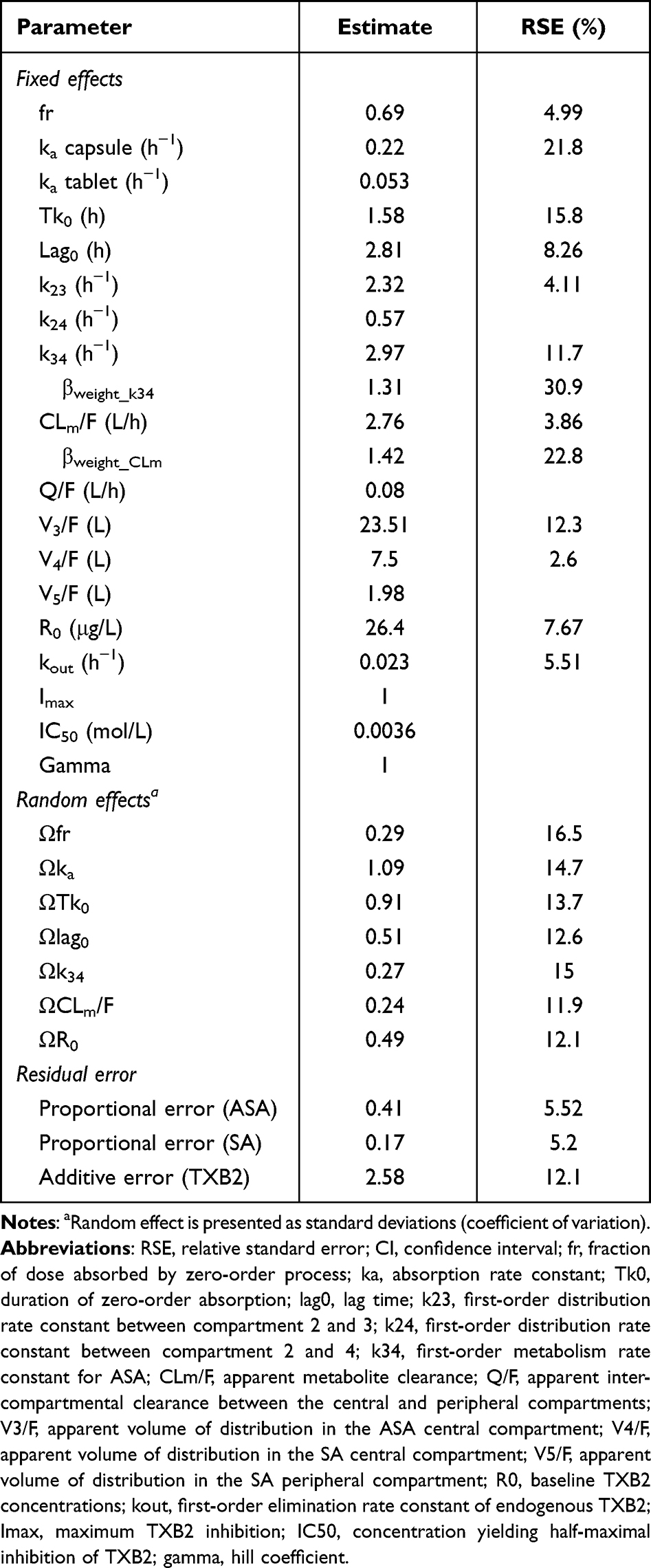 |
Table 1 Final Parameter Estimates of Population Pharmacokinetic-Pharmacodynamic Model of Enteric-Coated Aspirin |
Final Model Evaluation and Simulation
The performance of the final model was evaluated through both internal and external validations. The GOF plots indicated that the final model for enteric-coated aspirin was appropriate, with no significant trends observed in the residuals for ASA and SA (Figures S2 and S3). Although some trends in the residuals were observed in the TXB2 model, these were likely due to the limited number of time points for TXB2 concentration measurements (Figure S4). VPCs demonstrated good predictive performance of the developed model, indicating that it was appropriately specified and robust with acceptable precision (Figure S5). The observed plasma concentration data after the last dose were well within the 10th to 90th percentiles of the 1000 simulated replicates. Furthermore, individual predictions from the final PK-PD model closely matched the observed data.
For external validation, data were obtained from a published clinical study in which enteric-coated aspirin was administered at 80 or 160 mg/day for 7 days.17 Simulations were performed using the final model, and the predicted PK and PD concentration-time profiles were overlaid with observed external data. The results showed that the observed concentrations were well captured within the 5% to 95% prediction intervals of the simulated profiles for both dosage levels (Figure 2). These findings confirm that the model can adequately predict drug concentrations following both single and repeated (7-day) dosing of enteric-coated aspirin at 80 and 160 mg. Overall, the final model demonstrated good external predictability and robustness in characterizing the PK properties of enteric-coated aspirin.
 |
Figure 2 Simulated and external plasma concentration-time profile of acetylsalicylic acid (ASA) and salicylic acid (SA) after single and multiple administration of enteric-coated aspirin. (A) ASA after 80 mg single dose; (B) SA after 80 mg single dose; (C) ASA after 80 mg multiple dose; (D) SA after 80 mg multiple dose; (E) ASA after 160 mg single dose; (F) SA after 160 mg single dose; (G) ASA after 160 mg multiple dose; (H) SA after 160 mg multiple dose. Plots are presented in semi-log scale. Plots show observed external data (black dots), standard error (vertical solid line), 5th, 25th, 75th, 95th percentiles of simulated data (shaded area), and 50th percentiles of simulated data (solid line). |
Serum TXB2 levels from the simulated PD results were compared with external data obtained from 15 subjects, excluding one individual due to abnormally low baseline TXB2 levels (47 pmol/mL).17 In the external data, aspirin at 80 and 160 mg induced approximately 70% and 80% inhibition of TXB2, respectively, at 24 hours after the first dose.17 By day 7, prior to the final dose, inhibition levels increased to approximately 95% for 80 mg and 98–99% for 160 mg.17 The simulated TXB2 inhibition at steady state (day 7) also exceeded 80% for both doses, meeting the prespecified criterion for adequate inhibition. These results support the precision of the final model in characterizing the PD effects of enteric-coated aspirin at steady state (Figure 3). Although the enteric-coated capsule showed slightly lower TXB2 inhibition compared to the tablet formulation, the mean inhibition in both groups exceed 80%, indicating sufficient target engagement regardless of formulation (Figure 3).
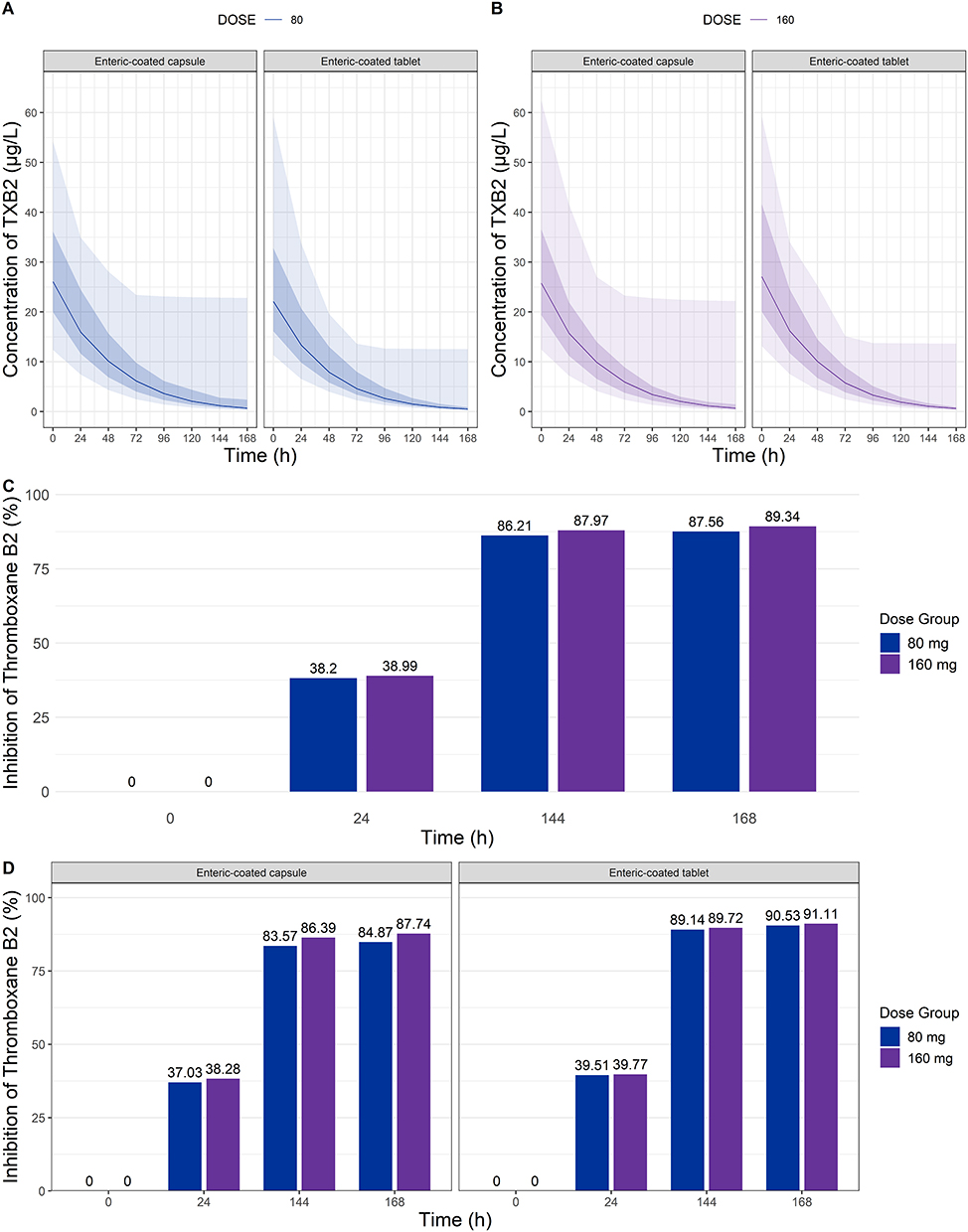 |
Figure 3 Simulated results of thromboxane B2 after multiple doses of enteric-coated capsule and tablet formulations of aspirin. (A) serum concentration-time profile after 80 mg dosing; (B) serum concentration-time profile after 160 mg dosing; (C) overall inhibition of thromboxane B2 (%) following 80 mg and 160 mg administration; (D) comparison of thromboxane B2 inhibition over time for each enteric-coated capsule and tablet formulation. Plots (A) and (B) display the 5th, 25th, 75th, and 95th percentiles of simulated data (shaded area), along with the 50th percentile (solid line). |
Discussion
This study developed a population PK-PD model for two different enteric-coated aspirin formulations to predict the PK and PD effects of aspirin, and to explore possible covariates in healthy Korean subjects. The PK of aspirin was adequately described by a model consisting of a one-compartment disposition model for ASA and a two-compartment model for SA, both with first-order elimination. The elimination of ASA was modeled as metabolism to SA occurring both before and after systemic absorption through the incorporation of a pre-systemic compartment. To characterize the absorption behavior of enteric-coated formulations, a dual absorption model was implemented, consisting of both first-order and zero-order processes with a lag time. The PD of aspirin was well described using a turnover model combined with an Emax function, with the IC50 fixed based on a previously published study.19
Covariate analysis showed that the inclusion of enteric-coated formulation on ka and body weight on k34 and CLm/F significantly improved the model fit (p-value < 0.005), with correlation test statistics of −3.13, 4.46, and 5.0, respectively. Body weight explained part of the IIV in k34 and CLm/F, consistent with finding from previously published studies.20,21 These results suggest that dose adjustments may be considered for individual with obesity or low body weight, where k34 and CLm/F may be increased or decreased, respectively. Additionally, the type of enteric-coated formulation influenced ka, with a higher absorption rate observed for the capsule (0.22 h−1) compared to the tablet (0.053 h−1). No other covariates were identified as significant predictors of the PK variability of enteric-coated aspirin.
The PK parameters estimated in this study were generally consistent with those observed in two clinical datasets used for model development (Study 1: capsule; Study 2: tablet) and previously published literature values. The CL/F of ASA, calculated from k34 and V3/F, was 69.8 L/h, which fell within the range of reported values from Study 1 (mean: 125.5 L/h), Study 2 (98.8 L/h), and the literature (87.5 L/h).22 The estimated central volume of distribution for ASA (V3/F, 23.5 L) was lower than those observed in the internal studies and literature, but still within a reasonable range considering IIV. Similarly, the CLm/F and volume of distribution of SA derived from V4/F and V5/F were 2.76 L/h and 9.48 L, respectively, aligning well with reported values from Study 1, Study 2, and the literature, which ranged between 3.7–5.2 L/h and 11.5–13.2 L.23
The performance of the final model was evaluated using an external dataset obtained from a published study.17 Since the final model was developed using only 100 mg data, external validation was conducted to evaluate its prediction accuracy for aspirin PK and PD across other dose ranges. The selection of 80 and 160 mg aspirin doses was based on evidence that daily doses of 75–150 mg effectively reduce serious vascular events, and that 80, 162.5, and 325 mg doses are approved by the US FDA for the prevention of cardiovascular events.24–26 External validation results for 80 mg and 160 mg aspirin demonstrated good agreement with the observed PK data, confirming the adequacy of the final model. In addition, the model successfully predicted TXB2 inhibition following 7-day repeated dosing and was consistent with the observed inhibition levels at steady state.
Simulated TXB2 inhibition profiles demonstrated comparable PD responses between the enteric-coated capsule and tablet formulations. As shown in Figure 3, the predicted TXB2 inhibition at steady state (168 hours) reached 87.7% (capsule) and 89.3% (tablet) for the 160 mg dose, and 84.9% (capsule) and 91.1% (tablet) for the 80 mg dose. These differences were minimal and unlikely to be clinically meaningful. Moreover, the time course of TXB2 inhibition was similar across formulations, indicating that the differences in absorption kinetics did not translate into significant differences in PD outcomes. These findings suggest that both formulations provide sufficient platelet COX-1 inhibition, supporting their interchangeable use in clinical practice under standard dosing regimens.
A few limitations of this study are that it does not include various data such as different dose range, especially for higher doses, older adults, or individuals with comorbidities. The final model was developed using 100 mg enteric-coated aspirin data from healthy subjects, aged 19 to 50 years, which may exclude some relevant variables that could influence PK and PD variability. Another limitation is that only the 0 h pre-dose on day 1 and 24 h post-dose on day 5 concentrations of TXB2 were used for the final model development. Therefore, only the PD effect of enteric-coated aspirin after multiple doses is expected to be adequately predicted. Further work is warranted to improve the final model with more variable data.
Conclusion
A population PK-PD model for two different enteric-coated aspirin formulations was adequately described by incorporating dual absorption, pre-systemic metabolism of ASA to SA, and a turnover model with an Emax function for TXB2 inhibition. Although the capsule formulation exhibited faster absorption compared to the tablet, these PK differences did not result in clinically meaningful differences in TXB2 inhibition at either 80 mg or 160 mg dose. This model can be used to evaluate formulation-dependent PK-PD characteristics of enteric-coated aspirin.
Acknowledgments
The clinical studies used in the population PK-PD analysis were sponsored by Hanmi Pharm. Co., Ltd, Seoul, Republic of Korea (Study 1) and Daewoong pharmaceutical Corp, Seoul, Republic of Korea (Study 2).
Funding
This work was supported by research fund of Chungnam National University.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Holinstat M. Normal platelet function. Cancer Metastasis Rev. 2017;36(2):195–198. doi:10.1007/s10555-017-9677-x
2. Khodadi E. Platelet function in cardiovascular disease: activation of molecules and activation by molecules. Cardiovasc Toxicol. 2020;20(1):1–10. doi:10.1007/s12012-019-09555-4
3. Altman R, Luciardi HL, Muntaner J, Herrera RN. The antithrombotic profile of aspirin. Aspirin resistance, or simply failure? Thromb J. 2004;2(1):1. doi:10.1186/1477-9560-2-1
4. Patrignani P, Filabozzi P, Patrono C. Selective cumulative inhibition of platelet thromboxane production by low-dose aspirin in healthy-subjects. J Clin Invest. 1982;69(6):1366–1372. doi:10.1172/Jci110576
5. Jourdi G, Marquis-Gravel G, Martin AC, Lordkipanidze M, Godier A, Gaussem P. Antiplatelet therapy in atherothrombotic diseases: similarities and differences across guidelines. Front Pharmacol. 2022;13:878416. doi:10.3389/fphar.2022.878416
6. Wu KK. Aspirin and salicylate - An old remedy with a new twist. Circulation. 2000;102(17):2022–2023. doi:10.1161/01.Cir.102.17.2022
7. Angiolillo DJ, Prats J, Deliargyris EN, et al. Pharmacokinetic and pharmacodynamic profile of a novel phospholipid aspirin formulation. Clin Pharmacokinet. 2022;61(4):465–479. doi:10.1007/s40262-021-01090-2
8. Reilly IAG, Fitzgerald GA. Inhibition of thromboxane formation invivo and exvivo - implications for therapy with platelet inhibitory drugs. Blood. 1987;69(1):180–186. doi:10.1182/blood.V69.1.180.180
9. Petrucci G, Rizzi A, Bellavia S, et al. Stability of the thromboxane B(2) biomarker of low-dose aspirin pharmacodynamics in human whole blood and in long-term stored serum samples. Res Pract Thromb Haemost. 2024;8(8):102623. doi:10.1016/j.rpth.2024.102623
10. Clerici B, Cattaneo M. Pharmacological efficacy and gastrointestinal safety of different aspirin formulations for cardiovascular prevention: a narrative review. J Cardiovasc Dev Dis. 2023;10(4). doi:10.3390/jcdd10040137
11. Hawthorne AB, Mahida YR, Cole AT, Hawkey CJ. Aspirin-induced gastric-mucosal damage - prevention by enteric-coating and relation to prostaglandin synthesis. Brit J Clin Pharmaco. 1991;32(1):77–83. doi:10.1111/j.1365-2125.1991.tb05616.x
12. Cox D, Maree AO, Dooley M, Conroy R, Byrne MF, Fitzgerald DJ. Effect of enteric coating on antiplatelet activity of low-dose aspirin in healthy volunteers. Stroke. 2006;37(8):2153–2158. doi:10.1161/01.STR.0000231683.43347.ec
13. Dubovska D, Piotrovskij VK, Gajdos M, Krivosikova Z, Spustova V, Trnovec T. Pharmacokinetics of acetylsalicylic-acid and its metabolites at low-doses - a compartmental modeling. Method Find Exp Clin. 1995;17(1):67–77.
14. Christiane Dings TL. Pharmacokinetic and pharmacodynamic modeling of acetylsalicylic acid and its major metabolite salicylic acid.
15. Ahn JE, Karlsson MO, Dunne A, Ludden TM. Likelihood based approaches to handling data below the quantification limit using NONMEM VI. J Pharmacokinet Phar. 2008;35(4):401–421. doi:10.1007/s10928-008-9094-4
16. Samson A, Lavielle M, Mentré F. Extension of the SAEM algorithm to left-censored data in nonlinear mixed-effects model:: application to HIV dynamics model. Comput Stat Data Anal. 2006;51(3):1562–1574. doi:10.1016/j.csda.2006.05.007
17. Cerletti C, Dell’Elba G, Manarini S, et al. Pharmacokinetic and pharmacodynamic differences between two low dosages of aspirin may affect therapeutic outcomes. Clin Pharmacokinet. 2003;42(12):1059–1070. doi:10.2165/00003088-200342120-00004
18. Rocca B, Tosetto A, Betti S, et al. A randomized double-blind trial of 3 aspirin regimens to optimize antiplatelet therapy in essential thrombocythemia. Blood. 2020;136(2):171–182. doi:10.1182/blood.2019004596
19. Kimura Y, Takano K, Satoh K, Aida K, Kobayashi T, Ozaki Y. Aspirin Half maximal inhibitory concentration value on platelet cyclooxygenase1 in severe type-2 diabetes mellitus is not significantly different from that of healthy individuals. Clin Appl Thromb-Hem. 2014;20(6):629–636. doi:10.1177/1076029613488934
20. Petrucci G, Zaccardi F, Giaretta A, et al. Obesity is associated with impaired responsiveness to once-daily low-dose aspirin and in vivo platelet activation. J Thromb Haemost. 2019;17(6):885–895. doi:10.1111/jth.14445
21. Mourikis P, Zako S, Dannenberg L, et al. Aspirin antiplatelet effects are associated with body weight. Vasc Pharmacol. 2020;125:106635.
22. Benedek IH, Joshi AS, Pieniaszek HJ, King SY, Kornhauser DM. Variability in the pharmacokinetics and pharmacodynamics of low dose aspirin in healthy male volunteers. J Clin Pharmacol. 1995;35(12):1181–1186. doi:10.1002/j.1552-4604.1995.tb04044.x
23. Nagelschmitz J, Blunck M, Kraetzschmar J, Ludwig M, Wensing G, Hohlfeld T. Pharmacokinetics and pharmacodynamics of acetylsalicylic acid after intravenous and oral administration to healthy volunteers. Clin Pharmacol. 2014;6:51–59. doi:10.2147/CPAA.S47895
24. Visseren FLJ, Mach F, Smulders YM, et al. 2021 ESC guidelines on cardiovascular disease prevention in clinical practice: developed by the task force for cardiovascular disease prevention in clinical practice with representatives of the European Society of Cardiology and 12 medical societies with the special contribution of the European Association of Preventive Cardiology (EAPC). Rev Esp Cardiol. 2022;75(5):429. doi:10.1016/j.rec.2022.04.003
25. Genus Lifesciences. YOSPRALA (aspirin; omeprazole) [package insert]. U.S. Food and Drug Administration.
26. HESP. DURLAZA (aspirin) [package insert]. U.S. Food and Drug Administration.
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.