Back to Journals » Lung Cancer: Targets and Therapy » Volume 10
Polo-like kinase 1 inhibition in NSCLC: mechanism of action and emerging predictive biomarkers
Authors Stratmann JA ![]() , Sebastian M
, Sebastian M
Received 16 March 2019
Accepted for publication 24 May 2019
Published 1 July 2019 Volume 2019:10 Pages 67—80
DOI https://doi.org/10.2147/LCTT.S177618
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sai-Hong Ou
Jan A Stratmann, Martin Sebastian
Department of Internal Medicine II, University Clinic of Frankfurt, 60596 Frankfurt, Germany
Abstract: Non-small cell lung cancer (NSCLC) is the leading cause of cancer death worldwide. Due to often unspecific disease symptoms, locally advanced or metastatic disease is diagnosed in the majority of all cases. Palliative treatment options comprise of conventional cytotoxic agents, immunotherapy with checkpoint inhibitors and the use of specific small-molecule tyrosine kinase inhibitors (TKI). However, these TKIs are mainly restricted to a small proportion of patients with lung cancer that harbor activating driver mutations. Still, the effectiveness and favorable safety profile of these compounds have prompted a systematic search for specific driver mechanisms of tumorigenesis and moreover the development of corresponding kinase inhibitors. In recent years, the Polo-like kinase (PLK) family has emerged as a key regulator in mitotic regulation. Its role in cell proliferation and the frequently observed overexpression in various tumor entities have raised much interest in basic and clinical oncology aiming to attenuate tumor growth by targeting the PLK. In this review, we give a comprehensive summary on the (pre-) clinical development of the different types of PLK inhibitors in lung cancer and summarize their mechanisms of action, safety and efficacy data and give an overview on translational research aiming to identify predictive biomarkers for a rational use of PLK inhibitors.
Keywords: non-small cell lung cancer, Polo-like kinase, targeted therapy, therapy resistance
Introduction
Non-small cell lung cancer (NSCLC) is the leading cause of cancer death worldwide.1 Due to often unspecific disease symptoms, locally advanced or metastatic disease is diagnosed in the majority of all cases.2 Among others, adenocarcinoma is the most common histopathological subtype and palliative treatment options comprise of conventional cytotoxic agents, immunotherapy with checkpoint-inhibitors and the use of specific small-molecule tyrosine kinase inhibitors (TKI). However, these TKIs are mainly restricted to a small proportion of patients with lung cancer that harbor activating driver mutations, such as activating EGFR mutations or alterations in ALK, ROS1 and BRAF genes. Still, the effectiveness and favorable safety profile of these compounds have prompted a systematic search for specific driver mechanisms of tumorigenesis and moreover the co-evolutionary development of corresponding kinase inhibitors. In the last two decades, the Polo-like kinase (PLK) family has emerged as a key regulator in mitotic regulation, being involved in the complex process from mitotic onset to its termination. The key role in cell proliferation and the frequently observed overexpression in various tumor entities have raised much interest in basic and clinical oncology aiming to attenuate tumor growth by targeting the PLK. In this review, we give a comprehensive summary on the (pre-) clinical development of the different types of PLK inhibitors in lung cancer and summarize their mechanisms of action, safety and efficacy data and give an overview on translational research aiming to identify predictive biomarkers for a rational use of PLK inhibitors.
Biological function and structure of Polo-like kinases and historical overview
The human homolog of the Polo gene was independently cloned by three research groups in 1993/1994.3–5 All groups reported a 603 amino-acid polypeptide with several nucleotide differences that were all classified as polymorphisms. The product of the human PLK1 gene is a 66KD serine/threonine kinase protein.5 Today, altogether 5 isoforms of PLK (PLK1-5) are known; however, PLK1 is by far the best characterized isoform (see Figure 1A).6–9 PLKs (with the exception of PLK5) contain a catalytic N-terminal serine/threonine kinase domain and a C-terminal tandem-Polo-box region with regulatory functions.10 The catalytic site incorporates most of the highly conserved hallmarks of serine/threonine protein kinases.11 The tandem Polo-boxes of the N-terminal domain are involved in substrate-binding and in determining the correct subcellular localization of PLK1.12
 |
Figure 1 Structure and function of the human Polo-like kinases: (A) Polo-like kinases in human cells. Schematic representation of the five identified PLKs in human cells. The open reading frame amino-acid lengths are shown on the right, the kinase domain is shown in red color with the corresponding amino-acid position. Polo-box domains are shown in blue color. (B) Schematic diagram of the cell cycle functions of PLK1. Abbreviations: PLK, Polo-like kinases; KD, kinase domain; PB, Polo-box domain; aa, amino acids. |
First insights on the cell-cycle-dependent expression of PLK1 were provided by Lake and Jelinek who showed that PLK1 mRNA is nearly absent in the G1 phase of the cell cycle, but reaccumulates in the S phase and reaches highest levels during the G2/M phase,3 linking its function to mitotic activity. Its role in mitosis was further elucidated in 1995, when Goldsteyn et al confirmed increased transcription at all stages of mitosis.13 They localized PLK1 juxtaposed to the spindle apparatus in confocal microscopy analyses and concluded that PLK1 plays a role in chromosome condensation, spindle dynamics and chromosome segregation. Aside of regulatory functions regarding mitosis onset, PLK1 was found to be involved in the assembly of key components of the contractile ring (eg, ECT2, RhoA GTPAse, CYK4) at the equatorial cortex during anaphase onset14,15 and finally in the exit process participating in controlling chromosome segregation and G1 phase entry.16,17 Other physiological roles of PLK1 have been recognized, involving telomere stabilization, extracellular matrix invasion and regulation of topoisomerase IIa in cell cycle progression (see Figure 1B).18–21 For example, Cyclin B1, a key component of the prophase initiation, was identified as an important target structure of PLK1, promoting its (Cyclin B1) nuclear translocation after phosphorylation.22 Activation of PLK1 in turn is a complex process, requiring phosphorylation of a conserved threonine residue (Thr 210) within the PLK1 kinase domain. The Aurora A kinase, a member of the Aurora serine/threonine kinase family, was found to phosphorylate PLK1 during G2/M phase in synergistic action with Bora, a known cofactor of Aurora A.23,24
Given the nature of PLK1 and its involvement in mitosis, unsurprisingly, Holtrich et al provided evidence, that expression in normal human tissue is limited to highly proliferative organs, such as the placenta, colon and the testis. However, the transcripts are detectable in tumors of various origins, including but not limited to lung cancer, colon and stomach cancer as well as non-Hodgkin lymphomas,4 firstly pointing to a possible relevance in malignant processes. Smith et al later showed that cells transformed with PLK1 were capable to produce tumors in nude mice.25 Liu and Erikson and others used vector-based siRNA and antisense oligonucleotides to deplete PLK1 in various cancer cell lines and found that transfected cells arrested in G2/M phase and became highly apoptotic.26–29 The formation of dumbbell-like chromatin and the presence of 4 N DNA content further suggested the inability of successfully transfected cells to completely separate their chromatin. PLK1-depleted cells enter mitosis, but accumulate in a pre-anaphase state with absence of focused spindle poles and chromosomes that do not become stably attached to the spindle apparatus.30 Interestingly, normal human epithelial cells transfected with siRNA showed no impaired survival.28 Conclusively, the preclinical data unequivocally pointed to a central role in regulating mitotic actions, which prompted a series of attempts to pharmacologically target PLK1.
Frequency of PLK1 expression in NSCLC and its prognostic relevance
In an early study on 111 resected lung cancer specimens from patients suffering from limited disease stages, a considerable transcription of PLK1 mRNA (PCR-based method) was documented in all but 4 patients. With a proportion of >80%, the majority of the study population consisted of male smokers with clinical stage I-II NSCLC. Additionally, high PLK1 expression was an independent risk factor for inferior survival in this cohort.31 Allera-Moreau et al performed gene expression profiling of 77 genes involved in DNA replication in 93 tumor specimens collected from chemo-naïve early-stage patients and analyzed its prognostic relevance. They could show that most genes analyzed were significantly deregulated compared to juxtaposed normal lung tissue. PLK1 was found to be overexpressed in 92% of all probes analyzed, and a transcriptive “replication signature” consisting of 5 specific genes involving PLK1 was associated with inferior relapse-free and overall survival.32 The prognostic relevance of PLK1 expression was also confirmed by Wang et al, who additionally found a positive correlation with the presence of lymph node metastases and advanced clinical stage (approximately 50% of all cases tested).33 Another study focused on squamous cell NSCLC and evaluated PLK1 expression by immunohistochemistry and PCR screening.34 PLK1 was upregulated in 72 of 132 tumor probes (55%) and overexpression markedly correlated with disease stage and tumor size. In line with previous data, high expression of PLK1 was a negative prognostic factor correlating with inferior survival in univariate and multivariate analysis.
Preclinical and clinical-pharmacological PLK-1 targeting in NSCLC
The biological function of PLK1 as a key regulator of mitosis and its expression in various tumor entities overall supported the potential role of PLK1 disruption in cancer therapy. Zhou et al performed PLK1 antisense oligonucleotide (ASO) (pcDNA3-PLK1) studies in A549 lung cancer cells and determined cell proliferation, cell cycle distribution and apoptosis.35 The number of viable cells was markedly reduced 48 hrs after successful transfection as compared with control cell lines. Flow cytometry revealed a significant increase of cells in G2/M phase and apoptosis in the transfected cell lines. Mixing studies furthermore revealed a sensitization to vinorelbine quantified by a reduction of the IC50 by more than 40% in the transfected cell lines, implying a synergistic effect of chemotherapy and PLK1 disruption. Another study was carried out with low-proliferative NCI H596 lung carcinoma cell lines. PLK1 function was blocked with an adenoviral delivery of a dominant-negative (dnPLK1) gene.36 The expression of the dnPLK1 caused significant apoptosis at relatively low wild-type:dominant-negative expression ratios despite low proliferation rates of the H596 cell line. In another study, in vitro cell growth of A549 lung cancer cell lines transfected with PLK1 ASOs was reduced by more than 50% with a moderate induction of G2/M phase arrest.29 Moreover, systemical treatment of A549 xenograft nude mice with bolus injections of PLK1 ASOs inhibited tumor growth by 70–86%. Kawata et al used a lung cancer xenograft model to determine PLK1 influence on metastatic tumor spread: A549 cells were inoculated into the murine spleen and subsequent liver metastasis was compared between control animals and mice treated with PLK1 small interfering RNA.37 Macroscopic analysis of mice exsanguinated 70 days after cancer cell inoculation showed a significantly lower volume of liver metastases. Finally, in another xenograft model (nude mice) with subcutaneously inoculated A594 cells, treatment with PLK1 silencing small hairpin DNA resulted in a significant delay in tumor growth compared to sham treatment, and the combination with gemcitabine caused a strong inhibition of tumor cell proliferation by enhanced apoptosis in an additive manner.38
In summary, disruption of PLK1 was feasible in preclinical models and showed encouraging anticancer activity, thus leading the way to the clinical evaluation of PLK1 inhibitors.
The development of small molecules to target PLK1
Inhibitory molecules of the kinase domain may be distinguished in ATP-competitive and non-ATP-competitive inhibitors, depending on their mechanism of action. The deep cavity of the PLK1 ATP binding site is an obvious target; however, the highly conserved structure of binding sites makes the identification of specific inhibitors a challenging endeavor due to potential off-target side effects.
The first specific inhibitor described in the literature is scytonemin, an extracellular matrix pigment synthesized by cyanobacteria. It is supposed to be an effective absorber of damaging ultraviolet rays, while allowing the transmittance of wavelength necessary for photosynthesis.39 This natural marine product was found to inhibit PLK1 in a dose-dependent manner while sparing toxic effects in nonproliferating cells, providing a possible template for the upcoming development of PLK1 inhibitors.40 However, scytonemin showed similar inhibitory effects on other kinases (CDK1, CHK1, MYT, PKC), and the inhibitory indices indicated additional non-competitive modes of action. An overview on PLK1 inhibitors that had been tested clinically in patients with lung cancer gives Table 1. A comprehensive summary enumerating all conducted trials with PLK1 inhibitors in lung cancer patients is depicted in Table 2.
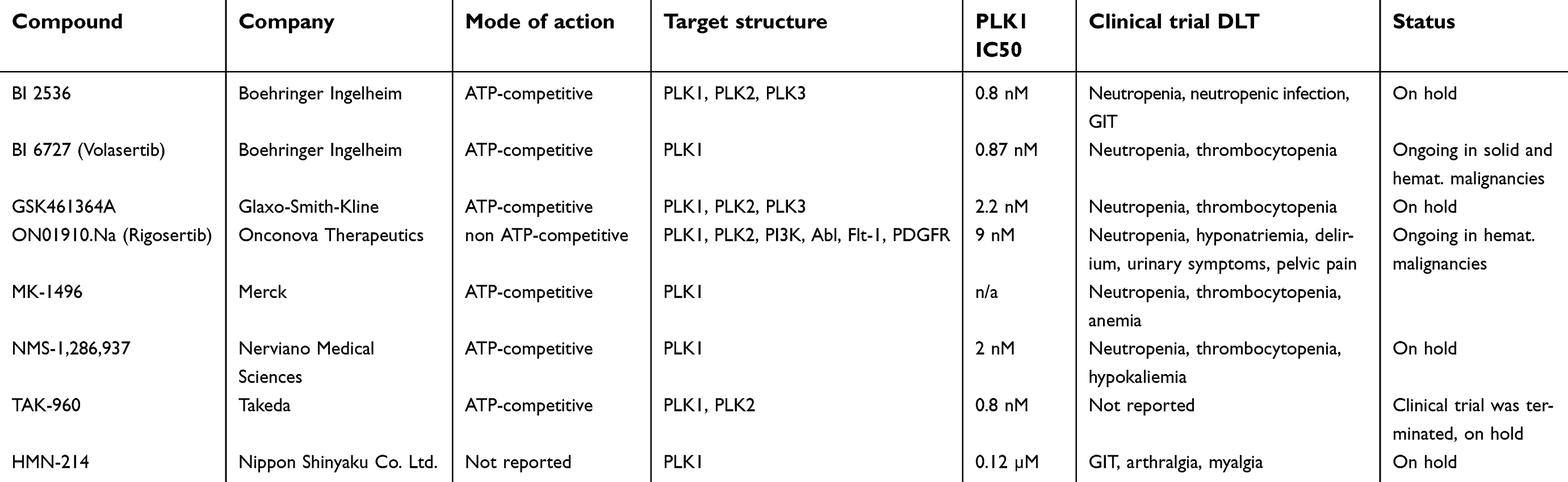 |
Table 1 PLK1 inhibitors invovled in clinical trials with lung cancer |
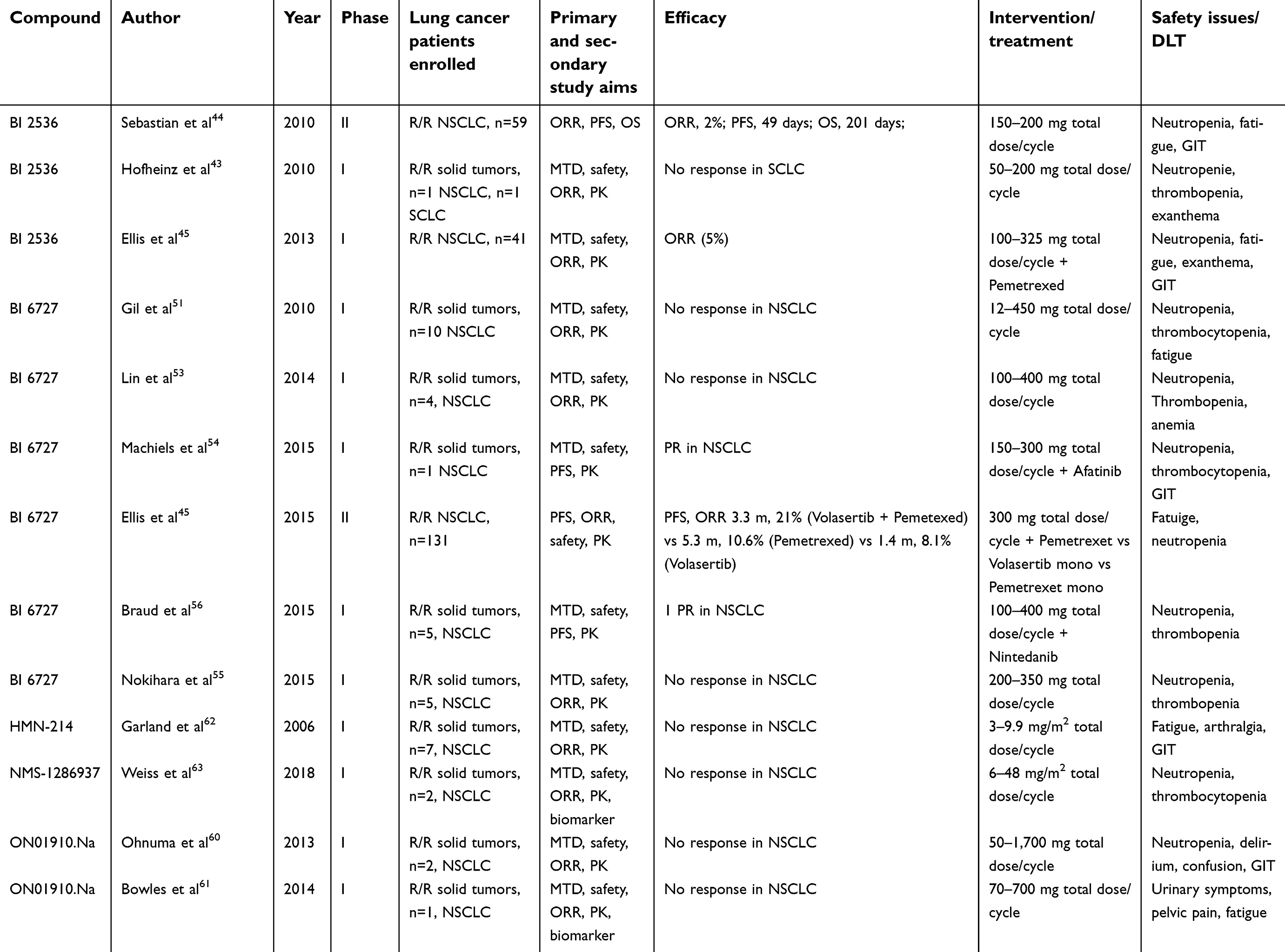 |
Table 2 Clinical trials involving PLK1 inhibitors and patients with lung cancer |
BI 2536
BI 2536 was a first-in-class highly selective ATP-competitive small molecule based upon a dihydropteridone derivative that accomplished inhibiting the enzymatic activity of PLK1 by blocking the ATP-binding site. This compound inhibits PLK1 with an at least 1.000-fold selectivity against a panel of other kinases and an IC50 of 0.8 nM. Irrespective of the tumor origin, the agent inhibits the proliferation of various cell lines in culture assays,41 including the KRAS mutated A549 in addition to NCI-A460 and the TP53 mutated NCI-520 lung cancer cell lines. Comparable to previous RNA-based silencing studies, BI 2536 induced mitotic arrest and cells accumulate 4 N DNA content, indicative of G2/M arrest. Subsequently, massive tumor cell death and apoptosis occurred after 48 hrs of treatment. The growth inhibitory potential was also tested in an A549 xenograft model by using a twice-weekly i.v. schedule, and excellent tolerability and meaningful tumor regression were observed.
A first in human Phase I open-label dose-escalating PLK1 inhibitor study was conducted in patients with refractory and/or metastatic solid tumors.42 A total of 40 patients were treated with at least one cycle of parenteral BI 2536 following a 3+3 escalation scheme starting with 25 mg on day 1 every 3 weeks (D1Q3W). Patients included suffered from colorectal and renal cancer as well as sarcoma among several other cancer entities. The inclusion of patients with lung cancer, however, was not reported. Dose-limiting toxicities were observed at the 250 mg dose level, comprising of neutropenic infection and neutropenia; therefore, the maximum tolerated dose was set to 200 mg for further enrollment. Other namable toxicities were gastrointestinal side effects, fatigue, arthralgia and γ-glutamyltransferase increase without additional signs of hepatic toxicity. Antitumor activity was encouraging with a reported disease control rate of 42% with one patient achieving a partial response.
A total of three clinical trials with BI 2536 were conducted in which patients with NSCLC were enrolled. A Phase I trial aimed to define the maximum tolerated dose of single-agent BI 2536 and overall safety in various cancer entities, including each one patient diagnosed with SCLC and NSCLC, respectively. Toxicity was comparable to previous data with mainly hematotoxic side effects and no new safety issues raised. The patient suffering from SCLC experienced disease stabilization for more than 2 cycles.43
Two clinical trials enrolled patients with relapsed or refractory NSCLC only, a Phase II trial evaluating the efficacy of single-agent BI 2536 and a Phase I trial aiming to determine the recommended Phase II dose of pemetrexed in combination with BI 2536. Both clinical trials showed only modest efficacy in NSCLC patients with tumor response limited to a few patients each. Sebastian and co-authors concluded that despite the objective response rates (ORR, 2%) following single-agent BI 2536 administration in R/R NSCLC patients being low, the capability of BI 2536 to achieve response rates with low treatment-induced mortality suggests clinical activity of Plk-1 inhibitors in relapsed NSCLC.44 Importantly, most of the adverse events affected the hematopoietic system without causing relevant clinical problems to the majority of patients, thus potentially warranting combination strategies. However, although not powered for response rate, the combination of BI 2536 with pemetrexed in the Phase I trial has similarly shown only modest anti-tumor activity with an ORR of 5% (n=2).45 Still, these two patients had a progression-free survival (PFS) of 20.5 months and a censored PFS of 20.1 months, raising the questions of valid biomarkers to more precisely allocate PLK1 inhibitory strategies.
BI 6727 (volasertib)
The second-generation PLK1 inhibitor volasertib (BI 6727), an additional dihydropteridinone derivative, is a potent and selective agent with a favorable pharmacokinetic profile.46 In comparison to BI 2536, volasertib shows improved tissue penetration leading to a high volume of distribution and a prolonged terminal half-life, which offers a theoretic therapeutic advantage, thus leading to prioritization of volasertib in clinical development. Preclinical efficacy was determined in several cell culture assays and its derivative xenograft models, including the lung cancer cell line NCI-H460. Furthermore, it was shown that volasertib (and BI 2536) was effective in EGFR mutant cell lines, and drug-induced cell death was increased in those gaining acquired resistance, especially due to epithelial-to-mesenchymal transition, thus providing a rationale for volasertib and EGFR TKI combination strategies.47–49 Therapeutical potential of volasertib in preclinical assays has also been demonstrated for SCLC cell lines, in which PLK1 inhibition significantly attenuated cell proliferation and induced apoptosis in a dose-dependent manner.50
The clinical development of volasertib in patients suffering from NSCLC consists of 6 clinical trials, 5 of which were Phase I trials enrolling various solid tumor entities in the relapsed or refractory state.51–56 Two of these trials evaluated increasing doses of volasertib in combination with other target therapy compounds, afatinib54 and nintedanib,56 respectively. In total, 25 patients with NSCLC were enrolled in these 5 trials, of which 2 patients achieved a partial remission as a best response. Transient hematologic side effects, fatigue and gastrointestinal toxicity were the most common dose-limiting adverse events; however, treatment was overall reported as well-tolerated.
A three-armed Phase II trial evaluating volasertib single agent or in combination with pemetrexed against pemetrexed monotherapy was conducted by Ellis et al. The trial enrolled 131 patients with relapsed or refractory NSCLC. Although the overall response rate of 21% was highest in patients treated in the combination trial arm compared to volasertib (8.1%) or pemetrexed (10.6%) monotherapy, PFS with the combination strategy (3.3 months) was inferior to pemetrexed single agent treatment. In an exploratory comparison between volasertib monotherapy and pemetrexed monotherapy, the HR was 2.045 (95% CI, 1.27–3.292; p=0.0027) favoring pemetrexed, although surprisingly the response rates in both single-agent trial arms were similar. The discordance between response rate and PFS was attributed to chance imbalances within this small clinical trial.
Although all these trials could convincingly demonstrate that the toxicity profile of volasertib was very well manageable, even in combination strategies, the overall efficacy in NSCLC patients was lower than expected.
ON01910 (rigosertib)
ON01910 was the first-of-its class non-ATP-competitive PLK1 inhibitor binding near or at the PLK1 substrate binding site, since substrates compete for its inhibitory activity.57 In vitro studies with normal human cell lines showed surprisingly low growth attenuation and were very resistant to the apoptotic effect of ON01910. In several tumor cell culture assays (including two lung cancer cell lines: A549, H157), however, ON01910 induced mitotic arrest characterized by spindle abnormalities leading to apoptotic death. Of interest, it was tested if co-culturing of tumor cells with sublethal amounts of ON01910 may induce resistance to the compound, but such cell lines could not be isolated, indicating that acquired resistance to ON01910 itself may cause a survival disadvantage. Furthermore, ON01910 has been identified as an effective inhibitor of the PI3K/Akt/mTOR pathway in preclinical series.58,59
The clinical development of ON01910 in NSCLC is based on two Phase I clinical trials, that enrolled in total of 3 NSCLC cases,60,61 all of which did not respond to this non-ATP-competitive agent. Aside of transient hematologic side effects, dose-limiting toxicities consisted of disturbances in electrolyte balances, pelvic pain and urinary tract symptoms in addition to the occurrence of neurocognitive dysfunction. These side effects differed to former experiences with PLK1 inhibitors, presumably owing to the different mechanism of action of ON01910. Priority of the clinical development of rigosertib is currently restricted to malignant hematologic diseases.
NMS-1286937/HMN-214
Two early-phase clinical trials had been conducted aiming to define the MTD and overall safety with additional PLK1 inhibiting compounds: HMN-214 and NMS-1286937.62,63 Altogether 9 lung cancer patients were recruited with no documented response in these patients. Safety and toxicity were overall comparable to other ATP-competitive PLK1 inhibitors.
Other ATP and non-ATP competitive PLK1 inhibitors
There is an ongoing list of ATP-competitive and non-competitive compounds in preclinical or early phase clinical development (eg, MLN0905,64 Ro3280,65 CFI-400945,66 CYC14067); however, patients with lung cancer have not been enrolled in trials conducted with these compounds; thus, detailed information on preclinical and/or trial results are not presented here. A list of ongoing clinical trials involving PLK1 inhibitors in solid malignancies depicts Table 3.
 |
Table 3 Active clinical trials with PLK1 inhibitors in solid malignancies/NSCLC |
The new class of polo-box-domain inhibitors
The highly conserved nature of the ATP-binding pocket and the anticipated or experienced side effects of its inhibition gave rise to a new class of PLK1 targeting compounds that target the PLK1 tandem polo-box domain (PBD). Of interest, unlike PLK1-3, PLK5 only contains a single PBD. Polo-box binding inhibitors fall into 2 categories, peptide-based and small-molecule based. Peptides primarily provide functional information on protein–protein interactions and give rise to the identification of surface structures and potential binding pockets for targeting the PBD, that might be allosterically masked in the absence of a peptide ligand. However, peptides are in general not cell-permeable and are susceptible to proteolysis. These inhibitors serve as proof-of-concept models to accelerate the development of pharmacological agents. Small molecule inhibitors on the other hand provide the advantages of cell permeability, are characterized by a high degree of selectivity and represent inhibitors for potential cancer-therapy.
The first phosphopeptide designed to target the PDB was PoloBoxtide optimal that was not specifically designed to target the PBD, but was co-identified with the regulatory domain of the PLKs themselves.12,68 The definition of the optimal binding motif of the PBD in these pivotal studies provided the basis for the development of PBD targeting agents. Other peptide-based inhibitors, PLHSpT, FDPPLHSpTA, and their modified sister compounds were able to induce mitotic arrest in a high proportion of cancer cells after microinjection in culture assays, supporting their mechanism of action in the context of malignancy.69 The first small-molecule inhibitor of the PBD was Poloxin that was found through systematic screening of chemical libraries.70 The compound induced mitotic arrest and apoptosis in HeLa cells with an IC50 of 4.8 µM and suppressed tumor growth in xenograft mouse models.71 A handful of comparable compounds have been developed/identified so far: Poloxin-2,72 Purpurogallin,69 Poloxipan,73 bg-34,74 Poloppin75 and Poloppin-2.75 To our knowledge, there is currently no clinical trial recruiting with this new type of PLK inhibitors. However, one of the main green tea polyphenols (epigallocatechin) was found to be an inhibitor of the PLK1 PBD,76 thus falling into this category of PLK inhibitors. The only clinical trial that was conducted in 17 patients with advanced lung cancer treated with green tea extract found low efficacy with no objective responses.77
The search for predictive biomarkers
The clinical activity of PLK1 inhibitors in NSCLC has only shown modest activity. Still, a small proportion of patients derives the meaningful clinical benefit that might be masked among a larger proportion of patients who fail to benefit from PLK1 inhibition. Therefore, the identification of predictive biomarkers might be the key to enrich patients who benefit from this class of inhibitors. Unfortunately, translational research and exploratory biomarker analysis were not part of the study objectives in most early phase clinical trials that had involved patients with NSCLC. Still, there is some preclinical and clinical evidence on molecular mechanisms that support the usefulness of PLK1 inhibition in specific (NSCL) cancer subentities.
Preclinical evidence for emerging biomarkers
It has been consistently shown that TP53 defective cancer cell lines were more prone to reduced survival than their wild-type counterparts.78–84 Although TP53 usually acts in an anticancer capacity, it was recently shown that functional TP53 reduces the sensitivity of cell lines to PLK1 inhibitors and maintains centrosome separation and completion of mitosis in the presence of PLK1 inhibition.85 Cells lacking TP53 show an increased proportion of abnormal spindle formation and arrest in G1 phase upon treatment with PLK1 inhibitors. Therefore, the presence of inactivated TP53 might serve as a predictive biomarker in NSCLC patients, that is present in approximately 50% of all cases.86
KRAS mutations are one of the most common genetic alterations in patients with NSCLC; however, its prognostic role as a single or co-mutation has extensively been investigated with inconsistent results (eg, reviewed in87). There have been attempts to therapeutically address KRAS mutated tumors (such as NSCLC) which have been proven to be largely refractory to those target treatment approaches. An RNAi screen performed by Luo et al identified PLK1 and components of the APC/C ubiquitin-conjugating holoenzyme as a survival critical cofactor in KRAS p.G12C cell lines and xenografts.88 Increased apoptosis in KRAS mutated cell lines compared to their wild-type counterparts was also seen after treatment with the PDB inhibitor Pollopin,75 BI 6727 and BI2536.47 Furthermore, it has been shown that ON01910 (rigosertib) has the ability to bind to the RAS-binding domain of the RAF family proteins and thus effectively blocking RAS downstream signaling.89 The PI3K-mTor pathway as a second key regulator of cell growth cross-talks with the RAS-RAF-ERK pathway and positively and negatively co-regulate each other.90 As mentioned above, rigosertib has been identified as an effective inhibitor of PI3K. In xenograft models of colorectal and lung cancer (A549 cell line xenograft), reduction in tumor growth after rigosertib treatment was accompanied by downregulation of RAS-RAF-ERK and PI3K-mTor pathways. This has also been shown for other preclinical models of hepatocellular carcinoma, head and neck and prostate cancers.79,91,92 Whether this effect is highly specific for rigosertib or is also traceable in other PLK1 inhibitors is not conclusively clarified; however, a direct targeting of PI3K has not been shown for the other compounds. In summary, tumors harboring activating KRAS mutations or show increased activity of RAS downstream signaling pathways are supposed to be more sensitive to PLK1 targeting. In case of rigosertib, (co-) activation of the PI3K pathway, as indicated by PIK3CA expression or PTEN loss (a tumor suppressor gene counteracting to the PI3K pathway), might additionally function as a predictive marker for tumor response in PLK1 inhibition. However, although KRAS and TP53 alterations preclinically indicate a sensitization to PLK1 inhibition, the high proportion of patients with NSCLC harboring these mutations and the low tumor response rates contradict a sufficient sensitivity of these markers.
Cyclin B1, a physiologic target structure of PLK1, is a key mitotic cyclin in the G2/M phase transition of the cell cycle, is overexpressed in various tumor entities and has consistently been described as a negative prognostic factor in NSCLC with a frequency of approximately 20%, particularly in squamous cell NSCLC.93–97 In one study, the dynamic reduction of mRNA transcription of cyclin B1 upon treatment with ON01910 positively correlated with response to PLK1 inhibition in patient-derived pancreatic cancer xenografts.98 Thus, although absolute expression of cyclin B1 did not conclusively predict tumor response in xenografts or in those patients who were subsequently treated with ON01910, early dynamic changes were highly predictive for treatment response. A similar relationship has been described in patients with hematologic malignancies treated with ON01910 and the dynamic changes of cyclin D1 expression measured by flow cytometry.99 However, this relationship has not been proven in lung cancer (models) yet. Other biomarkers with predictive potential have been evaluated in solid malignancies other than NSCLC. The centrosomal protein CEP55 is a downstream target of the MAPK pathway and a key regulator of cytokinesis.100 Its overexpression is associated with inferior survival in NSCLC and breast cancer.101 In xenograft models of triple negative breast cancer, the combined inhibition of MEK1/2 (AZD6244) and PLK1 (volasertib) substantially impacted tumor growth.100 However, the role of CEP55 as a potential biomarker for PLK1 inhibition in NSCLC has not been elucidated yet. Additionally, head and neck squamous cell cncer (HNSCC) cell lines and xenograft models with mutated AJUBA, presumably an essential activator of the Aurora Kinase A, were highly sensitive to knockdown or inhibition of PLK1.102 Presence of activating AJUBA mutations might therefore play a role as a predictive biomarker in head and neck cancers and other solid malignancies for successful PLK1 targeting.102
Finally, there is emerging evidence that PLK1 activation plays a key role as an acquired mechanism of resistance, particularly in the process of epithelial-to-mesenchymal transition (EMT). EMT is a complex molecular and cellular process involved in tissue remodeling that has been linked to tumor progression and metastasis and precludes a high plasticity of cancer cells.103 Importantly, EMT has been found to be associated with inherited and acquired drug resistance,104–106 in case of NSCLC particularly in refractoriness to platinum therapy and acquired resistance to EGFR TKIs.107–109 Presence of an EMT gene signature determined by analyzing 264 different genes in lung cancer cell lines predicted sensitivity to PLK1 inhibition.47 This could also be shown in two erlotinib-resistant cell lines with activating EGFR mutations that harbored an EMT gene profile.49 Of note, it was recently shown that NSCLC cell lines (HEK293) with epithelial gene signature and constitutive cMET activation were highly resistant to volasertib treatment, whereas PLK1 inhibition prevented cMET phosphorylation in mesenchymal isogeneic cell pairs leading to high apoptosis rates.110 The combination of cMET and PLK1 inhibition in this setting led to sustainable xenograft tumor regression possibly paving the way for combination strategies involving PLK1 and cMET targeting therapy. Conclusively, acquired resistance to target therapies or conventional cytotoxic agents involving EMT might be a domain of PLK1 inhibition in the future.
Clinical and patient-derived evidence for predictive biomarkers
The largest set of tumor samples for exploratory biomarker screening were obtained from the phase I trial conducted by Sebastian et al with BI 2536.44 Breitenbuecher and colleagues performed broad spectrum posthoc analyses with archival tumor samples from 47 patients using immunohistochemistry and DNA sequencing.111 Sequence analysis of all mutational hotspot exons of KRAS, EGFR, BRAF and PIK3CA was successfully conducted in 26 patients, immunohistochemistry analysis for the expression of PTEN, HER2, PLK, p-AKT and p-ERK was informative in 20 patients. Exploratory correlation analyses showed notably long PFS in some patients harboring KRAS mutations in the cohort of nonsquamous NSCLC, but this relationship failed to meet statistical significance. There was no survival difference in patients with KRAS-mutated lung cancer compared to the KRAS wildtype cohort. Partially owing to the small sample size, other outcome associations with genetic alterations could not be identified. When testing for PI3K/AKT/mTOR pathway activation indicated by positive ERK, AKT and (loss of) PTEN IHC, there was no significant relationship with PFS or OS outcomes.
In the Phase I trial in relapsed or refractory solid malignancies (NSCLC, n=1) conducted by Bowles et al, archival tumor tissue blocks from 32 participants were assessed with DNA sequencing for the presence of common mutations in 15 hotspot genes.61 Additionally, in a subset of patients, FISH analyses were performed to determine the PIK3CA and PTEN gene copy number. In a subset of patients with squamous cell cancers (n=6; NSCLC, n=1), a PI3K pathway activation via PIK3CA amplification and PTEN loss in combination with an inactivated TP53 was seen in those 2 patients with therapy response. However, others with these mutations failed to respond to therapy indicating that both mutations may be necessary but are not sufficient to induce a response to PLK1 inhibition.
Conclusions
The captivating position of PLK1 as a central key regulator of mitosis leads to numerous attempts to pharmacologically disrupt PLK1 signaling in various cancer entities, including lung cancer. Currently, three different types of small-molecule inhibitors – ATP-competitive, non-ATP competitive and PBD inhibitors – have found their way into clinical trials; however, most experiences were gained with the ATP-competitive compounds BI 2536 and BI 6727 (Volasertib). The overall toxicity of PLK inhibitors was judged as well manageable with dose-dependent hematotoxicity as the limiting side effect in most clinically tested compounds. The overall efficacy in patients with NSCLC however was limited and in the context of the rapidly broadening landscape of immunotherapy (combinations) and small-molecule strategies, further development of PLK1 inhibitors is currently primarily restricted to more promising areas, such as hematologic malignancies. Still, a small proportion of patients shows an enduring benefit of PLK1 inhibition, probably masked by a higher proportion of patients showing refractoriness to this small-molecule class. There have been few clinical attempts to define predictive biomarkers to enrich lung cancer patients that may derive increased benefit from PLK1 inhibition, but in summary, these suggested biomarkers performed weakly in predicting tumor responses in NSCLC patients and more research is needed to define the molecular frame of effective PLK1 targeting. In recent years, the role of PLK1 in acquired therapy resistance has been investigated and has been linked to resistance to cytotoxic agents as well as target therapies. In light of these observations, the future domain of PLK1 inhibitors in the treatment of lung cancer may be found in combination strategies to prevent early treatment failure and delay obligatory acquired resistance in advanced or metastatic disease. More studies are inevitably needed to define the optimal role of PLK1 inhibition in lung malignancies.
Disclosure
Dr Jan A Stratmann reports personal fees from Bristol-Myers & Squibb, personal fees from Novartis, outside the submitted work. Dr Martin Sebastian reports personal fees from Lilly, personal fees from Astra-Zeneca, personal fees from Bristol-Myers & Squibb, personal fees from Merck Sharp & Dohme, personal fees from Pfizer, personal fees from Takeda, personal fees from Roche, personal fees from AbbVie, personal fees from Boehringer-Ingelheim, personal fees from Celgene, personal fees from Novartis, outside the submitted work. The authors report no other conflicts of interests in this work.
References
1. Cheng T-YD, Cramb SM, Baade PD, Youlden DR, Nwogu C, Reid ME. The international epidemiology of lung cancer: latest trends, disparities, and tumor characteristics. J Thorac Oncol. 2016;11:1653–1671. doi:10.1016/j.jtho.2016.05.021
2. McPhail S, Johnson S, Greenberg D, Peake M, Rous B. Stage at diagnosis and early mortality from cancer in England. Br J Cancer. 2015;112(Suppl 1):S108–S115. doi:10.1038/bjc.2015.49
3. Lake RJ, Jelinek WR. Cell cycle- and terminal differentiation-associated regulation of the mouse mRNA encoding a conserved mitotic protein kinase. Mol Cell Biol. 1993;13(12):7793–7801. doi:10.1128/mcb.13.12.7793
4. Holtrich U, Wolf G, Bräuninger A, et al. Induction and down-regulation of PLK, a human serine/threonine kinase expressed in proliferating cells and tumors. Proc Natl Acad Sci U S A. 1994;91(5):1736–1740. doi:10.1073/pnas.91.5.1736
5. Hamanaka R, Maloid S, Smith MR, O’Connell CD, Longo DL, Ferris DK. Cloning and characterization of human and murine homologues of the Drosophila polo serine-threonine kinase. Cell Growth Differ. 1994;5(3):249–257.
6. Liby K, Wu H, Ouyang B, Wu S, Chen J, Dai W. Identification of the human homologue of the early-growth response gene Snk, encoding a serum-inducible kinase. DNA Seq. 2001;11(6):527–533.
7. Li B, Ouyang B, Pan H, et al. Prk, a cytokine-inducible human protein serine/threonine kinase whose expression appears to be down-regulated in lung carcinomas. J Biol Chem. 1996;271(32):19402–19408. doi:10.1074/jbc.271.32.19402
8. Martin C-A, Ahmad I, Klingseisen A, et al. Mutations in PLK4, encoding a master regulator of centriole biogenesis, cause microcephaly, growth failure and retinopathy. Nat Genet. 2014;46(12):1283–1292. doi:10.1038/ng.3122
9. de Cárcer G, Escobar B, Higuero AM, et al. Plk5, a polo box domain-only protein with specific roles in neuron differentiation and glioblastoma suppression. Mol Cell Biol. 2011;31(6):1225–1239. doi:10.1128/MCB.00607-10
10. Strebhardt K, Ullrich A. Targeting polo-like kinase 1 for cancer therapy. Nat Rev Cancer. 2006;6(4):321–330. doi:10.1038/nrc1841
11. Hanks SK, Quinn AM, Hunter T. The protein kinase family: conserved features and deduced phylogeny of the catalytic domains. Science. 1988;241(4861):42–52.
12. Elia AEH, Rellos P, Haire LF, et al. The molecular basis for phosphodependent substrate targeting and regulation of PLKs by the polo-box domain. Cell. 2003;115(1):83–95. doi:10.1016/S0092-8674(03)00725-6
13. Golsteyn RM, Mundt KE, Fry AM, Nigg EA. Cell cycle regulation of the activity and subcellular localization of PLK1, a human protein kinase implicated in mitotic spindle function. J Cell Biol. 1995;129(6):1617–1628. doi:10.1083/jcb.129.6.1617
14. Wolfe BA, Takaki T, Petronczki M, Glotzer M. Polo-like kinase 1 directs assembly of the HsCyk-4 RhoGAP/Ect2 RhoGEF complex to initiate cleavage furrow formation. PLoS Biol. 2009;7(5):e1000110. doi:10.1371/journal.pbio.1000110
15. Petronczki M, Glotzer M, Kraut N, Peters J-M. Polo-like kinase 1 triggers the initiation of cytokinesis in human cells by promoting recruitment of the RhoGEF Ect2 to the central spindle. Dev Cell. 2007;12(5):713–725. doi:10.1016/j.devcel.2007.03.013
16. Moshe Y, Boulaire J, Pagano M, Hershko A. Role of Polo-like kinase in the degradation of early mitotic inhibitor 1, a regulator of the anaphase promoting complex/cyclosome. Proc Natl Acad Sci U S A. 2004;101(21):7937–7942. doi:10.1073/pnas.0402442101
17. Hansen DV, Loktev AV, Ban KH, Jackson PK. PLK1 regulates activation of the anaphase promoting complex by phosphorylating and triggering SCFbetaTrCP-dependent destruction of the APC Inhibitor Emi1. Mol Biol Cell. 2004;15(12):5623–5634. doi:10.1091/mbc.e04-07-0598
18. Yan W, Yu H, Li W, et al. PLK1 promotes the migration of human lung adenocarcinoma epithelial cells via STAT3 signaling. Oncol Lett. 2018;16(5):6801–6807. doi:10.3892/ol.2018.9437
19. Li H, Wang Y, Liu X. PLK1-dependent phosphorylation regulates functions of DNA topoisomerase IIalpha in cell cycle progression. J Biol Chem. 2008;283(10):6209–6221. doi:10.1074/jbc.M709007200
20. Rizki A, Mott JD, Bissell MJ. Polo-like kinase 1 is involved in invasion through extracellular matrix. Cancer Res. 2007;67(23):11106–11110. doi:10.1158/0008-5472.CAN-07-2348
21. Wu Z-Q, Yang X, Weber G, Liu X. PLK1 phosphorylation of TRF1 is essential for its binding to telomeres. J Biol Chem. 2008;283(37):25503–25513. doi:10.1074/jbc.M803304200
22. Toyoshima-Morimoto F, Taniguchi E, Shinya N, Iwamatsu A, Nishida E. Polo-like kinase 1 phosphorylates cyclin B1 and targets it to the nucleus during prophase. Nature. 2001;410(6825):215–220. doi:10.1038/35065617
23. Seki A, Coppinger JA, Jang C-Y, Yates JR, Fang G. Bora and the kinase Aurora a cooperatively activate the kinase PLK1 and control mitotic entry. Science. 2008;320(5883):1655–1658. doi:10.1126/science.1157425
24. Macůrek L, Lindqvist A, Lim D, et al. Polo-like kinase-1 is activated by aurora A to promote checkpoint recovery. Nature. 2008;455(7209):119–123. doi:10.1038/nature07185
25. Smith MR, Wilson ML, Hamanaka R, et al. Malignant transformation of mammalian cells initiated by constitutive expression of the polo-like kinase. Biochem Biophys Res Commun. 1997;234(2):397–405.
26. Liu X, Erikson RL. Polo-like kinase (PLK)1 depletion induces apoptosis in cancer cells. Proc Natl Acad Sci U S A. 2003;100(10):5789–5794. doi:10.1073/pnas.1031523100
27. Liu X, Erikson RL. Activation of Cdc2/cyclin B and inhibition of centrosome amplification in cells depleted of PLK1 by siRNA. Proc Natl Acad Sci U S A. 2002;99(13):8672–8676. doi:10.1073/pnas.132269599
28. Spänkuch-Schmitt B, Bereiter-Hahn J, Kaufmann M, Strebhardt K. Effect of RNA silencing of polo-like kinase-1 (PLK1) on apoptosis and spindle formation in human cancer cells. J Natl Cancer Inst. 2002;94(24):1863–1877. doi:10.1093/jnci/94.24.1863
29. Spänkuch-Schmitt B, Wolf G, Solbach C, et al. Downregulation of human polo-like kinase activity by antisense oligonucleotides induces growth inhibition in cancer cells. Oncogene. 2002;21(20):3162–3171. doi:10.1038/sj.onc.1205412
30. Sumara I, Gimenez-Abian JF, Gerlich D, et al. Roles of polo-like kinase 1 in the assembly of functional mitotic spindles. Curr Biol. 2004;14(19):1712–1722. doi:10.1016/j.cub.2004.09.049
31. Wolf G, Elez R, Doermer A, et al. Prognostic significance of polo-like kinase (PLK) expression in non-small cell lung cancer. Oncogene. 1997;14(5):543–549. doi:10.1038/sj.onc.1200862
32. Allera-Moreau C, Rouquette I, Lepage B, et al. DNA replication stress response involving PLK1, CDC6, POLQ, RAD51 and CLASPIN upregulation prognoses the outcome of early/mid-stage non-small cell lung cancer patients. Oncogenesis. 2012;1:e30. doi:10.1038/oncsis.2012.29
33. Wang Z-X, Xue D, Liu Z-L, et al. Overexpression of polo-like kinase 1 and its clinical significance in human non-small cell lung cancer. Int J Biochem Cell Biol. 2012;44(1):200–210. doi:10.1016/j.biocel.2011.10.017
34. Li H, Wang H, Sun Z, Guo Q, Shi H, Jia Y. The clinical and prognostic value of polo-like kinase 1 in lung squamous cell carcinoma patients: immunohistochemical analysis. Biosci Rep. 2017;37:BSR20170852. doi:10.1042/BSR20170852
35. Zhou Q, Su Y, Bai M. Effect of antisense RNA targeting Polo-like kinase 1 on cell growth in A549 lung cancer cells. J Huazhong Univ Sci Technol Med Sci. 2008;28(1):22–26. doi:10.1007/s11596-008-0106-9
36. Cogswell JP, Brown CE, Bisi JE, Neill SD. Dominant-negative polo-like kinase 1 induces mitotic catastrophe independent of cdc25C function. Cell Growth Differ. 2000;11(12):615. http://cgd.aacrjournals.org/cgi/content/full/11/12/615
37. Kawata E, Ashihara E, Maekawa T. RNA interference against polo-like kinase-1 in advanced non-small cell lung cancers. J Clin Bioinforma. 2011;1(1):6. doi:10.1186/2043-9113-1-6
38. Zhao X-Y, Nie C-L, Liang S-F, Yuan Z, Deng H-X, Wei Y-Q. Enhanced gemcitabine-mediated cell killing of human lung adenocarcinoma by vector-based RNA interference against PLK1. Biomed Pharmacother. 2012;66(8):597–602. doi:10.1016/j.biopha.2012.01.003
39. Sinha RP, Häder D-P. UV-protectants in cyanobacteria. Plant Sci. 2008;174(3):278–289. doi:10.1016/j.plantsci.2007.12.004
40. Stevenson CS, Capper EA, Roshak AK, et al. The identification and characterization of the marine natural product scytonemin as a novel antiproliferative pharmacophore. J Pharmacol Exp Ther. 2002;303(2):858–866. doi:10.1124/jpet.102.036350
41. Steegmaier M, Hoffmann M, Baum A, et al. BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Curr Biol. 2007;17(4):316–322. doi:10.1016/j.cub.2006.12.037
42. Mross K, Frost A, Steinbild S, et al. Phase I dose escalation and pharmacokinetic study of BI 2536, a novel Polo-like kinase 1 inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2008;26(34):5511–5517. doi:10.1200/JCO.2008.16.1547
43. Hofheinz R-D, Al-Batran S-E, Hochhaus A, et al. An open-label, phase I study of the polo-like kinase-1 inhibitor, BI 2536, in patients with advanced solid tumors. Clin Cancer Res. 2010;16(18):4666–4674. doi:10.1158/1078-0432.CCR-10-0318
44. Sebastian M, Reck M, Waller CF, et al. The efficacy and safety of BI 2536, a novel PLK-1 inhibitor, in patients with stage IIIB/IV non-small cell lung cancer who had relapsed after, or failed, chemotherapy: results from an open-label, randomized phase II clinical trial. J Thorac Oncol. 2010;5(7):1060–1067. doi:10.1097/JTO.0b013e3181d95dd4
45. Ellis PM, Chu QS, Leighl N, et al. A phase I open-label dose-escalation study of intravenous BI 2536 together with pemetrexed in previously treated patients with non-small-cell lung cancer. Clin Lung Cancer. 2013;14(1):19–27. doi:10.1016/j.cllc.2012.04.003
46. Rudolph D, Steegmaier M, Hoffmann M, et al. BI 6727, a Polo-like kinase inhibitor with improved pharmacokinetic profile and broad antitumor activity. Clin Cancer Res. 2009;15(9):3094–3102. doi:10.1158/1078-0432.CCR-08-2445
47. Ferrarotto R, Goonatilake R, Yoo SY, et al. Epithelial-mesenchymal transition predicts polo-like kinase 1 inhibitor-mediated apoptosis in non-small cell lung cancer. Clin Cancer Res. 2016;22(7):1674–1686. doi:10.1158/1078-0432.CCR-14-2890
48. Crystal AS, Shaw AT, Sequist LV, et al. Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science. 2014;346(6216):1480–1486. doi:10.1126/science.1254721
49. Wang Y, Singh R, Wang L, et al. Polo-like kinase 1 inhibition diminishes acquired resistance to epidermal growth factor receptor inhibition in non-small cell lung cancer with T790M mutations. Oncotarget. 2016;7(30):47998–48010. doi:10.18632/oncotarget.10332
50. Wang Y, Wu L, Yao Y, Lu G, Xu L, Zhou J. Polo-like kinase 1 inhibitor BI 6727 induces DNA damage and exerts strong antitumor activity in small cell lung cancer. Cancer Lett. 2018;436:1–9. doi:10.1016/j.canlet.2018.08.007
51. Gil T, Schöffski P, Awada A, et al. Final analysis of a phase I single dose-escalation study of the novel polo-like kinase 1 inhibitor BI 6727 in patients with advanced solid tumors. J Clin Oncol. 2010;28(15_suppl):3061. doi:10.1200/jco.2010.28.15_suppl.3061
52. Schöffski P, Awada A, Dumez H, et al. A phase I, dose-escalation study of the novel Polo-like kinase inhibitor volasertib (BI 6727) in patients with advanced solid tumours. Eur J Cancer. 2012;48(2):179–186. doi:10.1016/j.ejca.2011.11.001
53. Lin -C-C, Su W-C, Yen C-J, et al. A phase I study of two dosing schedules of volasertib (BI 6727), an intravenous polo-like kinase inhibitor, in patients with advanced solid malignancies. Br J Cancer. 2014;110(10):2434–2440. doi:10.1038/bjc.2014.195
54. Machiels J-P, Peeters M, Herremans C, et al. A phase I study of volasertib combined with afatinib, in advanced solid tumors. Cancer Chemother Pharmacol. 2015;76(4):843–851. doi:10.1007/s00280-015-2860-2
55. Nokihara H, Yamada Y, Fujiwara Y, et al. Phase I trial of volasertib, a Polo-like kinase inhibitor, in Japanese patients with advanced solid tumors. Invest New Drugs. 2016;34(1):66–74. doi:10.1007/s10637-015-0300-0
56. de Braud F, Cascinu S, Spitaleri G, et al. A phase I, dose-escalation study of volasertib combined with nintedanib in advanced solid tumors. Ann Oncol. 2015;26(11):2341–2346. doi:10.1093/annonc/mdv354
57. Gumireddy K, Reddy MVR, Cosenza SC, et al. ON01910, a non-ATP-competitive small molecule inhibitor of PLK1, is a potent anticancer agent. Cancer Cell. 2005;7(3):275–286. doi:10.1016/j.ccr.2005.02.009
58. Prasad A, Khudaynazar N, Tantravahi RV, Gillum AM, Hoffman BS. ON 01910.Na (rigosertib) inhibits PI3K/Akt pathway and activates oxidative stress signals in head and neck cancer cell lines. Oncotarget. 2016;7(48):79388–79400. doi:10.18632/oncotarget.12692
59. Xu F, He Q, Li X, et al. Rigosertib as a selective anti-tumor agent can ameliorate multiple dysregulated signaling transduction pathways in high-grade myelodysplastic syndrome. Sci Rep. 2014;4:7310. doi:10.1038/srep07310
60. Ohnuma T, Lehrer D, Ren C, et al. Phase 1 study of intravenous rigosertib (ON 01910.Na), a novel benzyl styryl sulfone structure producing G2/M arrest and apoptosis, in adult patients with advanced cancer. Am J Cancer Res. 2013;3(3):323–338.
61. Bowles DW, Diamond JR, Lam ET, et al. Phase I study of oral rigosertib (ON 01910.Na), a dual inhibitor of the PI3K and PLK1 pathways, in adult patients with advanced solid malignancies. Clin Cancer Res. 2014;20(6):1656–1665. doi:10.1158/1078-0432.CCR-13-2506
62. Garland LL, Taylor C, Pilkington DL, Cohen JL, von Hoff DD. A phase I pharmacokinetic study of HMN-214, a novel oral stilbene derivative with polo-like kinase-1-interacting properties, in patients with advanced solid tumors. Clin Cancer Res. 2006;12(17):5182–5189. doi:10.1158/1078-0432.CCR-06-0214
63. Weiss GJ, Jameson G, von Hoff DD, et al. Phase I dose escalation study of NMS-1286937, an orally available Polo-Like Kinase 1 inhibitor, in patients with advanced or metastatic solid tumors. Invest New Drugs. 2018;36(1):85–95. doi:10.1007/s10637-017-0491-7
64. Duffey MO, Vos TJ, Adams R, et al. Discovery of a potent and orally bioavailable benzolactam-derived inhibitor of Polo-like kinase 1 (MLN0905). J Med Chem. 2012;55(1):197–208. doi:10.1021/jm2011172
65. Wang -N-N, Li Z-H, Zhao H, et al. Molecular targeting of the oncoprotein PLK1 in pediatric acute myeloid leukemia: RO3280, a novel PLK1 inhibitor, induces apoptosis in leukemia cells. Int J Mol Sci. 2015;16(1):1266–1292. doi:10.3390/ijms16011266
66. Mason JM, Lin DCC, Wei X, et al. Functional characterization of CFI-400945, a Polo-like kinase 4 inhibitor, as a potential anticancer agent. Cancer Cell. 2014;26(2):163–176. doi:10.1016/j.ccr.2014.05.006
67. Moureau S, Pohler E, Kroboth K, et al. Therapeutic potential of novel PLK1 inhibitor CYC140 in esophageal cancer and acute leukemia. European Journal of Cancer. 2016;69:S117. doi:10.1016/S0959-8049(16)32948-3
68. Elia AEH, Cantley LC, Yaffe MB. Proteomic screen finds pSer/pThr-binding domain localizing PLK1 to mitotic substrates. Science. 2003;299(5610):1228–1231. doi:10.1126/science.1079079
69. Yun S-M, Moulaei T, Lim D, et al. Structural and functional analyses of minimal phosphopeptides targeting the polo-box domain of polo-like kinase 1. Nat Struct Mol Biol. 2009;16(8):876–882. doi:10.1038/nsmb.1628
70. Reindl W, Strebhardt K, Berg T. A high-throughput assay based on fluorescence polarization for inhibitors of the polo-box domain of polo-like kinase 1. Anal Biochem. 2008;383(2):205–209. doi:10.1016/j.ab.2008.08.014
71. Yuan J, Sanhaji M, Krämer A, et al. Polo-box domain inhibitor poloxin activates the spindle assembly checkpoint and inhibits tumor growth in vivo. Am J Pathol. 2011;179(4):2091–2099. doi:10.1016/j.ajpath.2011.06.031
72. Scharow A, Raab M, Saxena K, et al. Optimized PLK1 PBD inhibitors based on poloxin induce mitotic arrest and apoptosis in tumor cells. ACS Chem Biol. 2015;10(11):2570–2579. doi:10.1021/acschembio.5b00565
73. Reindl W, Yuan J, Krämer A, Strebhardt K, Berg T. A pan-specific inhibitor of the polo-box domains of polo-like kinases arrests cancer cells in mitosis. Chembiochem. 2009;10(7):1145–1148. doi:10.1002/cbic.200900059
74. Srinivasrao G, Park J-E, Kim S, et al. Design and synthesis of a cell-permeable, drug-like small molecule inhibitor targeting the polo-box domain of polo-like kinase 1. PLoS One. 2014;9(9):e107432. doi:10.1371/journal.pone.0107432
75. Narvaez AJ, Ber S, Crooks A, et al. Modulating protein-protein interactions of the mitotic polo-like kinases to target mutant KRAS. Cell Chem Biol. 2017;24(8):1017–1028.e7. doi:10.1016/j.chembiol.2017.07.009
76. Shan H-M, Shi Y, Quan J. Identification of green tea catechins as potent inhibitors of the polo-box domain of polo-like kinase 1. ChemMedChem. 2015;10(1):158–163. doi:10.1002/cmdc.201402284
77. Laurie SA, Miller VA, Grant SC, Kris MG, Ng KK. Phase I study of green tea extract in patients with advanced lung cancer. Cancer Chemother Pharmacol. 2005;55(1):33–38. doi:10.1007/s00280-004-0859-1
78. Guan R, Tapang P, Leverson JD, Albert D, Giranda VL, Luo Y. Small interfering RNA-mediated Polo-like kinase 1 depletion preferentially reduces the survival of p53-defective, oncogenic transformed cells and inhibits tumor growth in animals. Cancer Res. 2005;65(7):2698–2704. doi:10.1158/0008-5472.CAN-04-2131
79. Anderson RT, Keysar SB, Bowles DW, et al. The dual pathway inhibitor rigosertib is effective in direct patient tumor xenografts of head and neck squamous cell carcinomas. Mol Cancer Ther. 2013;12(10):1994–2005. doi:10.1158/1535-7163.MCT-13-0206
80. Degenhardt Y, Greshock J, Laquerre S, et al. Sensitivity of cancer cells to PLK1 inhibitor GSK461364A is associated with loss of p53 function and chromosome instability. Mol Cancer Ther. 2010;9(7):2079–2089. doi:10.1158/1535-7163.MCT-10-0095
81. McKenzie L, King S, Marcar L, et al. p53-dependent repression of polo-like kinase-1 (PLK1). Cell Cycle. 2010;9(20):4200–4212. doi:10.4161/cc.9.20.13532
82. Danovi D, Folarin A, Gogolok S, et al. A high-content small molecule screen identifies sensitivity of glioblastoma stem cells to inhibition of polo-like kinase 1. PLoS One. 2013;8(10):e77053. doi:10.1371/journal.pone.0077053
83. Liu X, Lei M, Erikson RL. Normal cells, but not cancer cells, survive severe PLK1 depletion. Mol Cell Biol. 2006;26(6):2093–2108. doi:10.1128/MCB.26.6.2093-2108.2006
84. Sur S, Pagliarini R, Bunz F, et al. A panel of isogenic human cancer cells suggests a therapeutic approach for cancers with inactivated p53. Proc Natl Acad Sci U S A. 2009;106(10):3964–3969. doi:10.1073/pnas.0813333106
85. Smith L, Farzan R, Ali S, Buluwela L, Saurin AT, Meek DW. The responses of cancer cells to PLK1 inhibitors reveal a novel protective role for p53 in maintaining centrosome separation. Sci Rep. 2017;7(1):16115. doi:10.1038/s41598-017-16394-2
86. Steels E, Paesmans M, Berghmans T, et al. Role of p53 as a prognostic factor for survival in lung cancer: a systematic review of the literature with a meta-analysis. Eur Respir J. 2001;18(4):705–719. doi:10.1183/09031936.01.00062201
87. Ferrer I, Zugazagoitia J, Herbertz S, John W, Paz-Ares L, Schmid-Bindert G. KRAS-Mutant non-small cell lung cancer: from biology to therapy. Lung Cancer. 2018;124:53–64. doi:10.1016/j.lungcan.2018.07.013
88. Luo J, Emanuele MJ, Li D, et al. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell. 2009;137(5):835–848. doi:10.1016/j.cell.2009.05.006
89. Athuluri-Divakar SK, Vasquez-Del Carpio R, Dutta K, et al. A small molecule RAS-mimetic disrupts RAS association with effector proteins to block signaling. Cell. 2016;165(3):643–655. doi:10.1016/j.cell.2016.03.045
90. Mendoza MC, Er EE, Blenis J. The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem Sci. 2011;36(6):320–328. doi:10.1016/j.tibs.2011.03.006
91. Liu XS, Song B, Elzey BD, et al. Polo-like kinase 1 facilitates loss of Pten tumor suppressor-induced prostate cancer formation. J Biol Chem. 2011;286(41):35795–35800. doi:10.1074/jbc.C111.269050
92. Dietrich P, Freese K, Mahli A, Thasler WE, Hellerbrand C, Bosserhoff AK. Combined effects of PLK1 and RAS in hepatocellular carcinoma reveal rigosertib as promising novel therapeutic “dual-hit” option. Oncotarget. 2018;9(3):3605–3618. doi:10.18632/oncotarget.23188
93. Ye C, Wang J, Wu P, et al. Prognostic role of cyclin B1 in solid tumors: a meta-analysis. Oncotarget. 2016;8(2):2224–2232. doi:10.18632/oncotarget.13653
94. Soria JC, Jang SJ, Khuri FR, et al. Overexpression of cyclin B1 in early-stage non-small cell lung cancer and its clinical implication. Cancer Res. 2000;60(15):4000–4004.
95. Cooper WA, Kohonen-Corish MRJ, McCaughan B, Kennedy C, Sutherland RL, Lee CS. Expression and prognostic significance of cyclin B1 and cyclin A in non-small cell lung cancer. Histopathology. 2009;55(1):28–36. doi:10.1111/j.1365-2559.2009.03331.x
96. Arinaga M, Noguchi T, Takeno S, et al. Clinical implication of cyclin B1 in non-small cell lung cancer. Oncol Rep. 2003;10(5):1381–1386.
97. Yoshida T, Tanaka S, Mogi A, Shitara Y, Kuwano H. The clinical significance of Cyclin B1 and Wee1 expression in non-small-cell lung cancer. Ann Oncol. 2004;15(2):252–256. doi:10.1093/annonc/mdh073
98. Jimeno A, Chan A, Cusatis G, et al. Evaluation of the novel mitotic modulator ON 01910.Na in pancreatic cancer and preclinical development of an ex vivo predictive assay. Oncogene. 2009;28(4):610. doi:10.1038/onc.2008.424
99. Olnes MJ, Shenoy A, Weinstein B, et al. Directed therapy for patients with myelodysplastic syndromes (MDS) by suppression of cyclin D1 with ON 01910.Na. Leuk Res. 2012;36(8):982–989. doi:10.1016/j.leukres.2012.04.002
100. Kalimutho M, Sinha D, Jeffery J, et al. CEP55 is a determinant of cell fate during perturbed mitosis in breast cancer. EMBO Mol Med. 2018;10:9. doi:10.15252/emmm.201708566
101. Jiang C, Zhang Y, Li Y, et al. High CEP55 expression is associated with poor prognosis in non-small-cell lung cancer. OTT. 2018;11:4979–4990. doi:10.2147/OTT.S165750
102. Zhang M, Singh R, Peng S, et al. Mutations of the LIM protein AJUBA mediate sensitivity of head and neck squamous cell carcinoma to treatment with cell-cycle inhibitors. Cancer Lett. 2017;392:71–82. doi:10.1016/j.canlet.2017.01.024
103. Legras A, Pécuchet N, Imbeaud S, et al. Epithelial-to-mesenchymal transition and MicroRNAs in lung cancer. Cancers (Basel). 2017;9:8. doi:10.3390/cancers9080101
104. Heery R, Finn SP, Cuffe S, Gray SG. Long non-coding RNAs: key regulators of epithelial-mesenchymal transition, tumour drug resistance and cancer stem cells. Cancers (Basel). 2017;9:4. doi:10.3390/cancers9040038
105. Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol. 2017;14(10):611–629. doi:10.1038/nrclinonc.2017.44
106. Du B, Shim JS. Targeting Epithelial-Mesenchymal Transition (EMT) to overcome drug resistance in cancer. Molecules. 2016;21:7. doi:10.3390/molecules21070965
107. Brozovic A. The relationship between platinum drug resistance and epithelial-mesenchymal transition. Arch Toxicol. 2017;91(2):605–619. doi:10.1007/s00204-016-1912-7
108. Xiao D, He J. Epithelial mesenchymal transition and lung cancer. J Thorac Dis. 2010;2(3):154–159. doi:10.3978/j.issn.2072-1439.2010.02.03.7
109. Hashida S, Yamamoto H, Shien K, et al. Acquisition of cancer stem cell-like properties in non-small cell lung cancer with acquired resistance to afatinib. Cancer Sci. 2015;106(10):1377–1384. doi:10.1111/cas.12749
110. Singh R, Peng S, Viswanath P, et al. Non-canonical cMet regulation by vimentin mediates PLK1 inhibitor-induced apoptosis. EMBO Mol Med. 2019;11:5. doi:10.15252/emmm.201809960
111. Breitenbuecher F, von Pawel J, Sebastian M, et al. Comprehensive biomarker analyses in patients with advanced or metastatic non-small cell lung cancer prospectively treated with the polo-like kinase 1 inhibitor BI2536. Oncol Res Treat. 2017;40(7–8):435–439. doi:10.1159/000475503
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.