Back to Journals » Journal of Blood Medicine » Volume 10
Polycythemia Vera-Associated Complications: Pathogenesis, Clinical Manifestations, And Effects On Outcomes
Authors Cuthbert D, Stein BL
Received 18 June 2019
Accepted for publication 18 September 2019
Published 18 October 2019 Volume 2019:10 Pages 359—371
DOI https://doi.org/10.2147/JBM.S189922
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin Bluth
Danielle Cuthbert,1 Brady Lee Stein2
1McGaw Medical Center of Northwestern University, Department of Internal Medicine, Chicago, IL 60611, USA; 2Northwestern University Feinberg School of Medicine, Division of Hematology/Oncology, Department of Medicine, Chicago, IL 60611, USA
Correspondence: Brady Lee Stein
Northwestern University Feinberg School of Medicine, Division of Hematology/Oncology, Department of Medicine, 645 N. Michigan Avenue, Suite 1020, Chicago, IL 60611, USA
Tel +1 312 695 6832
Fax +1 312 695 7814
Email [email protected]
Abstract: Polycythemia vera is a Philadelphia-negative chronic myeloproliferative neoplasm, characterized by erythrocytosis, which is unique, compared to essential thrombocytosis and primary myelofibrosis. Though longevity can usually be expected, vascular morbidity is associated with this condition, as well as a propensity to evolve into myelofibrosis (post-PV MF) and acute myeloid leukemia. In addition, patients can have a pronounced symptom burden. Herein, contributors to the symptomatic burden, as well as the thrombotic and transformative tendencies are reviewed. From a symptom perspective, some are explained by cytokine release, others by microvascular complications, whereas certain symptoms can herald disease evolution. Thrombosis has multifactorial contributors, including but not limited to gender, and inflammatory stress; investigators have recently hypothesized that microparticles and Neutrophil Extracellular Trap Formations may add to thrombotic burden. Finally, we examine the progression to post-PV MF as well as leukemic transformation, highlighting well-established risk factors including age and leukocytosis, certain treatments, and the presence of “non-driver” mutations.
Keywords: polycythemia vera, symptomatic burden, thrombosis, myelofibrosis, leukemic transformation
Introduction
Polycythemia vera (PV) is a Philadelphia-negative chronic myeloproliferative neoplasm (MPN), along with essential thrombocytosis (ET) and primary myelofibrosis (PMF). Dr. Louis Henri Vaquez was the first to describe PV as a disease state, which was further characterized by Dr. William Osler in 1903.1 In 1951, Dr. William Dameshek classified ET, PV and PMF as “Myeloproliferative Disorders” since they shared common features.2 Three key driver mutations have been subsequently described in MPNs – JAK2, CALR and MPL – all of which act to constitutively activate the JAK-STAT pathway.3–6 JAK2 V617F is most prevalent in PV, compared to ET and PMF (99% vs 60%).7 In 2007, the World Health Organization (WHO) updated diagnostic and classification criteria for MPNs, with a later revision in 2016 to include new clinical, morphological and molecular genetic parameters.8 Current criteria focus on elevated hemoglobin, hematocrit or red cell mass levels, bone marrow changes including hypercellularity and pleomorphic changes, JAK2 mutational status, and subnormal erythropoietin levels. There is some disagreement about the diagnostic criteria, discussed elsewhere.9 Treatment of PV is aimed at reducing symptom burden, and typically includes aspirin and phlebotomy, in addition to cytoreductive therapies, hydroxyurea, pegylated interferon and ruxolitinib.10
This paper focuses on the disease complications associated with PV, highlighting pathophysiology and risk factors. The reader is directed to other comprehensive reviews regarding diagnosis and management.11 We begin with a review of the PV symptom burden, which can be attributed to cytokine release, microvascular disturbance, or can herald disease evolution. Next we discuss thrombosis, a hallmark complication of PV, reviewing the role of gender, inflammation, and novel pathophysiological contributors, including microparticles and Neutrophil Extracellular Trap Formations (NETs). We then shift to examine disease progression to myelofibrosis with a review of risk factors including advanced age, leukocytosis, JAK2 allele burden and other non-driver mutations. Finally, we end with a discussion of leukemic conversion, with an emphasis on the prognostic implications of mutations and risk from certain treatments.
Polycythemia Vera Symptom Burden
PV symptoms can have a significant impact on patients’ overall quality of life (QOL), first demonstrated by an important survey in 2007.12 Importantly, symptom burden is independent from risk classification and varies by gender. For example, a 2017 study by Geyer et al evaluated the association between gender, disease characteristics and symptom burden among 2,006 MPN patients.13 Study results revealed that while men had higher rates of thrombocytopenia, transfusion requirements and shorter disease duration, women experienced more severe and more frequent symptoms, particularly abdominal and microvascular symptoms. It is noted that self-reporting of symptoms and diagnosis can be a limitation of MPN QOL studies, and could account for overlap in symptoms between ET, PV, and MF. However, an important point from this body of work, spanning more than a decade, is the symptom burden can be significant not only in MF, but also in ET and PV. Historically, symptom burden has been underappreciated in ET and PV. We view PV symptoms within three broad categories, including those due to cytokine release, microvascular disturbance (we discuss macrovascular thrombosis in a subsequent section), or disease evolution.
Cytokine Excess
It has been well established that patients with MPNs, including PV, have significantly higher cytokine levels compared to the general, healthy population. This increased predisposition to systemic inflammation is thought to be in part due to the JAK2 V617F driver mutation.14 A 2011 analysis by Vaidya et al evaluated the cytokine profile of PV patients (n=76) compared to a PMF cohort (n=92) and healthy controls (n=35).15 Patients with PV had higher levels of MIP-1α, eotaxin, GM-CSF, IFN- α and IFN-γ, compared to both PMF and healthy patients. Patients with both PV and PMF had elevated levels of IL-1RA, IL-7, HGF, MIG, and VEGF. Authors also evaluated correlations between cytokine levels and clinical outcomes, concluding the following associations – platelet count (MIG), arterial thrombosis (VEGF, MIP-1α, IFN- γ), decreased survival (HGF, IFN- α, GM-CSF), among others. A 2015 review by Mondet et al demonstrated that the majority of cytokine expression occurs in PV and PMF, while ET patients had relatively low levels of inflammatory markers.16 Their findings also highlighted similar differences between PV and PMF cytokine profiles compared to the prior study, but did note that PV patients expressed higher levels of IL-5, IL-11 and IL-23, as well.
Cytokine excess may explain such PV-associated symptoms as fatigue, night sweats, fever, bone pain and pruritus. These symptoms are prevalent in PV. According to a 2007 study by Mesa et al which sought to analyze the symptoms of 1179 MPN patients, fatigue was the most common symptom and endorsed by 85% of PV patients (n=344).12 For some, fatigue resulted in significant barriers to physical activity and low quality of life ratings overall. As an example, PV patients scored 46.5 out of 100 on the Functional Assessment of Cancer Therapy-Anemia (FACT-An) questionnaire, where 0 indicates low QOL, and 100 is consistent with a high QOL. Additional symptomatic burden included 65% (n= 263) with pruritus and 43% (n=174) suffering from bone pain.
Aquagenic pruritus (AP), is another hallmark symptom experienced by PV, and can lead to substantial suffering. A 2013 study by Siegel et al focused on the characteristics of AP in 441 patients and its overall effects on quality of life by distributing a patient directed questionnaire.17 They found that 68% (n=301) of PV patients suffered from AP, which can often be the first manifestation of PV, with 65% of all patients experiencing symptoms on average 2.9 years before being diagnosed with the disease. Patients typically described AP as itching (71.8%, n=216), tickling (20.9%, n=63), stinging (30.6%, n=92), burning (17.9%, n=54), among other descriptors. 44 patients (14.6%) described their AP symptoms and pain as unbearable. Itching is most severe on the chest, back, medial side of arms and ventral legs. The survey also asked patients about the association between water contact and AP, with 137 (45.5%) experiencing worse symptoms with warm water compared to cold, and 114 patients (37.9%) noting no difference. In the majority (78.4%) of patients, AP started less than ten minutes after water exposure. Study authors used the 100-point European Organization for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (EORTC QLQ-C30) to assess QOL metrics in PV patients with and without AP. Study findings revealed that AP patients had overall poorer QOL (56/100 vs 64/100). Moreover, they also experienced more fatigue, dyspnea, pain, as well as issues pertaining to emotional, cognitive and social functioning.
The exact mechanism whereby pruritus develops is not completely understood, but several theories have been proposed. A 1987 Dermatology study by Steinman found that PV patients with water-induced pruritus had higher levels of histamine as well as cutaneous fibrinolytic activity, which is typically seen in high levels in patients with inflammatory vasculitides such as scleroderma.18 Jackson et al analyzed skin mast cells, basophils and histamine levels in 13 PV patients.19 Their study results found that pruritus was associated with high levels of mast cells, but not with basophil or histamine levels. A 2009 study by Ishii et al focused on the association between mast cells and pruritogenesis in MPN patients, by analyzing pruritogenetic factor release in MPN patients compared to normal controls.20 Study findings revealed that MPN-specific mast cells produced greater levels of inflammatory markers including histamine, leukotrienes, IL-31, as well as higher amounts of CD34+ cells. Study authors overall concluded that MPN mast cells possess unique inflammatory properties compared to healthy control that result in increased pruritogenesis in this population. Vannucchi’s group evaluated the role of basophil activation among MPN patients, especially as it relates to JAK2 allele burden and symptom burden.21 A total of 78 JAK2 V617F –positive PV patients, 70 individuals with ET and 22 with PMF were included in their analysis. Using CD63+ as a cellular surface biomarker for basophils, study results showed that basophil levels were higher in PV patients compared to ET or PMF individuals (p<0.0001 and p=0.02, respectively). Moreover, there was a strong correlation between JAK2 allele burden (less than or greater than 50 percent) and mean basophil count (82.8±48.9 versus 155.5±109.7 x 106/L respectively; p=0.002). Overall, study authors found that levels of circulating CD63+ basophils and JAK2 allele burden were significantly higher in patients with aquagenic pruritus compared to those without this symptom.
Microvascular Complications
Microvascular symptoms of PV are also common and include headaches, visual disturbances, dizziness, numbness, decreased concentration, mood disturbances and sexuality problems. A 2011 analysis by Scherber et al sought to analyze symptomatic burden in MPNs through two assessment tools - the 18-item Myeloproliferative Neoplasm Symptom Assessment Form (MPN-SAF), and the Brief Fatigue Inventory (BFI).22 Their analysis revealed the following prevalence of the most common vascular symptoms among a cohort of 145 PV patients, including – headache (52.2%), dizziness (52.1%), numbness (66.2%), concentration difficulties (61.2%), sad mood (65%) and sexuality difficulties (56.8%). Of note, patients with PV experienced more severe headaches and concentration problems compared to the ET and MF cohorts.
A 2016 analysis by Geyer et al focused on sexuality symptoms as an indicator of MPN symptom burden and overall QOL.23 A total of 1971 MPN patients, 682 of whom had PV, were included and completed the MPN-SAF Sexuality Score as well as EORTC-QLQ C30 questionnaires. The MPN-SAF Sexuality Score assesses several domains on a scale of 0 (absent symptoms) to 10 (worse imaginable). Overall study conclusions revealed that 64% of MPN patients experience sexual dysfunction, with 43% rating their difficulties as severe. Furthermore, compared to healthy age-matched controls, MPN patients had worse sexual ratings according to the MPN-SAF questionnaire (MPN‐SAF score of 3.6 vs 2.0, P<0.001). More specifically, PV patients had an average MPN-SAF Sexuality score of 3.61, compared to 4.38 among PMF individuals, and 3.12 in the ET cohort. Additional contributing factors included age greater than 65 years old, cytopenias, frequent transfusion requirements, and exposure to certain treatments including glucocorticoids and immunomodulators. Overall, sexual dysfunction was concluded to be multifactorial and related to microvascular disturbances, disease-related complications and treatment, as well as other indicators of poor QOL including impaired social, cognitive and emotional functioning.
These microcirculatory disturbances may be explained by increased blood viscosity or platelet hypersensitivity. Erythromelalgia is a vasomotor complication of PV that manifests as warmth, erythema and burning pain in the extremities.24 In addition to pain, patients frequently experience hand and/or feet paresthesias. Although the pathogenesis is incompletely understood, it is thought to be partially mediated by platelet hypersensitivity. In 1985, Michiels et al studied the pathophysiology of erythromelalgia via skin punch biopsies which revealed arteriolar inflammation, fibromuscular proliferation and thrombotic occlusions.24 Their team saw resolution of pain and reversal of microvascular complications with aspirin and indomethacin but not other therapeutic agents, thereby highlighting the important role of platelet-mediated inflammation and microcirculatory occlusion in the pathogenesis of erythromelalgia.
Symptoms That May Suggest Disease Evolution
According to the aforementioned 2007 study by Mesa et al, among 405 PV patients, 49% (n=198) suffered from night sweats, 13% (n=52) had fevers, and 10% (n=40) endorsed unexplained weight loss.12 In 2016, Dr. Mesa and his colleagues published the multi-center MPN Landmark Study which analyzed MPN patients’ assessments of overall disease burden, QOL, activities of daily living and work productivity, using the previously mentioned MPN-SAF questionnaire.25 A total of 813 individuals (MF, n = 207; PV, n = 380; ET, n = 226) completed the survey. Among PV patients, the following incidences of constitutional symptoms were reported – night sweats (45%), weight loss (12%), and fevers (5%). Interestingly, there was no significant difference in MPN-SAF ratings among PV patients with high versus low prognostic risk scores (16.2 versus 16.8). Reduced QOL was endorsed by 66% of PV patients, despite a substantial proportion of those patients having low prognostic risk scores (62%) and low symptom severity (33%). Furthermore, 10% of PV patients felt that their activities of daily living were limited by pain a great deal, 63% expressed that their disease affected their family/social life, and 37% had reduced work hours.25
While these symptoms can be present in lower risk, newly diagnosed patients, they are more often observed as the disease progresses;12 onset later in the disease can may be an indicator of progression to myelofibrosis.
Over time, PV patients develop progressive splenomegaly. Splenomegaly in MPN is often a result of extramedullary hematopoiesis due to bone marrow dysfunction and is more common in individuals with fibrotic disease.26,27 A 2014 review by Geyer et al evaluated the symptom severity and clinical variables of 1334 PV patients with use of the MPN-SAF patient survey, with ratings on a scale of 0 (absent symptoms) to 10 (worse imaginable).28 The subgroups analyzed included patients who had palpable splenomegaly (PV-S), failed hydroxyurea (PV-HU), required ongoing phlebotomy (PV-P), or had all three features (PV-HUPS). Study findings revealed that PV-S patients experienced more early satiety (3.3) and pruritus (3.8) compared to the other groups. PV-HUPS patients had worse patient-reported symptoms and QOL overall.
Thrombosis
Thrombosis is a well-known complication of PV that can result in significant morbidity and mortality. It is estimated that major thrombotic events are present in approximately 34 to 39% of PV patients at the time of diagnosis.29 The mechanisms involved in the pathogenesis of thrombosis are multifaceted, and involve several cell types, prothrombotic factors and inflammatory markers. A recent January 2019 paper by Bar-Natan and Hoffman describes a pro-inflammatory MPN milieu – a delicate interplay between vessel endothelium, cell adhesion molecules (including integrins and selectins), platelet/red blood cell interactions, and the presence of JAK2 V617F mutations.30 Some of the well-established risk factors for PV-associated thrombosis include advanced age, disease duration, prior history of thrombosis, erythrocytosis, leukocytosis, and JAK2 V617F allele burden (Table 1).31
 |
Table 1 Unique Risk Factors For Thrombotic Complications |
Interestingly, MPN patients also have higher rates of thrombosis in atypical sites. For example, there can be involvement of the abdominal vasculature, including the portal (PVT), hepatic (Budd-Chiari Syndrome), splenic and mesenteric veins.32 A 2012 meta-analysis by Smalberg et al reviewed the prevalence of MPNs in splanchnic vein thrombosis (SVT), including Budd-Chiari (n=1062) and PVT (n=855) patients.32 In Budd-Chiari patients, 41% had MPNs, with PV patients comprising 53% of the MPN cohort. Among patients diagnosed with PVT, 31.5% had an underlying MPN disorder, with subtype analysis showing 27.5% who had PV (95% CI 19.0–38.1). Moreover, PV was found to be more prevalent in Budd-Chiari than PVT (P=0.001). Overall, study authors conclude that these findings highlight a need for additional workup, such as JAK2 mutation testing, as part of the routine workup of SVT.
A recent review by Spivak highlighted the pathophysiology, clinical significance, and treatment of hepatic vein thrombosis (HVT) among PV patients, particularly young women.9 Moreover, the review proposes that HVT occurs due to increased blood viscosity from an increased RCM, and resultant nitrous oxide (NO) scavenging by hemoglobin molecules. As a result, it is suggested that phlebotomy is the cornerstone of management. Further, the concept of plasma volume expansion is discussed, which can have a masking effect, obscuring recognition and aggressive management of erythrocytosis.
There are other selected contributors to the development of thrombotic complications, including gender differences, inflammatory markers (hs-CRP and PTX3), and the role of microparticles and neutrophilic extracellular traps (NETs).
Gender And Vascular Complications
It has been shown the clinical course can vary between men and women living with MPNs. For example, women are more likely to have ET, whereas PMF is more common in males.13 Additional studies have shown that PV manifests at a younger age in women, and women may have lower JAK2 V617F allele burdens compared to men.33 Thrombosis burden and complications are not an exception to gender differences that exist among MPN patients.
A 2011 retrospective study analyzed the association between gender and vascular complications among 270 JAK2 V617F –positive individuals with MPN.34 Within the patient cohort, 164 were women, and 63% (n=104) had PV. There was no statistical difference in the rate of vascular complications among subtypes of MPN. Women were diagnosed with MPN at younger ages, with a median age difference of 6 years (P=0.001), had lower median white blood cell counts (9.5 vs 13.2 x 109/L, P=0.02) and lower rates of JAK2 V617F allele burden compared to men (53% vs 63%, P=0.05). The prevalence of chronic diseases including hypertension and diabetes mellitus was similar between genders, however women had lower rates of dyslipidemia (9% vs 25%, P=0.001) and tobacco use (22% vs 39%, P=0.01). Despite these differences, study findings revealed no statistically significant difference in vascular complications between women and men (27% vs 18%, P=0.09). However, among women who had a presenting vascular event during their MPN diagnosis (n=15), 60% (n=9) of these events were abdominal venous thromboses (AVT). This was statistically significant compared to men who presented with AVT at diagnosis (60% vs 14%, P=0.04).
A subsequent retrospective study in 2013 that focused on age-specific disease characteristics of PV patients had similar findings.35 120 patients aged ≤45 years old were compared to a cohort of 85 individuals ≥65 years old. Among 32 younger individuals with vascular events, these predominantly involved the venous system (n=18, 56%). Of a total of 18 patients with SVT, 16 were in the younger patient cohort (P=0.0056), and 16 were female. Hence, from the above findings we can presume that despite significant risk factors, younger women with PV have a higher incidence of AVT. Similar findings were demonstrated in a 2017 analysis by How et al that focused on the clinical and molecular risk factors for MPN-associated SVT.36 Their review highlighted key risk factors including younger age, female sex, JAK2 V617F mutations and the presence of other hypercoagulable conditions. Study authors conclude that there is a unique interplay between activated blood cells and the splanchnic endothelial environment in MPN disorders.
In 2011, Colaizzo and colleagues sought to evaluate whether there are sex-specific genetic differences that alter the natural course of JAK2 V617F positive MPNs.37 More specifically, study authors questioned whether gender impacts JAK2 somatic and acquired mutation status, and the propensity to develop SVT. Among a total of 180 patients with SVT, this included 95 women and 85 men. 38 patients (21.1%; 95% CI 15.8–27.7) had MPNs – 12 PV patients (8 women vs 4 men), 9 individuals with ET (5 women vs 4 men), and 17 MF cases (9 women vs 8 men). Overall, 43 patients were carriers of the JAK2 V617F mutation, with a higher frequency in women (n=29, 30.5%; 95% CI 22.1–40.4) compared to men (n=14, 16.5%; 95% CI 10.0–25.9). Moreover, women carrying the rs12343867 CC genotype, a specific 46/1 haplotype, demonstrated higher rates of JAK2 V617F mutations compared to those without this specific genotype (60.0% [95% CI 31.2–83.3] vs 26.8% [95% CI 18.4–37.4]; OR: 4.1; 95% CI 1.1–14.9). Overall, study authors point toward a likely association between female sex, JAK2 V617F mutations, 46/1 haplotypes, and an increased risk of developing abdominal venous thrombosis.
Inflammatory Markers – hs-CRP And Pentraxin
Inflammation is a well-known catalyst for thrombus formation. There are several fundamental cell types, signaling proteins and other key biomarkers involved in the inflammatory cascade and resultant clot formation.30 Pentraxins are a group of multimeric proteins that function as pattern recognition proteins, and have been described as key regulators of our innate immune systems.38 Two specific pentraxins, high sensitivity C-reactive protein (hs-CRP) and pentraxin-3 (PTX3) have historically been studied in cardiovascular disease with a focus on atherosclerosis.
A 2011 study by Barbui et al evaluated the role of both hs-CRP and PTX3 in thrombotic complications among patients with ET and PV.39 A total of 244 patients were included – 173 ET patients, and 71 individuals with PV. Patients had serial blood work obtained at consecutive office visits over a median time of 5.3 years from diagnosis. 97% (n=69) of PV patients had a JAK2 V617F mutation, compared to 63% (n=108) of the ET cohort. Hs-CRP and PTX3 values were analyzed based on tertiles. Study results revealed that individuals with the highest levels of hs-CRP (≥3 mg/L) had higher rates of major thrombosis compared to the lowest quartile (36% vs 17%, P=0.01), as well as an increased likelihood of JAK2 V617F allele burden greater than 50% (31% vs 8%, P=0.003). After accounting for confounders during a multivariate analysis, the odds ratio of thrombus formation remained significantly higher in patients with hs-CRP levels above 3mg/L. Interestingly, an inverse relationship was observed between PTX3 values and thrombotic episodes. Individuals with PTX3 levels in the first tertile (<2.5mg/L) had more thrombotic events compared to those with levels >4.5mg/L (39% vs 25%, P=0.045), whereas the highest tertile carried the lowest risk of thrombosis. These findings were re-demonstrated in the multivariate analysis. Similar to hs-CRP, higher levels of PTX3 correlated with an increased JAK2 V617F allele burden greater than 50% (27% vs 9%, P=0.01). Overall, major thrombosis events were more prevalent within the highest hs-CRP cohort and lowest PTX3 tertile.
A subsequent 2017 analysis by Lussana et al investigated the association between MPN driver mutations and inflammatory biomarkers including hs-CRP and PTX3.40 172 PV patients and 305 individuals with ET were included. Results revealed a significant correlation between JAK2 V617F homozygous status and high levels of PTX3 (OR 1.93, 95% CI 1.44–2.59, P< 0.0001) among both disease groups, after adjusting for factors in a multivariate model. There was no significant association between hs-CRP levels and driver mutational status. Focusing on thrombotic outcomes, this paper found similar findings compared to the aforementioned study – higher levels of PTX3 correlated with lower rates of thrombosis (OR 0.57, 95% CI 0.31–1.04), whereas elevated hs-CRP values was associated with increased thrombotic events (OR 2.57, 95% CI 1.39–4.75). Though of interest, these markers are not yet used in routine clinical practice to risk stratify MPN patients.
Microparticles
Microparticles (MP) are 0.1 to 1 μm circulating membrane fragments commonly bound to tissue factor (TF) that serve as prothrombotic biomarkers in cardiovascular disease and several solid tumor malignancies.41 MPs are typically released into the circulation after being activated by different cell types, especially platelets and endothelial cells.41
A 2010 study by Duchemin et al examined MP activity among 44 MPN patients compared to controls.42 Study results found that MPN patients had significantly higher levels of MPs (24.0 ± 9.0 nM vs 10.6 ± 4.4 nM, P< 0.001). They subsequently evaluated thrombin generation in the presence of thrombomodulin (TM), an important cofactor that inactivates thrombin, thereby acting as an anticoagulant. Findings revealed that MPs were associated with less thrombin inhibition (20.1 ± 9.5% vs 28.4 ± 11.8%, P< 0.001), indicating that these patients have lower sensitivities to TM and a higher risk of thrombosis. To further illustrate this point, after plasma was filtrated through a 0.22 μm membrane, there was an increased sensitivity to TM. Overall, study authors conclude that MPs result in “TM-resistance” in patients, thereby supporting its role as a procoagulant.
A 2012 pilot study compared MP levels between 47 MPN patients (ET=13, PV=18, MF=12) and healthy individuals.43 Overall, there were similar levels of TF-bearing MPs between MPN patients and controls (22.7% vs 14.7%, P=0.16). However, MPN patients demonstrated higher levels of TF-bearing MPs derived from CD41a (median 7.0% vs 2.0%, P=0.009), indicating that these particles were more likely to originate from platelets, and may result in an increased risk of thrombosis.
In 2017, Zhang et al sought to evaluate the role of various subtypes of MPs and their association with thrombosis, splenomegaly and JAK2 V617F mutation status.41 The study included a total of 92 patients with MPN (ET=60, PV=20, MF=12). Thrombotic complications were seen in 23 patients, with increased MP levels compared to the non-thrombosis group (Platelet-derived MPs (PMPs): 1989.7+2,023.7/µL vs 617.7+1,169.5, P<0.05). Similarly, among MPN patients with splenomegaly (n=50), MP levels were higher compared to the non-splenomegaly cohort (PMPs: 1,447.5+1,873.1/µL vs 381.1±656.8/µL, P< 0.05). Comparable results were found with higher PMP levels among JAK2 V617F positive (n=50) versus JAK2 V617F negative (n=42) groups (P<0.05).
A 2017 study by Taniguchi et al similarly evaluated the association between plasma MPs and thrombosis among 59 MPN patients.44 Similar to the aforementioned studies, TF+MPs were significantly higher in patients with thrombotic complications compared to those without (33.8/µL vs 47.2/µL, P=0.02). Interestingly, study authors then examined TF+MP levels in patients who had received cytoreductive therapy compared to those who had not. Results revealed that individuals who had not received cytoreductive treatment had higher levels of TF+MPs (101.2/µL vs 42.5/µL, P< 0.001).
While the role of MPNs has been illuminated in translational studies, prognostic implications from prospective, controlled clinical studies are not available.
Neutrophil Extracellular Trap Formations
Neutrophil Extracellular Traps (NETs) are key entities of the innate immune systems that defend host organisms against bacteria and other pathogens.45 They are comprised of fundamental factors including neutrophils, DNA and granular proteins that synergistically result in an increased inflammatory state. NETs activation and release results in a process called NETosis, ultimately culminating in apoptosis. Therefore, their role has been implicated in the pathophysiology of disease states including sepsis, acute respiratory distress syndrome, glomerulonephritis and cystic fibrosis. Moreover, NETs are linked to a greater thrombotic tendency as a result of their pro-inflammatory properties. However, similar to MPs, NET formation cannot be assessed in routine clinical practice.
In 2016, Wolach et al examined the relationship between NETs and thrombus formation among 14 MPN patients compared to age-matched controls.46 They found that neutrophils from individuals with all subtypes of MPN had significantly higher levels of NETs compared to controls (P=0.003). They subsequently incubated healthy neutrophils with ruxolitinib in-vitro, and noted decreased levels of NETosis in these cells. Study authors then used JAK2 V617F knock-in mice to evaluate the association between NETs and thrombosis. Overall, JAK2 V617F mutated mice (n=11) have an increased propensity for NETs formation and higher rates of thrombosis after two hours compared to wild-type (n=8) mice (45% vs 0%, P=0.04). Interestingly, treatment with ruxolitinib was associated with less thrombotic events in mice models. Wolach and colleagues extrapolated their findings in a 2018 analysis, which commented on overall similar findings – higher NETs formation among MPN patients compared to healthy controls (P=0.0006), and development of a NET-rich, prothrombotic phenotype among JAK2 mutated mice with decreased rates of thrombosis after receiving ruxolitinib.47
Progression To Myelofibrosis
Fibrotic evolution to MF highlights an important disease transformation with significant impacts on morbidity and mortality among PV patients. According to the International Working Group for Myeloproliferative Neoplasms Research and Treatment (IWG-MRT) consensus criteria, the diagnosis of post-PV MF requires a bone marrow fibrosis grade ≥2 (3-point scale) or ≥3 (4-point scale), in addition to two clinical features such as anemia, splenomegaly, constitutional symptoms, need for phlebotomy and/or cytoreductive therapy, or a leukoerythroblastic smear.48 In 2014, a group led by Passamonti developed a prognostic model (MYSEC-PM) to predict survival in post-PV and post-ET MF patients.49 Variables in the model included: age >65 years, time to MF progression >15 years, previous thrombosis, constitutional symptoms, hemoglobin <10 g/dL, and circulating blast ≥1%. The predictive value of MYSEC-PM was confirmed in a validation cohort (P=0.01).
The frequency of post-PV MF has been estimated to be 4.9 to 6% at 10 years, and 6 to 14% at 15 years.50,51 According to a 2015 review by Cerquozzi and Tefferi, the literature reveals an incidence of MF transformation of less than 10% at a disease duration of less than 10 years.50 When analyzing the incidence among different ethnic groups, a 2015 analysis by Bai et al showed higher rates of post-PV MF among Chinese PV patients – 27.4%, 39.9% and 61.1% at 10, 15 and 20 years, respectively.52 There have been several prognostic factors associated with disease transformation to MF among PV patients, including age, leukocytosis, JAK2 V617F allele burden, and non-driver mutational status (Table 2). Even after adjusting for these and other variables, progression to MF shortens survival rates among PV patients (HR 2.17, 95% CI 1.27–3.72, P=0.005).53
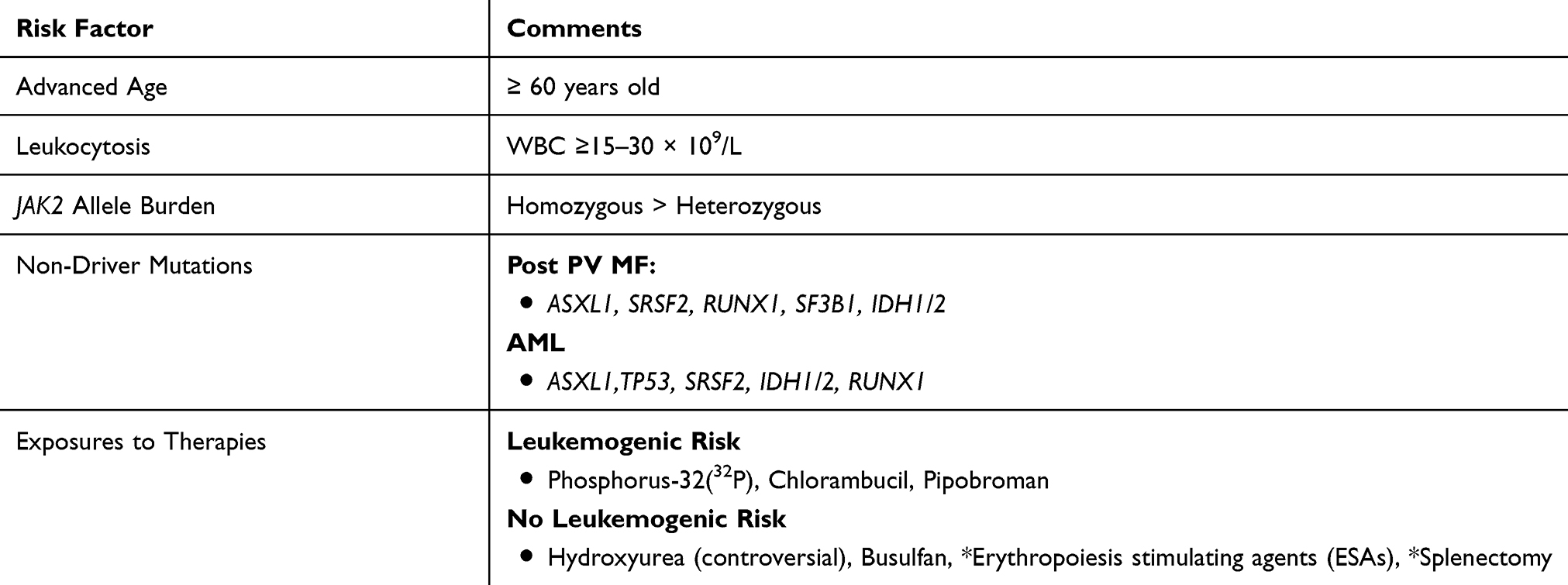 |
Table 2 Risk Factors For Post-PV MF And Leukemic Transformation |
Advanced Age And Leukocytosis
Advanced age, typically defined as ≥ 60 years old, is a well-established risk factor for disease progression and adverse outcomes among PV patients.1 In 2007, Gangat et al focused on the prognostic relevance of various factors on disease outcomes among 459 PV patients.54 Of these, 11.8% (n=54) patients developed post-PV MF. Interestingly, among this cohort, the only statistically significant variable was age ≥60 years. In 2008, Passamonti et al developed a prognostic model to predict survival in post-PV MF patients.53 Their cohort included 68 patients who with disease transformation to MF. Results showed a median age at diagnosis of 65 years, with a range of 44 to 81 years. A 2013 international study by Tefferi et al analyzed the survival and prognosis of 1545 PV patients.1 In a multivariate analysis, overall survival was adversely affected by older age. Study findings revealed a hazard ratio of 8.5 (95% CI 5.9–12.7) among individuals ≥67 years old, and 2.9 (95% CI 1.9–4.4) for ages 57–66 years. A 2013 study evaluated the age-related differences in characteristics and outcomes among a cohort of 204 PV patients – 120 patients ≤ 45 years old and 84 patients ≥65 years or older.36 12.7% of patients (n=26) had MF transformation, with a median age of 54 years among younger individuals and 77.5 years in the older population. There were similar incidences of MF progression among the younger cohort compared to the older (15% vs 10%, P=0.29). However, the median number of years to disease progression was statistically longer among younger patients (20 years vs 8 years, P=0.035) compared to the elderly cohort.
Leukocytosis is associated with multiple disease complications among MPNs, including increased risk of arterial and venous thrombosis, myocardial infarction, MF transformation and evolution to leukemia.53 Passamonti and colleagues highlighted similar findings specifically focusing on post-PV MF patients. Study results revealed that leukocyte count ≥ 15 × 109/L at PV diagnosis was a significant risk factor for the development of post-PV MF (P= 0.002). Moreover, once diagnosed with MF, leukocytosis greater than 30 × 109/L was a useful predictor of decreased survival. Overall, their dynamic prognostic score focused on three variables – leukocyte count ≥30 × 109/l., hemoglobin ≤100 g/l and platelet count ≤100 × 109/l. They conclude that the presence of any of these three risk factors results in a 4.2 fold increase in mortality. In the aforementioned 2013 international study by Tefferi et al, a leukocyte count ≥15 × 109/l among PV patients was associated with a HR of 1.8 (95% CI 1.1–2.8) in the multivariate survival analysis.1 Unfortunately, there is no proof that control of leukocytosis can prevent progression.
JAK2 Allele Burden
The JAK/STAT pathway plays an integral role in normal hematopoiesis. However, a gain-of-function mutation in JAK2 leads to constitutively activated JAK/STAT signaling, thereby giving rise to myeloid neoplasms.3 This hypersensitivity mutation is a hallmark feature of PV, with 99% of patients harboring the JAK2 V617F mutation.7 Moreover, there has been a correlation between higher mutant allele burden, often characterized as homozygous versus heterozygous, and worse disease outcomes. In 2006, Tefferi et al studied the clinical phenotype of wild-type, heterozygous and homozygous JAK2 phenotypes among a cohort of 63 PV patients.55 The JAK2 mutant allele was detected in 92% (n=58) patients, with a 21% frequency of homozygosity. Compared to heterozygous mutation carriers, homozygous individuals had higher hemoglobin levels (P=0.001) and experienced more pruritus symptoms (69% vs 38%, P=0.04). Focusing on disease evolution, homozygous patients had a higher rate of fibrotic transformation (23% vs 2%, P=0.009).
A 2007 study by Vannucchi et al sought to define the clinical profile of homozygous versus heterozygous JAK2 mutations among a cohort of PV and ET patients.56 The study population included a total of 962 patients, 323 with PV and 639 with ET. The PV group had a JAK2 mutation homozygosity frequency of 32.2%, compared to 2.2% among ET patients. Findings revealed that homozygosity was associated with more severe symptoms and worse disease outcomes, including higher rates of cardiovascular events, leukocytosis, splenomegaly, and aquagenic pruritus. Focusing on disease evolution, 15 PV patients transformed to a myeloid metaplasia (MMM), with an increased frequency among homozygous carriers (11.5% vs 1.4%, P<0.001).
A subsequent 2010 prospective study by Passamonti’s group focused on the role of JAK2 V617F allele burden on disease outcomes and complications in 338 PV patients.57 Of these, 94.7% (n=320) were carriers of the JAK2 V617F mutation. Individuals were divided into two groups – low mutant burden (<50%) and high-mutant burden (≥50%). There were a total of eight patients with disease progression to post-PV MF, all of who were classified as high-mutant burden carriers. Overall, study authors conclude that mutant allele burden significantly correlated with the risk of MF transformation (P=0.029), even in the multivariable analysis (P=0.03).
Novel Non-Driver Mutations
There is increasing awareness that additional non-driver somatic mutations may add to prognostication in PV. A 2016 paper by Tefferi et al highlighted the presence of such additional mutations and their prognostic implications.58 Using a myeloid neoplasm-relevant 27-gene panel, this group identified adverse variants/mutations that affected overall, leukemia-free and myelofibrosis-free survival. A total of 316 patients (n=133 PV, n=183 ET) at Mayo Clinic were included as the primary cohort, with an additional 215 patients at an Italian center used as an external validation group. Of the original Mayo Cohort with PV, 53% (n=70) patients possessed variants other than the traditional driver mutations, with TET2 and ASXL1 as the most common. ASXL1 co-segregated with IDH2 (P=0.003) and KIT (P=0.02), and TET2 correlated closely with SH2B3 (P=0.01). Focusing on prognosis and disease outcomes, ASXL1, SRSF2 and IDH2 were associated with worse outcomes. On a multivariable analysis, ASXL1 (HR 2.2, 95% CI 1.1–4.3) and SRSF2 (HR 6.1, 95% CI 2.0–19.2) were significantly correlated with inferior overall survival. For myelofibrosis-free survival, there was an association with SRSF2 and RUNX1 on the univariate analysis, however only SRSF2 (HR 27.2, 95% CI 2.7–274.3) remained significant on the multivariate analysis. Similar prognostic contributions were shown in the Italian group – overall survival: ASXL1 (HR 2.41, 95% CI 1.3–4.5) and SRSF2 (HR 3.84, 95% CI 1.2–12.6), myelofibrosis-free survival: ASXL1 (HR 1.9, 95% CI 1.2–3.6). Overall, study authors conclude that these sequence variants possess important prognostic relevance for PV patients.
A subsequent 2018 paper by Senín et al sought to evaluate the prognostic value of non-driver mutations in JAK2 V617F patients.59 JAK2 V617F allele burden and next generation sequencing (NGS) of non-driver genes was analyzed in 100 PV and ET patients with long-term molecular follow-up. A total of 84 additional somatic mutations were found in the patient cohort, with the lowest frequency seen in patients who did not progress to MF or AML (18% mutation frequency, 1.7 new mutations x 100 person-years). The highest variant allele frequency was seen among alterations in TET2, DNMT3A, TP53, and ASXL1. Overall, median survival was lower in patients with additional non-driver mutations compared to those without mutations (5 years vs 24 years, P= 0.04), with statistical significance seen among the following gene mutations; DNMT3A (P= 0.02), SRSF2 (P< 0.001), SF3B1 (P= 0.048), IDH1/2 (P< 0.001), and RUNX1 (P= 0.003). 24 patients (18 PV and 6 ET) had MF transformation, with higher rates seen among individuals with non-driver mutations (40% vs 23%, P=0.002). The genes associated with a higher chance of MF progression included SF3B1 (P= 0.02) and IDH1/2 (P< 0.0001). These findings highlight the prognostic relevance of non-driver mutations to MF transformation and overall survival. Although not currently used in mainstream clinical practice, their future impact is promising.
Transformation To Leukemia
MPN blast-phase (MPN-BP) is defined as the presence of 20% or more blasts in the bone marrow or peripheral blood.60 Patients with PV typically transition to MPN-BP through an intermediate MF phase as highlighted above, but direct transformation to AML can be observed. The incidence of progression is correlated with disease duration, with 2.3% and 7.9% of PV patients with leukemic conversion at 10 and 20 years, respectively.1 MPN-BP carries a poor prognosis, with a median survival of 1.5 to 2.5 months in untreated patients.61 Inherent risk factors for this progression include several that have already been highlighted, including advanced age, leukocytosis, cytokine excess and mutational status. Therapy-related neoplasms have been associated with certain therapies including Phosphorus-32(32P), and alkylating agents such as chlorambucil, and pipobroman.62 Moreover, individuals treated with more than one cytoreductive therapy have a greater likelihood of developing leukemia (18.2% vs 3.3%, P=0.0002).62 The potential leukemogenicity of hydroxyurea (HU) has been a contentious area of debate.63–66 (Table 2).
Various mutations have been associated with leukemic evolution of PV. The aforementioned analysis by Senín and colleagues also focused on non-driver somatic mutations associated with AML development.59 They found a 13.5% probability of AML transformation at 15 years, with significantly higher rates among individuals with mutations present during first sample compared to those without (33% vs 3.6%, P<0.0001). Among 12 patients who progressed to AML (two through a MF phase), the following somatic mutations were significantly associated with disease transformation: ASXL1 (P< 0.0001), TP53 (P= 0.01), SRSF2 (P< 0.0001), IDH1/2 (P< 0.0001), and RUNX1 (P< 0.0001). Moreover, after accounting for confounding variables in a multivariate analysis, the presence of these mutations was associated with an increased risk of AML (HR 12.2, 95% CI 2.6–57.1, P= 0.001). In the previously discussed 2016 study by Tefferi et al focusing on non-driver mutations, univariate analysis showed significant correlations between leukemia-free survival with SRSF2, IDH2, and RUNX1 among the PV cohort.58 On multivariate analysis, SRSF2 (HR 74.5, 95% CI 4.4–1261.7) and IDH2 (HR, 55.5, 95% CI 3.5–887.4) remained statistically significant.
Conclusion
PV can present with an array of clinical manifestations, and a symptom burden that often causes significant morbidity. Several studies have highlighted worsened quality of life experienced by PV patients, due to a multitude of constitutional symptoms, aquagenic pruritus and microvascular complications. PV patients often experience some level of emotional or social dysfunction, in addition to significant sexual dysfunction that impairs interpersonal relationships.
Morbidity and mortality in PV can stem from thrombotic complications; historically, risk classification has relied upon age and prior thrombosis history. However, there is an association between gender and thrombosis. Specifically, abdominal thromboses are more commonly observed in women compared to men. Inflammation plays a role in symptoms and thrombosis. Biomarkers of interest include hs-CRP and PTX3. The pathophysiology of thrombosis is multifaceted, and beyond demographics, inflammatory markers and cell counts, microparticles and neutrophil extracellular trap formations (NETs) may play a role.
Several risk factors are common for both myelofibrotic and leukemic progression, including age, leukocytosis, and novel “non-driver” mutations. However, we also highlight a correlation between JAK2 allele burden and MF progression. In some cases, therapies worsen the natural history of MPNs, rather than modify in a positive manner.
In all, there have been substantial discoveries relating to MPN-disease states, including PV, other the past several years. A challenge for the field is to validate novel surrogates that may not only characterize the symptomatic burden, but identify patients at risk for a worsening outcome. With a greater understanding of the pathophysiology of clinical manifestations as well as risk factors for disease transformation, in the future, hopefully we can provide optimal, targeted care to PV patients in order to palliate symptoms, improve overall quality of life, and increase overall survival for these individuals.
Disclosure
Dr Brady Lee Stein is a consultant for Apexx Oncology, Celgene, and Incyte, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Tefferi A, Rumi E, Finazzi G, et al. Survival and prognosis among 1545 patients with contemporary polycythemia vera: an international study. Leukemia. 2013;(27):1874–1881. doi:10.1038/leu.2013.163
2. Dameshek W. Some speculations on the myeloproliferative syndromes. Blood. 1951;6:372–375.
3. James C, Ugo V, Le Couédic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434(7037):1144–1148. doi:10.1038/nature03546
4. Pardanani AD, Levine RL, Lasho T, et al. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood. 2006;15:3472. doi:10.1182/blood-2006-04-018879
5. Nangalia J, Massie CE, Baxter EJ, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013;369:2391. doi:10.1056/NEJMoa1312542
6. Klampfl T, Gisslinger H, Harutyunyan AS, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369:2379. doi:10.1056/NEJMoa1311347
7. Tefferi A. Myeloproliferative neoplasms: A decade of discoveries and treatment advances. Am J Hematol. 2016;91:50–58. doi:10.1002/ajh.24221
8. Barbui T, Thiele J, Gisslinger H, et al. The 2016 WHO classification and diagnostic criteria for myeloproliferative neoplasms: document summary and in-depth discussion. Blood Cancer J. 2018;8(2):15. doi:10.1038/s41408-018-0054-y
9. Spivak JL. How I Treat Polycythemia Vera. Blood. 2019;134(4):341–352. doi:10.1182/blood.2018834044
10. Vannucchi AM, Kiladjian JJ, Griesshammer M, et al. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N Engl J Med. 2015;372:426–435. doi:10.1056/NEJMoa1409002
11. Spivak JL. Polycythemia vera: myths, mechanisms, and management. Blood. 2002;100:4272–4290. doi:10.1182/blood-2001-12-0349
12. Mesa RA, Niblack J, Wadleigh M, et al. The burden of fatigue and quality of life in myeloproliferative disorders (MPDs): an international internet-based survey of 1179 MPD patients. Cancer. 2007;109:68–76. doi:10.1002/cncr.22365
13. Geyer HL, Kosiorek H, Dueck AC, et al. Associations between gender, disease features and symptom burden in patients with myeloproliferative neoplasms: an analysis by the MPN QOL International Working Group. Haematologica. 2017;102:85–93. doi:10.3324/haematol.2016.158808
14. Pourcelot E, Trocme C, Mondet J, Bailly S, Toussaint B, Mossuz P. Cytokine profiles in polycythemia vera and essential thrombocythemia patients: clinical implications. Exp Hematol. 2014;42(5):360–368. doi:10.1016/j.exphem.2014.01.006
15. Vaidya R, Sulai N, Rozell SA, et al. Comprehensive plasma cytokine profiling in polycythemia vera: comparison with myelofibrosis and clinical correlates. Blood. 2011;118:3850. doi:10.1182/blood-2011-02-334870
16. Mondet J, Hussein K, Mossuz P. Circulating cytokine levels as markers of inflammation in Philadelphia negative myeloproliferative neoplasms: diagnostic and prognostic interest. Mediators Inflamm. 2015;2015:670580. doi:10.1155/2015/125380
17. Siegel FP, Tauscher J, Petrides PE. Aquagenic pruritus in polycythemia vera: characteristics and influence on quality of life in 441 patients. Am J Hematol. 2013;88(8):665–669. doi:10.1002/ajh.23474
18. Steinman HK. Water-induced pruritus. Clin Dermatol. 1987;5(3):41–48. doi:10.1016/s0738-081x(87)80007-x
19. Jackson N, Burt D, Crocker J, Boughton B. Skin mast cells in polycuthaemia vera: relationship to the pathogenesis and treatment of pruritus. Br J Dermatol. 1987;116:21–29. doi:10.1111/j.1365-2133.1987.tb05787.x
20. Ishii T, Wang J, Zhang W, et al. Pivotal role of mast cells in pruritogenesis in patients with myeloproliferative disorders. Blood. 2009;113(23):5942–5950. doi:10.1182/blood-2008-09-179416
21. Pieri L, Bogani C, Guglielmelli P, et al. The JAK2V617 mutation induces constitutive activation and agonist hypersensitivity in basophils from patients with polycythemia vera. Haematologica. 2009;94(11):1537–1545. doi:10.3324/haematol.2009.007047
22. Scherber R, Dueck AC, Johansson P, et al. The myeloproliferative neoplasm symptom assessment form (MPN-SAF): international prospective validation and reliability trial in 402 patients. Blood. 2011;118:401–408. doi:10.1182/blood-2011-02-334870
23. Geyer HL, Andreasson B, Kosiorek HE, et al. The role of sexuality symptoms in myeloproliferative neoplasm symptom burden and quality of life: an analysis by the MPN QOL International Study Group. Cancer. 2016;122(12):1888–1896. doi:10.1002/cncr.30013
24. Michiels JJ, Abels J, Steketee J, et al. Erythromelalgia caused by platelet-mediated arteriolar inflammation and thrombosis in thrombocythemia. Ann Intern Med. 1985;102:466–471. doi:10.7326/0003-4819-102-4-466
25. Mesa R, Miller CB, Thyne M, et al. Myeloproliferative neoplasms (MPNs) have a significant impact on patients’ overall health and productivity: the MPN Landmark survey. BMC Cancer. 2016;16:167. doi:10.1186/s12885-016-2208-2
26. Wolf BC, Banks PM, Mann RB, Nieman RS. Splenic hematopoiesis in polycythemia vera: a morphologic and immunohistologic study. Am J Clin Pathol. 1988;89:69–75. doi:10.1093/ajcp/89.1.69
27. Song MK, Park BB, Uhm JI. Understanding Splenomegaly in Myelofibrosis: association with Molecular Pathogenesis. Int J Mol Sci. 2018;19(3):898. doi:10.3390/ijms19030898
28. Geyer HL, Scherber RM, Dueck AC, et al. Symptom severity and clinical variables of polycythemia vera patients with splenomegaly, phlebotomy requirements and/or hydroxyurea use: a retrospective evaluation of 1334 patients. Blood. 2014;124:1848.
29. Tefferi A, Elliott M. Thrombosis in myeloproliferative disorders: prevalence, prognostic factors, and the role of leukocytes and JAK2V617F. Semin Thromb Hemost. 2007;33(4):313–320. doi:10.1055/s-2007-976165
30. Bar-Natan M, Hoffman R. New insights into the causes of thrombotic events in patients with myeloproliferative neoplasms raise the possibility of novel therapeutic approaches. Haematologica. 2019;104(1):3–6. doi:10.3324/haematol.2018.205989
31. Barbui T, Finazzi G, Falanga A. Myeloproliferative neoplasms and thrombosis. Blood. 2013;122:2176–2184. doi:10.1182/blood-2013-03-460154
32. Smalberg JH, Arends LR, Valla DC, Kiladjian JJ, Janssen HL, Leebeek FW. Myeloproliferative neoplasms in Budd-Chiari syndrome and portal vein thrombosis: a meta-analysis. Blood. 2012;120(25):4921–4928. doi:10.1182/blood-2011-09-376517
33. Stein BL, Wiliams DM, Wang NY, et al. Sex differences in the JAK2V617F allele burden in chronic myeloproliferative disorders. Haematologica. 2010;95(7):1090–1097. doi:10.3324/haematol.2009.014407
34. Stein BL, Rademaker A, Spivak JL, Moliterno AR. Gender and vascular complications in the JAK2 V617F-positive myeloproliferative neoplasms. Thrombosis. 2011;2011:874146. doi:10.1155/2011/874146
35. Stein BL, Saraf S, Sobol U, et al. Age-related differences in disease characteristics and clinical outcomes in polycythemia vera. Leuk Lymphoma. 2013;54(9):1989–1995. doi:10.3109/10428194.2012.759656
36. How J, Zhou A, Oh ST. Splanchnic vein thrombosis in myeloproliferative neoplasms: pathophysiology and molecular mechanisms of disease. Ther Adv Hematol. 2017;8(3):107–118. doi:10.1177/2040620716680333
37. Colaizzo D, Tiscia GL, Bafunno V, et al. Sex modulation of the occurrence of jak2 v617f mutation in patients with splanchnic venous thrombosis. Thromb Res. 2011;128(3):233–236. doi:10.1016/j.thromres.2011.03.024
38. Du Clos TW. Pentraxins: structure, function, and role in inflammation. ISRN Inflamm. 2013;2013:379040. doi:10.1155/2013/379040
39. Barbui T, Carobbio A, Finazzi G, et al. Inflammation and thrombosis in essential thrombocythemia and polycythemia vera: different role of C-reactive protein and pentraxin 3. Haematologica. 2011;96(2):315–318. doi:10.3324/haematol.2010.031070
40. Lussana F, Carobbio A, Salmoiraghi S, et al. Driver mutations (JAK2V617F, MPLW515L/K or CALR), pentraxin-3 and C-reactive protein in essential thrombocythemia and polycythemia vera. J Hematol Oncol. 2017;10:54. doi:10.1186/s13045-017-0425-z
41. Zhang W, Qi J, Zhao S, et al. Clinical significance of circulating microparticles in Ph− myeloproliferative neoplasms. Oncol Lett. 2017;14(2):2531–2536. doi:10.3892/ol.2017.6459
42. Duchemin J, Ugo V, Ianotto JC, Lecucq L, Mercier B, Abrgrall JF. Increased circulating procoagulant activity and thrombin generation in patients with myeloproliferative neoplasms. Thromb Res. 2010;126(3):238–242. doi:10.1016/j.thromres.2010.06.025
43. Stein BL, McMahon B, Weiss I, Kwaan HC. Tissue-factor bearing microparticles and thrombotic risk in the myeloproliferative neoplasms. Blood. 2012;120:1145.
44. Taniguchi Y, Tanaka H, Luis EJ, et al. Elevated plasma levels of procoagulant microparticles are a novel risk factor for thrombosis in patients with myeloproliferative neoplasms. Int J Hematol. 2017;106(5):691–703. doi:10.1007/s12185-017-2302-5
45. Delgado-Rizo V, Martínez-Guzmán MA, Iñiguez-Gutierrez L, et al. Neutrophil extracellular traps and its implications in inflammation: an overview. Front Immunol. 2017;8:81. doi:10.3389/fimmu.2017.00081
46. Wolach O, Sellar RS, Martinod K, et al. Thrombosis in myeloproliferative neoplasms is linked to increased Neutrophil Extracellular Trap (NET) formation. Blood. 2016;128:633. doi:10.1182/blood-2016-06-724161
47. Wolach O, Sellar RS, Martinod K, et al. Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasms. Sci Transl Med. 2018;10(436):eaan8292. doi:10.1126/scitranslmed.aao4496
48. Barosi G, Mesa RA, Thiele J, et al. International Working Group For Myelofibrosis Research And Treatment (IWG-MRT). Leukemia. 2008;22:437–438. doi:10.1038/sj.leu.2404914
49. Passamonti F, Alessandro V, Domenica C, et al. A new international multicenter‐based model to predict survival in myelofibrosis secondary to polycythemia and thrombocythemia: the mysec prognostic model (MYSEC‐PM). Blood. 2014;124:1826.
50. Cerquozzi S, Tefferi A. Blast transformation and fibrotic progression in polycythemia vera and essential thrombocythemia: a literature review of incidence and risk factors. Blood Cancer J. 2015;5:e366. doi:10.1038/bcj.2015.95
51. Passamonti F, Rumi E, Pungolino E, et al. Life expectancy and prognostic factors for survival in patients with polycythemia vera and essential thrombocythemia. Am J Med. 2004;117(10):755–761. doi:10.1016/j.amjmed.2004.06.032
52. Bai J, Ai L, Zhang L, Yang F, Zhou Y, Xue Y. Incidence and risk factors for myelofibrotic transformation among 272 Chinese patients with JAK2-mutated polycythemia vera. Am J Hematol. 2015;90(12):1116–1121. doi:10.1002/ajh.24191
53. Passamonti F, Rumi E, Caramella M, et al. A dynamic prognostic model to predict survival in post–polycythemia vera myelofibrosis. Blood. 2008;111:3383–3387. doi:10.1182/blood-2007-11-121434
54. Gangat N, Strand J, Li CY, Wu W, Pardanani A, Tefferi A. Leucocytosis in polycythaemia vera predicts both inferior survival and leukaemic transformation. Br J Haematol. 2007;138(3):354–358. doi:10.1111/j.1365-2141.2007.06674.x
55. Tefferi A, Lasho TL, Schwager SM, et al. The clinical phenotype of wild‐type, heterozygous, and homozygous JAK2V617F in polycythemia vera. Cancer. 2006;106:631–635. doi:10.1002/cncr.21730
56. Vannucchi AM, Antonioli E, Guglielmelli P, et al. Clinical profile of homozygous JAK2 617V>F mutation in patients with polycythemia vera or essential thrombocythemia. Blood. 2007;110:840–846. doi:10.1182/blood-2006-12-064287
57. Passamonti F, Rumi E, Pietra D, et al. A prospective study of 338 patients with polycythemia vera: the impact of JAK2 (V617F) allele burden and leukocytosis on fibrotic or leukemic disease transformation and vascular complications. Leukemia. 2010;24(9):1574–1579. doi:10.1038/leu.2010.148
58. Tefferi A, Lasho TL, Guglielmelli P, et al. Targeted deep sequencing in polycythemia vera and essential thrombocythemia. Blood Adv. 2016;1(1):21–30. doi:10.1182/bloodadvances.2016000216
59. Senín A, Fernández-Rodríguez C, Bellosillo B. Non-driver mutations in patients with JAK2V617F-mutated polycythemia vera or essential thrombocythemia with long-term molecular follow-up. Ann Hematol. 2018;97(3):443–451. doi:10.1007/s00277-017-3193-5
60. Mesa RA, Verstovsek S, Cervantes F, et al. International Working Group for Myelofibrosis Research and Treatment (IWG- MRT). Primary myelofibrosis (PMF), post polycythemia vera myelofibrosis (post-PV MF), post essential thrombocythemia myelofibrosis (post-ET MF), blast phase PMF (PMF-BP): consensus on terminology by the international working group for myelofibrosis research and treatment (IWG-MRT). Leuk Res. 2007;31(6):737–740. doi:10.1016/j.leukres.2006.12.002
61. Yogarajah M, Tefferi A. Leukemic transformation in myeloproliferative neoplasms: a literature review on risk, characteristics, and outcome. Mayo Clin Proc. 2017;92:1118–1128. doi:10.1016/j.mayocp.2017.05.010
62. Finazzi G, Caruso V, Marchioli R, et al. Acute leukemia in polycythemia vera. An analysis of 1638 patients enrolled in a prospective observational study. Blood. 2005;105:2664–2670. doi:10.1182/blood-2004-09-3426
63. Beer PA, Delhommeau F, Lecouédic JP, et al. Two routes to leukemic transformation following a JAK2 mutation–positive myeloproliferative neoplasm. Blood. 2010;115(14):2891–2900. doi:10.1182/blood-2009-08-236596
64. Kiladjian JJ, Chevret S, Dosquet C, Chomienne C, Rain JD. Treatment of polycythemia vera with hydroxyurea and pipobroman: final results of a randomized trial initiated in 1980. J Clin Oncol. 2011;29:3907–3913. doi:10.1200/JCO.2011.36.0792
65. Marchioli R, Finazzi G, Landolfi R, et al. Vascular and neoplastic risk in a large cohort of patients with polycythemia vera. J Clin Oncol. 2005;23:2224–2232. doi:10.1200/JCO.2005.07.062
66. Björkholm M, Derolf AR, Hultcrantz M, et al. Treatment-related risk factors for transformation to acute myeloid leukemia and myelodysplastic syndromes in myeloproliferative neoplasms. J Clin Oncol. 2011;29:2410–2415. doi:10.1200/JCO.2011.34.7542
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.