Back to Journals » Hepatic Medicine: Evidence and Research » Volume 9
PNPLA3 expression and its impact on the liver: current perspectives
Authors Bruschi FV, Tardelli M, Claudel T, Trauner M
Received 4 August 2017
Accepted for publication 20 September 2017
Published 6 November 2017 Volume 2017:9 Pages 55—66
DOI https://doi.org/10.2147/HMER.S125718
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Gerry Lake-Bakaar
Francesca Virginia Bruschi, Matteo Tardelli, Thierry Claudel, Michael Trauner
Hans Popper Laboratory of Molecular Hepatology, Division of Gastroenterology & Hepatology, Department of Internal Medicine III, Medical University of Vienna, Austria
Abstract: A single-nucleotide polymorphism occurring in the sequence of the human patatin-like phospholipase domain-containing 3 gene (PNPLA3), known as I148M variant, is one of the best characterized and deeply investigated variants in several clinical scenarios, because of its tight correlation with increased risk for developing hepatic steatosis and more aggressive part of the disease spectrum, such as nonalcoholic steatohepatitis, advanced fibrosis and cirrhosis. Further, the I148M variant is positively associated with alcoholic liver diseases, chronic hepatitis C–related cirrhosis and hepatocellular carcinoma. The native gene encodes for a protein that has not yet a fully defined role in liver lipid metabolism and, according to recent observations, seems to be divergently regulated among distinct liver cells type, such as hepatic stellate cells. Therefore, the aim of this review is to collect the latest data regarding PNPLA3 expression in human liver and to analyze the impact of its genetic variant in human hepatic pathologies. Moreover, a description of the current biochemical and metabolic data pertaining to PNPLA3 function in both animal models and in vitro studies is summarized to allow a better understanding of the relevant pathophysiological role of this enzyme in the progression of hepatic diseases.
Keywords: adiponutrin, liver disease, genetic polymorphism, gene expression, metabolism
Introduction on PNPLA3: origin and tissue-specific expression
PNPLA3 (alternatively known as adiponutrin-ADPN and calcium-independent phospholipase A2 epsilon) belongs to a group of lipid-metabolizing enzymes, first identified in potato tubers, known as the patatin-like phospholipase domain-containing (PNPLA) family.1,2 In contrast to the other members of the PNPLA family, which show nonspecific lipid acyl hydrolase activity and are soluble proteins,3 PNPLA3 is tightly associated with membranes and lipid droplets.4 PNPLA3 was described for the first time in 2001, as a nutritionally regulated transcript, strongly induced during differentiation of the murine cell line 3T3-L1 into mature adipocytes. The human adiponutrin gene sequence is located in the long arm (q) of chromosome 22 at position 13.31 and encodes for a 481-amino-acid-length transcript, with a predicted molecular weight of 52.8 kDa.5 Among the nine members of the PNPLA family, PNPLA3 shows the highest homology with PNPLA2, also known as adipose triglyceride (TG) lipase, ATGL or desnutrin (alternatively known as calcium-independent phospholipase A2 zeta). ATGL plays a critical role in TG metabolism by mediating a rate-limiting step in TG hydrolysis.6 Both adiponutrin and desnutrin/ATGL contain the conserved structure motive of the patatin-like domain, which comprises a β-sheet sandwiched between two α-helices and includes a consensus serine lipase motif (Gly-X-Ser-X-Gly), but different from classical lipases by employing a catalytic dyad (Ser-Asp), as opposed to a catalytic triad to affect hydrolysis.2,7,8
Remarkably, gene expression analysis indicated that the human PNPLA3 is expressed in a multitude of tissues, but the highest level was found in the liver, followed by retina, skin, adipose tissue, kidney, brain and spleen.9,10 In contrast, the relative expression levels in mice show that white and brown adipose tissues5 had significantly higher expression levels than the liver.8 Moreover, with regard to liver cellular compartments, murine hepatocytes displayed higher amount of PNPLA3 compared to hepatic stellate cells (HSCs),9 whereas human HSCs show more than 2 fold higher expression compared to hepatocytes,10,11 further underlying the profound discrepancy between the two species. In addition, it has been shown that PNPLA3 protein localization was strongly associated with cellular membranes and lipid droplets (LDs).4,8
Molecular mechanisms regulating PNPLA3 expression
Of note, as its closest homolog PNPLA2 (ATGL), PNPLA3 expression is changed in altered metabolic status, such as obesity,5,12,13 but in contrast to ATGL its expression is regulated by nutritional intake, in both liver and adipose tissue.5,8,14,15 Indeed, in hepatocytes PNPLA3 expression is highly induced by carbohydrate feeding and insulin. The mouse PNPLA3 promoter displays two specific consensus sites for the carbohydrate responsive-element binding protein (ChREBP) and the sterol regulatory element-binding protein 1c (SREBP-1c). SREBP-1c is the major SREBP-1 isoform in the liver, which is regulated in response to fasting and refeeding.16,17 SREBP-1c also promotes fatty acid synthesis by activating several genes in the biosynthetic pathway. Insulin enhances SREBP-1c gene expression and its protein cleavage, therefore favoring its binding to sterol-response element18 in the promoter of target genes.19 PNPLA3 mRNA levels were increased in SREBP-1c transgenic mice with an expression pattern resembling that of fatty acid synthase (FAS).9 Upregulation of SREBP-1c by carbohydrate feeding is mediated by liver X receptor (LXR), as it was discovered that PNPLA3 mRNA levels increase 15-fold within 24 h of treatment with the LXR agonist, T0901317. Furthermore, experiments indicated that SREBP-1 interacts directly with a response element in the first intron of PNPLA3 gene9 and overexpression of SREBP-1c mouse hepatocytes led, as a direct consequence, to an increase in PNPLA3 gene expression.20
In human hepatocarcinoma cells (HuH-7), addition of either palmitic, stearic or oleic acid to culture medium increased PNPLA3 protein abundance. This post-translational increase in PNPLA3 levels remained after blocking TG synthesis with a specific inhibitor of acylCoA-synthase, suggesting that the nutritional control of PNPLA3 is due to a feed-forward loop where SREBP-1c increases transcription of PNPLA3 and genes involved in fatty acid synthesis, thus resulting in PNPLA3 protein stabilization9 (Figure 1). In addition to SREBP-1c, it was shown that overexpression of ChREBP correlated with an increase of adiponutrin/PNPLA3 mRNA levels in mouse liver and isolated murine hepatocytes. However, in human hepatoma cells a recombinant adenovirus expressing mouse SREBP1-c enhanced human PNPLA3 expression, with FAS and stearoyl-CoA desaturase-1 (SCD-1) gene expression following a similar pattern of regulation, whereas overexpression of human ChREBP had no effect on PNPLA3. Additionally, silencing of endogenous murine ChREBP in hepatocytes abolished PNPLA3 transcription in response to glucose.20 Furthermore, Rae-Whitcombe et al analyzed a 3100 bp human PNPLA3 promoter fragment with luciferase reporter assays and found an upregulation of reporter activity upon glucose treatment, while a more recent study showed that silencing of ChREBP abolished induction of PNPLA3 mRNA by glucose in immortalized human hepatocytes.21 Conversely, coexpressing rat ChREBP did not induce the transcription; this coupled with the mouse data would seem to indicate a requirement for the cofactor Mlx22 to achieve full transcriptional activity in human cells.23 In conclusion, ChREBP controls PNPLA3 mRNA levels in mouse liver and isolated murine hepatocytes, whereas the specific ChREBP response element is missing in the human PNPLA3 promoter. In contrast, in vivo and in vitro approaches proved that both human and murine SREBP-1c lead to PNPLA3 upregulation, in response to carbohydrate feeding.
 | Figure 1 Mechanism of induction of PNPLA3. Notes: Carbohydrate intake increases insulin secretion from the pancreas, thus leading to heterodimerization of LXR with RXR in the liver. These two nuclear receptors bind and induce the expression of SREBP-1c, which in turn increases gene related to fatty acid synthesis (FAS, ELOVL6, ACC, SCD-1) and PNPLA3 expression. Accumulation of long-chain fatty acids, especially C18:0 and C18:1, prevents degradation of PNPLA3 via the proteasome and stabilizes the protein, thus increasing its abundance. Abbreviations: FAS, fatty acid synthase; ACC, acetyl-CoA carboxylase; SCD, stearoyl-CoA desaturase; LXR, liver X receptor; PNPLA3, patatin-like phospholipase domain-containing 3 gene; RXR, retinoid X receptor; SREBP-1c, sterol regulatory element-binding protein 1c; Ub, ubiquitin. |
Molecular analysis of Western-type diets-fed mice showed a 50-fold higher PNPLA3 expression in hepatocytes after 2 weeks that persisted until the end of the 6 weeks feeding period. Interestingly, glucose-6-phosphate dehydrogenase (G6PD) and PNPLA3 followed similar expression pattern.24 G6PD is a cytosolic enzyme involved in the pentose phosphate pathway. G6PD transforms glucose-6-phosphate into 6-phosphoglucono-d-lactone and NADP to NADPH, which is subsequently used for the biosynthesis of fatty acids, making it a key enzyme involved in the synthesis of TGs from glucose (lipogenesis).25 Moldes et al26 reported that infusion of insulin significantly stimulated PNPLA3 mRNA expression in adipose tissue. Additionally, Kastrouni et al27 detected increased activity of G6PD in adipose tissue upon insulin administration, which coincided with elevated G6PD transcription. Moreover, similar to PNPLA3,24 G6PD expression levels decreased significantly in the liver and adipose tissue of fasted mice.28 Combined, these findings suggest that the mRNA expression levels of PNPLA3 and G6PD are tightly controlled by the same upstream regulators, at least in mice.
Regarding other liver cell types, recent reports have shown that co-treatment of retinol and palmitic acid in human HSCs markedly resulted in a dose-dependent decrease in PNPLA3 protein expression, in line with the increase of LDs amount. Conversely, depletion of retinol and palmitic acid resulted in robust increase of PNPLA3 protein amount. HSCs stimulated with insulin displayed also a robust increase in PNPLA3 protein content, which was lost using siRNA-targeting adiponutrin/PNPLA3.10
Notably, recent data published from our group have highlighted the contribution of PNPLA3 to achieve an activated phenotype in human HSCs. In details, it has been reported for the first time that PNPLA3 was tightly and significantly induced both at protein and mRNA levels after 15 days from primary human cells isolation, suggesting a close link with HSC activation. Moreover, PNPLA3 transient downregulation was accompanied by decreased expression of the two main profibrogenic markers, Collagen1α1 and α-smooth muscle actin (SMA).11 Interestingly, we also found that the human PNPLA3 expression colocalizes with the profibrogenic marker α-SMA in pathological biopsies (from nonalcoholic steatohepatitis [NASH] patients), compared to healthy liver, further enhancing that PNPLA3 might have a pivotal function in HSCs rather than in hepatocytes, in humans.11
PNPLA3 enzymatic role
Regarding the enzymatic role of this protein, PNPLA3 displayed various and even apparently opposing features. As its upregulation depends on refeeding stimuli, it would be reasonable to associate adiponutrin toward lipogenic genes. In vitro studies with recombinant human PNPLA3 in Sf9 cells have shown that adiponutrin possesses both TG hydrolytic and diacylglycerol (DAG) transacylase activities.4,14,15,29 Remarkably, human recombined PNPLA3 catalyzes the hydrolysis of the three major glycerolipids, triacylglycerols (TAGs), DAGs and monoacylglycerols, with higher preference for TAGs and for oleic acid as the fatty acid moiety. In addition, recent studies revealed that human and murine recombinant PNPLA3 acts also as acyl-CoA-dependent lysophosphatidic acid acyltransferase, thus favoring the synthesis of phosphatidic acid toward TAGs.30 Nevertheless, these interesting evidences were obtained using recombinant purified protein, whereas both overexpression of PNPLA3 in cells and in mouse livers did not modify the overall cellular triacylglycerol (TAG) content, supporting the idea that its primary function might not be related to TAG metabolism.4,8 Moreover, knockout strategies also failed to verify any of the investigated and reported PNPLA3 activities, because total deletion of PNPLA3 at cellular and mouse levels did not display any significant difference compared to expressing controls either in liver and adipose tissue. Indeed, it has been described that mice lacking PNPLA3 displayed no alterations in body weight, adipose mass, insulin sensitivity, fatty liver development or energy homeostasis.15,31 Altogether, both in vitro and in vivo approaches did not elucidate the full enzymatic role of PNPLA3 and suggest that it has a prime role in energy homeostasis and fat metabolism, at least in mice.
Hepatic functions of PNPLA3 genetic variant reported in vitro and in vivo
The attention of the scientific community on PNPLA3 significantly rose in 2008, when a cohort study was performed on a population including a wide range of ethnicities, known as the Dallas Heart Study. This publication has reported the tight correlation between a genetic variant of PNPLA3, rs738409 C>G and the increased risk for developing hepatic steatosis and inflammation, then followed by separated confirmations in other cohort and in different liver diseases (Figure 2). The PNPLA3 variant is a single-base polymorphism, which leads to the switch from a cytosine (C) to a guanine (G), resulting in an amino acid substitution of an isoleucine (I) to a methionine (M) at the position 148 (I148M) of the coding sequence. In vivo data and computer models have pointed out that this polymorphism is located in the active site of the enzyme, which might alter the substrate access to the catalytic dyad and consecutively resulting in a loss of function of the native protein.4 In vitro evidences using recombinant proteins have reported that fatty acids accumulation increased proportionally to the increase of TAGs, and the hydrolase activity was reduced in cells expressing the I148M PNPLA3.4 To explore the molecular and physiological implications of the I148M PNPLA3, several mechanistic studies using different technics have been performed in the last years. In particular, cultured hepatocytes overexpressing either the wild type (WT) or the I148M PNPLA3 isoforms revealed reduced fatty acid hydrolysis and increased TG accumulation in cells carrying the genetic variant, thus suggesting a loss of function compared to WT carriers.4 Another study on human hepatocytes carrying the PNPLA3 variant has reported a decrease in very-low-density-lipoprotein release, indicating that the impact of this genetic variant affects several pathways considered to interact and significantly contribute to hepatic steatosis.32
 | Figure 2 PNPLA3 and risk factors in the progression of liver disease. Notes: PNPLA3 is an independent risk factor for progression of liver injury from steatosis to steatohepatitis, cirrhosis and hepatocellular carcinoma. Abbreviations: FFA, free fatty acids; HCV, hepatitis C virus; HSC, hepatic stellate cell; IR, insulin resistance; PNPLA3, patatin-like phospholipase domain-containing 3 gene; RXR, retinoid X receptor; VLDL, very-low-density lipoprotein. |
In vivo approaches using transgenic (knock-in) mice have revealed that overexpression of the human genetic variant of PNPLA3 in mouse liver causes hepatic steatosis when the animals were challenged with high-sucrose diet,33,34 suggesting an impaired hydrolysis of TAG because of altered remodeling of hepatocyte LDs. Additionally, knock-in mice carrying in the mouse coding sequence the mutation at position 148 did not display differences under control diet, whereas hepatic steatosis was found only in mice overexpressing the I148M PNPLA3 under fatty liver-inducing diet,34 further suggesting a discrepancy in the regulation of the PNPLA3 gene among human and mouse. Recently, a study from the same group proposed an alternative mechanism to undercover the differences between WT and I148M PNPLA3. Based on their observations, they suggest the accumulation of I148M PNPLA3 on LDs as first sign of lipid metabolism impairment. Thereafter, the clearance of the protein is altered because of decreased ubiquitination, subsequently resulting in retarded proteasome degradation and diminished TAGs mobilization from LDs.35
Interestingly, in vivo findings have underlined the contribution of an altered hepatic long-chain fatty acids composition toward a proinflammatory condition, such as the enrichment of n-6/n-3 polyunsaturated fatty acids (PUFAs), thus promoting de novo lipogenesis in the liver.36,37 In this regard, the PNPLA3 polymorphism I148M has been positively associated with increased n-6 PUFA in hepatic fat content, in a cohort of youths,38 which was further investigated in adults with discrepancies in the n-6 PUFA and increased amount of α-linolenic acid (18:3) n-3,39 suggesting a possible role of this enzyme in hepatic fat remodeling.
In the context of hepatic fibrosis, a recent study from our group pointed out for the first time that the PNPLA3 I148M resulted in increased release of proinflammatory and profibrogenic cytokines, known to be active key players of the fibrotic process, such as RANTES (CCL5), MCP-1 (CCL2), IL-8 and GM-CSF. Notably, this augmented secretion of proinflammatory soluble mediators was significantly confirmed in both human primary HSC I148M carriers and overexpressing I148M LX-2, in unstimulated conditions.11 Interestingly, culture medium collected from cells expressing the PNPLA3 genetic variant have been shown to recruit higher amount of immune cells, such as monocytes, compared to their WT PNPLA3 carriers. Remarkably, it has also been shown that retinol content of both primary and overexpressing HSCs was significantly lowered in cells carrying the I148M PNPLA3, which is not in line with the previous hypothesis of the genetic variant as retinyl palmitate lipase. A putative difference might be in the experimental strategy, because previous authors loaded HSCs with retinyl-palmitate mixture in advance,10,40 while in our study cells were not pretreated, thus reflecting the different outcome observed in vitro. In addition, molecular analysis further confirmed that the retinoid X receptor and retinoic acid receptor signaling was completely abolished in cells expressing the PNPLA3 variant, thus supporting that the lack of retinoid ligands for these nuclear receptors might not play a role in characterizing the active phenotype of I148M-expressing HSCs.11 In contrast, in vivo studies reported that the PNPLA3 variant carriers with nonalcoholic fatty liver disease (NAFLD) show lower fasting circulating concentrations of retinol, and obese patients with the I148M had lower fasting circulating concentrations of retinol.41 Furthermore, observations in patients carrying the variant allele42 displayed elevated hepatic retinyl-palmitate content and reduced ratio of retinol/retinyl-palmitate, thus suggesting a possible implication of the PNPLA3 variant in retinol metabolism in vivo.
Thereafter, our data on HSCs revealed that I148M PNPLA3 impacts on several molecular mechanisms regulating the phenotype of activated cells, such as decreased peroxisome proliferator activated receptor gamma signaling and enhanced c-Jun activated kinase/activator protein 1 (JNK/AP-1) pathway,11 thus suggesting that the lack of in vivo findings in animal models, which resemble the human situation, might be due to different patterns of protein expression and specific liver cell type contribution.
Therefore, consistent with our observation in human HSCs and with previous reports in hepatocytes in vivo and in vitro (summarized in Figure 3), we might hypothesize that the role of PNPLA3 genetic variant in liver disease mirrors a complex interplay between the different hepatic cell types, thus leading to a different contribution and features toward severe pathologies.
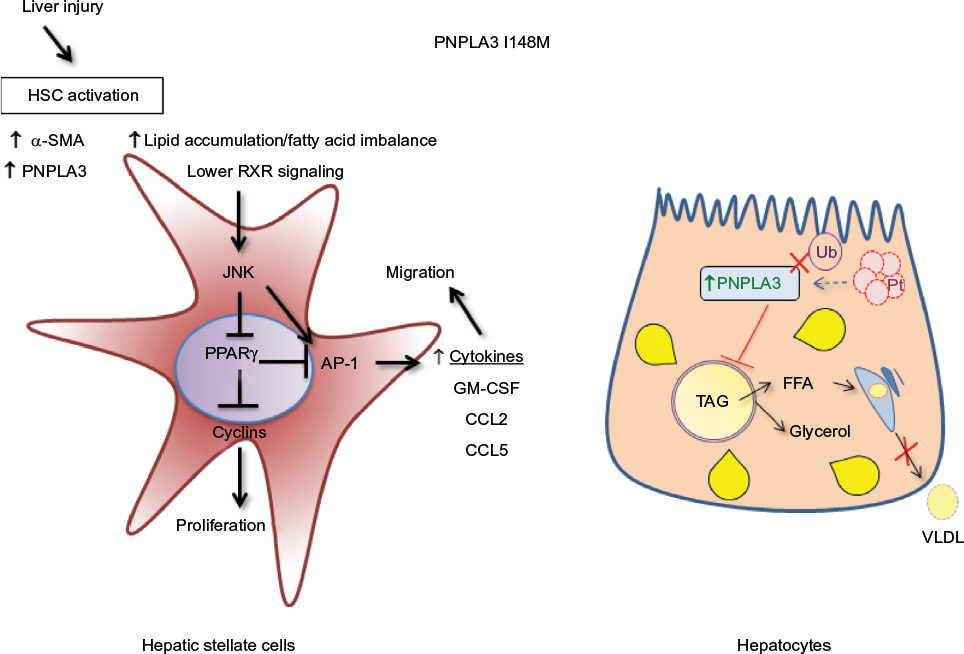 | Figure 3 Differential molecular mechanisms of PNPLA3 I148M in HSCs and hepatocytes. Notes: HSCs are depicted in the left panel, in which activation and increase of proinflammatory FFAs directly activates JNK/AP-1 pathway, increasing HSCs proliferation and secretion of inflammatory mediators. On the right side, a different scenario is represented in regard to hepatocytes: ubiquitination of PNPLA3 does not take place because of the I148M variant, and this results in an increase in PNPLA3 and FFA content, because the proteasome is not able to degrade PNPLA3 protein efficiently. The augmented amount of FFAs cannot be released in the form of VLDL from the hepatocytes to the blood stream. Abbreviations: AP-1, activator protein 1; FFA, free fatty acids; HSC, hepatic stellate cell; JNK, c-Jun activated kinase; LXR, liver X receptor; PPARγ, peroxisome proliferator activated receptor gamma; PNPLA3, patatin-like phospholipase domain-containing 3 gene; RXR, retinoid X receptor; SMA, smooth muscle actin; TAG, triacylglycerol; VLDL, very-low-density lipoprotein; Ub, ubiquitin. |
Recently, a rare human PNPLA3 genetic variant, known as rs2294918 E434K PNPLA3, has been investigated in relation to I148M polymorphism. Donati et al have highlighted that the concomitant presence of these two genetic variants on PNPLA3 attenuates the effects of the I148M PNPLA3 alone, by diminishing its accumulation on LDs and decreasing liver damage (lower alanine transaminase levels).43
Clinical evidences connecting the I148M PNPLA3 to progression toward severity of liver diseases
Nonalcoholic fatty liver disease
NAFLD is a multifactorial disorder frequently associated with obesity, insulin resistance (IR) and features of the metabolic syndrome.44 It represents a group of hepatic disorders characterized by accumulation of TGs in hepatocytes. The initial stage of NAFLD is hepatic steatosis, which might progress to NASH, characterized by hepatocyte damage (ballooning), inflammatory infiltrate and the initial presence of fibrotic lesions. In case of persistent injury, it might lead to cirrhosis, a chronic organ impairment, which predisposes to cancer and requires liver transplantation.45 Notably, hepatocellular carcinoma (HCC) is also increasingly observed in pre-cirrhotic stages of NASH.46
The clinical study by Romeo et al provided the first significant association between a genetic variant of the PNPLA3 gene, reported as rs738409 C>G, and the risk of NAFLD development.47 The frequency of the PNPLA3 I148M was higher in Hispanic population and lower in European Americans and African Americans. Intriguingly, there was no association between the PNPLA3 genotype and other risk factors for NAFLD, such as obesity and IR.47 Remarkably, in the last 10 years, association of I148M mutation of PNPLA3 with hepatic fat has been independently and robustly replicated in multiple studies including individuals from different ethnicities and geographical regions.48
Additional studies have focused on searching an association between the I148M PNPLA3 and NAFLD severity, risk of steatohepatitis and progression to fibrosis. The first evidence came from the observations made by Sookoian et al,49 which demonstrated a robust link between I148M PNPLA3 and NAFLD severity, as determined by histological assessment of liver biopsies, independent of body mass index (BMI), age, sex and insulin sensitivity. Thereafter, the PNPLA3 genetic variant was associated not only with higher risk for simple steatosis, but also NASH, fibrosis and cirrhosis development in different populations and ethnicities (Japanese,50 Americans,51 Italians52,53 and Malaysians54), thus remarkably underlying that I148M acts as an independent risk factor in human NAFLD pathogenesis55–57 (Table 1; Figure 2). Notably, recurrence of steatosis and NASH after liver transplantation was independent from host but significantly linked to donor genotype. In particular, higher prevalence of steatosis and exclusively NASH cases developed in recipients who received the homozygous for the I148M allele, further suggesting that it is in the liver where PNPLA3 exerts its key physiological action.58
 | Table 1 Number of the studies evaluating the association between PNPLA3 I148M and liver damage Abbreviations: ALD, alcoholic fatty liver disease; ALT, alanine transaminase; AST, aspartate transaminase; CHB, chronic hepatitis B; CHC, chronic hepatitis C; HCC, hepatocellular carcinoma; HS, hepatic steatosis; IBD, inflammatory bowel disease; LB, liver biopsy; NAFLD, nonalcoholic fatty liver disease; NASH, nonalcoholic steatohepatitis; T2DM type 2 diabetes mellitus. |
Interestingly, recent reports59 have additionally pointed out an important difference between “metabolic NAFLD” and “PNPLA3 NAFLD,” suggesting a difference in terms of lipotoxic free fatty acids, mainly saturated fatty acids and harmful ceramides as determinants for IR and D2M-driven NAFLD. The genetic “PNPLA3 NAFLD” is rather characterized by polyunsaturated TGs, confirming a profound and marked discrepancy in origin of fatty liver diseases.
Alcoholic liver disease
Alcohol-related cirrhosis still represents one of the leading causes of death in western countries.60 A first study released in 2009 highlighted a close correlation between the I148M PNPLA3 and increased risk for alcohol-induced cirrhosis along with the Hispanic ancestry.61 Lately, several individual studies, meta-analysis and, recently, genome-wide association study further robustly associated the increased risk for developing alcohol-related cirrhosis with the PNPLA3 genetic variant.62–64 Along with rapid disease progression and advanced risk for developing HCC in a background of alcohol-associated liver diseases,65 a recent study pointed out that the presence of the I148M PNPLA3 should be considered as a risk factor for severe alcoholic hepatitis and impacts negatively on the medium-term survival of these patients, independent of drinking behavior.66
Chronic hepatitis C
Steatosis has been reported to negatively influence the natural history of chronic hepatitis C (CHC) being associated with more aggressive histological features, faster progression of fibrosis and poorer response to therapy.67,68 Recent studies have shown that I148M PNPLA3 genotype influences steatosis and/or fibrosis development in CHC patients. In 2011, Valenti et al69 compared two independent series of Italian patients with CHC and showed for the first time that PNPLA3 genetic variant influences steatosis development independent of age, sex, BMI, diabetes, alcohol intake and viral genotype. In the same study, PNPLA3 promoted fibrosis and risk of developing HCC in patients with CHC. The results were confirmed in a study including 537 Caucasian CHC patients.70 Additional clinical findings linked the presence of the PNPLA3 genetic variant to higher risk for advanced fibrosis in patients with CHC.71,72 However, the contribution of PNPLA3 variant to severity of hepatitis C virus (HCV) infections is still controversial and perhaps not direct, because there was no link between the genetic PNPLA3 polymorphism, the viral genotype and the development of hepatic steatosis in subsequent studies.73,74
Interestingly, a report in a cohort of HIV/HCV co-infected patients demonstrated that liver fibrosis and cirrhosis progression accelerated, suggesting a possible influence of genetic factors contributing to the variability of these pathologies.75 In particular, a recent study conducted in a larger population clearly defined the role of I148M PNPLA3 as an independent risk factor for fibrosis progression, resulting in higher cirrhosis occurrence in HIV/HCV co-infected patients.76 Molecular data have shown that HCV alters dramatically lipid metabolism in infected hepatocytes,77 suggesting a possible link between I48M PNPLA3 in remodeling LDs and thus favoring fat accumulation leading to steatosis. However, further mechanistic studies are needed to verify this hypothesis.
Regarding hepatitis B, there is a marked correlation between PNPLA3 genetic variant and liver fat and inflammation, even though there is not with fibrosis development.78,79
Liver disease with other etiologies
Clinical observations have reported that primary sclerosis cholangitis patients carrying the I148M genetic variant have reduced survival, compared to their WT counterparts. Moreover, this association was restricted to men with severe dominant stenosis.80
In addition, a recent study on Wilson disease patients showed that the I148M PNPLA3 genotype was significantly associated to increased steatosis prevalence.81
Carriers of the I148M genotype affected by inflammatory bowel disease shad significantly more hepatic steatosis and higher levels of transaminase enzymes.82
Furthermore, this PNPLA3 genetic variant resulted was a risk factor for increased hepatotoxicity in a children cohort in remission after therapy for acute lymphoblastic leukemia (ALL)83 and B-cell ALL.84
Hepatocellular carcinoma
HCC is mainly a consequence of chronic viral hepatitis B and C in developing countries,85 whereas in Europe and North America ~45% of HCCs are caused by alcohol consumption.86 The first proven association between the PNPLA3 genetic variant and hepatic cancer emerged in the study from Valenti et al,69 which was confirmed by later studies, underlying the tight connection between hepatic carcinogenesis and HCV-related cirrhosis. Notably, a meta-analysis of European patients with cirrhosis robustly established a correlation between I148M polymorphism and HCC, with stronger association with alcoholic liver disease (ALD) than CHC-related cirrhosis,87 thus suggesting that this genetic variant exerts a profound influence on processes triggering carcinogenesis beyond the pathologic background. With regard to NAFLD, several studies significantly supported the strong association of the I148M variant and the risk of HCC.88–90 Moreover, the relationship between PNPLA3 genotype and the occurrence of HCC in the presence of cirrhosis is robust when analyzed in NAFLD and ALD background and was also confirmed by recent meta-analysis.57 Retrospective studies supported this association in patients with cirrhosis due to CHC, although in case–control studies the link with higher risk of HCC in HCV background was not consistent.73
Conclusions
There is a growing worldwide interest in NAFLD, a disease commonly affecting up to 40% individuals of our population with further predicted increases. We now have insights into the genetic basis of ethnic differences in the prevalence of NAFLD. One of these genetic variations, the rs738409 I148M variant of PNPLA3 gene, is strongly associated with NAFLD in different populations, and convincingly linked to enhanced liver disease severity, such as fibrosis cirrhosis and even cancer, suggesting that rs738409 could be considered as an independent risk factor for liver dysfunction, beyond its metabolic background (ALD, CHC).
Defining the precise PNPLA3 function in order to unravel novel therapies for the treatment of NAFLD will be the key of future research.
Disclosure
The authors report no conflicts of interest in this work.
References
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.