Back to Journals » Journal of Asthma and Allergy » Volume 14
PM2.5 Aggravated OVA-Induced Epithelial Tight Junction Disruption Through Fas Associated via Death Domain-Dependent Apoptosis in Asthmatic Mice
Authors He X, Zhang L, Hu L, Liu S, Xiong A, Wang J, Xiong Y, Li G
Received 30 August 2021
Accepted for publication 3 November 2021
Published 20 November 2021 Volume 2021:14 Pages 1411—1423
DOI https://doi.org/10.2147/JAA.S335590
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Luis Garcia-Marcos
Xiang He,1,2,* Lei Zhang,1,2,* Lingjuan Hu,1,3,* Shengbin Liu,1,2 Anying Xiong,1,2 Junyi Wang,1,2 Ying Xiong,4 Guoping Li1,2
1Laboratory of Allergy and Precision Medicine, Chengdu Institute of Respiratory Health, The Third People’s Hospital of Chengdu, Affiliated Hospital of Southwest Jiaotong University, Chengdu, 610031, People’s Republic of China; 2Department of Pulmonary and Critical Care Medicine, Chengdu Third People’s Hospital Branch of National Clinical Research Center for Respiratory Disease, Affiliated Hospital of ChongQing Medical University, Chengdu, 610031, People’s Republic of China; 3Department of Respiratory Disease, Renshou County People’s Hospital, Renshou, 620550, People’s Republic of China; 4Department of Pulmonary and Critical Care Medicine, Sichuan Friendship Hospital, Chengdu, 610000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Guoping Li; Ying Xiong Email [email protected]; [email protected]
Background: Exposure to air pollutants cause exacerbation of asthma, but the experimental evidence and the mechanisms still need to be collected and addressed.
Methods: Asthma model was constructed by ovalbumin (OVA) combined with or without airborne fine particulate matter 2.5 (PM2.5) exposure. Lung sections were stained by hematoxylin-eosin staining (H&E) and Masson’s trichrome. RNA-seq and gene set enrichment analysis (GSEA) was performed to identify the key pathway. TdT mediated dUTP Nick End Labeling (TUNEL) assay, real-time qPCR, Western blot, immunofluorescence and lentivirus transfection were applied for mechanism discovery.
Results: In this study, we found PM2.5 aggravated airway inflammation in OVA-induced asthmatic mice. RNA-seq analysis also showed that epithelial mesenchymal transition (EMT) was enhanced in OVA-induced mice exposed to PM2.5 compared with that in OVA-induced mice. In the meantime, we observed that apoptosis was significantly increased in asthmatic mice exposed to PM2.5 by using GSEA analysis, which was validated by TUNEL assay. By using bioinformatic analysis, Fas associated via death domain (FADD), a new actor in innate immunity and inflammation, was identified to be related to apoptosis, EMT and tight junction. Furthermore, we found that the transcript and protein levels of tight junction markers, E-cadherin, zonula occludens (ZO)-1 and Occludin, were decreased after PM2.5 exposure in vivo and in vitro by using RT-qPCR and immunofluorescence, with the increased expression of FADD. Moreover, down-regulation of FADD attenuated PM2.5-induced apoptosis and tight junction disruption in human airway epithelial cells.
Conclusion: Taken together, we demonstrated that PM2.5 aggravated epithelial tight junction disruption through apoptosis mediated by up-regulation of FADD in OVA-induced model.
Keywords: asthma, epithelial tight junction disruption, PM2.5, FADD, apoptosis
Introduction
Asthma is a common, long-term, chronic inflammatory disease of airway, characterized by airway hyper-responsiveness (AHR), variable airflow obstruction, and cellular inflammation, followed by tissue repair and regeneration, which affects millions of people worldwide.1 It can be triggered by a hypersensitivity reaction following the interaction of genetic and specific environmental factors, such as aeroallergens. Airway epithelial barrier is the first line, which defenses against pathogens and restricts trans-epithelial crossing of inhaled allergens. Epithelial barrier function is maintained by formation of adherens junctions and tight junctions. Adherens junctions, which contain the transmembrane protein E-cadherin, are critical for maintaining apical-basolateral polarization and adhesion to neighbouring cells.2 Tight junctions (TJs) are located at the most apical part of the intracellular junctional complex between adjacent epithelia.3 TJs are comprised of proteins such as occludin, and claudins, and zonula occludens (ZO)-1.4 These molecules mediate the organization of transmembrane proteins in the membrane and their subsequent attachment to the cytoskeleton, and maintain a size- and ion-selective barrier, regulating the permeability of the epithelium.5,6
FADD, consisting of the N-terminal death effector domain (DED) and C-terminal death domain (DD), is an adaptor that bridges death receptor (DR) signaling to the caspase cascade. FADD has been implicated in numerous signaling pathways, including those for cell cycle regulation, immune signaling, and autophagy as well as physiological outcome inducing inflammation, cell proliferation, embryonic and immune cell development and tumorigenesis.7 At early times, Occludin is localized at the death-inducing signaling complex and can be immune-precipitated with FADD, a member of this complex.
Airborne fine particulate matter 2.5 (PM2.5) is the most common component of air pollution in the developing world. As an important environmental risk factor, PM2.5, has been considered to be a great threat to human health. There is a strong association between inhalation of airborne particulate matter (PM) and human respiratory and cardiovascular diseases.8,9 Long-term exposure to PM2.5 has been associated with both asthma development and increased asthma severity.10–12 However, the existing experimental data do not sufficiently explain how exposure to PM2.5 could increase the risk of asthma, and the involved mechanisms still need to be addressed. This study investigated the detrimental effects of PM2.5 on the exacerbation of asthma. The epithelial tight junction disruption by PM2.5 through FADD-dependent apoptosis likely contributed directly to the development of asthma.
Materials and Methods
Preparation of PM2.5
PM2.5 was collected as described according to the previous publication.13,14 Briefly, PM2.5 samples were collected from October to December 2018 using an air sampler on the open-air roof of a ten-story building of the third people’s Hospital of Chengdu. To eliminate the contamination, the filters were preheated at 550°C. Then, balanced and stabilized before sampling (20±0.5°C, 40±5% RH). The filter containing the sample was cut into a size of 1 cm×1 cm, immersed in sterilized ddH2O, and eluted by ultrasonic shock for 3 times. The eluent was filtered with medical gauze. It is then freeze-dried by vacuum freeze-drying apparatus and sealed at −80°C for preservation. Before use, PM2.5 particles were prepared into particulate suspension with a concentration of 10 mg/mL with phosphate buffered saline (PBS).
Animal Experiments
Healthy female C57BL/6 mice (6–8 weeks old) were purchased from Chengdu Da Shuo Laboratory Animal Co., Ltd. This study was approved by Animal Ethics Committee of Southwest Jiaotong University. All experimental procedures and animal care were carried out under the guidance of the Ethics Committee in order to minimize the suffering of animals (SWJTU-2107-004(SWJTU)). The mice were randomly divided into control group, PM2.5-exposed group (PM2.5 group), asthma model group (OVA group), and PM2.5-exposed asthma group (OVA + PM2.5 group). To establish the asthmatic group, the mice were intraperitoneally sensitized on days 0 and 7 with 25 μg of OVA and 2 mg of aluminum hydroxide, challenged with an intranasal administration of 20 μg of OVA from day 14 to 21. For PM2.5 exposure group, the mice were persistently instilled intranasally with 100 μg PM2.5 for 7 consecutive days. For OVA + PM2.5 group, the mice were treated with 100 μg PM2.5 during OVA challenge. The control group received PBS during sensitization and challenge phases instead of OVA. On day 22, all the mice were sacrificed for tissue collection and analyzed.
Cell Culture and Transfection
BEAS-2B or human bronchial epithelial (HBE) cells were cultured with high-glucose Dulbecco’s Modified Eagle’s Medium (DMEM) containing 10% fetal bovine serum (FBS) (Gibco, USA), grown at 37°C in with 5% CO2 condition. BEAS-2B cell line was purchased from GeneChem. The culture medium was changed every 1–2 days. When the cells reached 80% confluence in the plastic plates, PM2.5 was exposed to the culture at the concentration of 62 μg/cm2 for 3 hrs, 6 hrs, 12 hrs, 24 hrs and 48 hrs. In some experiments, cells were treated with 40 μg/mL house dust mite (HDM) alone or together with PM2.5 for 24 hours. Then, the activation of the relevant signaling pathways or the expression of the relevant proteins were assessed. shFADD lentiviruses were packed by Genechem as previously described and transduced into BEAS-2B using multiplicity of infection (MOI) 80 (after cells had grown to 30–50% confluence).15 After 72 hours, the FADD expression was evaluated by qPCR and immunofluorescence staining.
Histology
Lung tissues were isolated and immersed in a 10% formaldehyde solution for fixation. The fixed tissues were dehydrated in ascending series of ethanol, and embedded in paraffin. The sections (3 μm) of the lung specimens were stained with standard hematoxylin-eosin staining (H&E) and Masson’s trichrome to assess histology. The pathological changes of lung airway, alveolar wall thickness and inflammatory cell infiltration were observed with a microscope (IX73, Olympus, Japan). The degree of peribronchial and perivascular inflammation was scored from 0 to 5 according to our previous methods, with approximately 10 areas scored in total.16
RNA Isolation and Quantitative Real-Time PCR
BEAS-2B cells were treated with PM2.5 at different time points (3 hrs, 6 hrs, 12 hrs, 24 hrs and 48 hrs). The total RNA was isolated using the Fastpure Cell/Tissue Total RNA Isolation Kit (Vazyme, China) according to the manufacturer’s instructions. The purified RNA was synthesized using HiScript IIQ RT SuperMix for qPCR (Vazyme, R223–01) for the first strand cDNA. The quantitative real-time PCR analyses were performed with duplicate samples using ChamQ Universal SYBR qPCR Master Mix (Vazyme, China). The data was calculated by using 2-ΔΔCT, ACTIN as the internal reference gene.
TUNEL Assay
Apoptotic cells were detected using the TUNEL Apoptosis Assay Kit (Solarbio) according to the manufacturer’s instructions. Briefly, after treatment, cells were fixed with 4% formaldehyde. 50 µL of TUNEL working solution was added to each sample, incubated at 37°C for 30–60 minutes. Removed TUNEL working solution, and washed the cells 1–2 times with PBS, added 100 uL Reaction buffer. Fluorescence images were taken with CSU-W1 confocal microscopy (OLMPUS, Japan).
Immunofluorescence Staining
Immunofluorescence was performed as previously described.17 Briefly, frozen lung tissue sections (5μm) and cells were fixed with ice-cold methanol and permeabilized in PBS containing 0.25% Triton X-100. Then blocked for 30 min with 10% goat serum. Specimens were then incubated with antibodies against E-cadherin (Abcam, USA), ZO-1 (Cell Signaling Technology, USA), Occludin (Abcam, USA) and FADD (Bioss, China). TRITC-conjugated secondary Ab was used to probe the primary antibodies. Nuclei were stained with 4′-6-diamidino-2-phenylindole dihydrochloride (DAPI, Solarbio). Fluorescence images were taken with CSU-W1 confocal microscopy (OLMPUS, Japan) analyzed with CellSens Application Suite Software.
Data Collection and Gene Set Enrichment Analysis (GSEA)
The Cancer Genome Atlas (TCGA) lung adenocarcinoma paired normal samples were downloaded from Xena datahub (https://xenabrowser.net/, TCGA Pan-Cancer cohort). The published datasets, GSE108134, was obtained from public database Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/). The fold changes of severe asthma versus moderate asthma were calculated for GSEA analysis by using the limma R package, and a data matrix including genes log2 fold change values were used as the input data. Gene set enrichment analysis was performed with GESA 3.0 according to previous publications.18
Western Blotting
Cells were homogenized and re-suspended in radioimmunoprecipitation assay (RIPA) buffer with protease inhibitor cocktail. The protein concentrations were determined with a microplate reader (Molecular Device). The proteins were denatured by boiling in SDS loading buffer, separated by 10% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) at 100–120V for 90min and electroblotted onto 0.22-μm microporous polyvinylidene difluoride (PVDF) membrane with 250 mA current for 1–2 hrs using a wet transfer method. Subsequently, the membranes were blocked in 5% (w/v) non-fat milk in Tris-buffered saline with 0.1% (v/v) Tween-20 (TBST) for 1 hr at room temperature, followed by incubation with primary antibodies overnight at 4°C. The membranes were washed with TBST and incubated with the appropriate secondary antibodies for 1 hr at room temperature. After washing with TBST, the proteins recognized by the antibodies against β-actin, E-cadherin, ZO-1, and Occludin were visualized by Immobilon ECL Ultra Western HRP Substrate (Merck Millipore, Billerica, MA, USA).
Impedance Spectroscopy
The control cells and shFADD cells were seeded on polycarbonate Transwell inserts (24 well, 8.0 μm pore size), placed in the CellZscope module and cultured in a humidified incubator (37°C, 5% CO2). Resistance values were recorded using an automated cell monitoring system, CellZscope (nanoAnalytics, UK) until a plateau was reached wherein PM2.5 was added on top of the monolayer. TEER values, expressed in Ω cm2, were recorded in real-time over a 24-hours period post-addition of PM2.5. All experiments were carried out in triplicate and repeated at least three times.
Statistical Analysis
The data were analyzed using SPSS 17.0 software (SPSS, Chicago, IL, USA). Quantitative data are presented as the mean ± standard deviation (SD). Statistically significant differences in the mean values were identified by one-way ANOVA, followed by Dunnett’s t-test in GraphPad Prism 7. Two-group comparisons were analyzed by using the Tukey-Kramer post-test or Dunnett’s T3. The differences were considered significant when p < 0.05. Graphs were made in GraphPad Prism 7 (GraphPad Software Inc., LaJolla, CA).
Results
PM2.5 Exposure Aggravated Airway Inflammation and Epithelial Mesenchymal Transition in Asthmatic Mice
Loss of epithelial junctions increased susceptibility towards pathogens and allergen as a consequence of exposure to different environmental factors.19 To determine the effect of PM2.5 exposure on allergic airway inflammation, we established an OVA-induced mice model exposed to PM2.5 (Figure 1A). HE staining showed that PM2.5 exposure severely increased infiltration of inflammatory cells in OVA-challenged mice compared with those in other groups (Figure 1B). These data also demonstrated that PM2.5 exposure significantly aggravated airway inflammation for OVA-induced allergic responses. In the meantime, the percentage of collagen volume fraction (CFV) in OVA-challenged mice exposed to PM2.5 was higher than those in other groups (Figure 1B). In addition, we applied RNA-Seq to gain insights into the gene expression profiling in different groups. The analysis of aberrantly expressed genes between OVA group and OVA plus PM2.5 group identified a total of 996 differentially expressed genes, with 409 genes up-regulated and 587 genes down-regulated (|Log2 fold change| ≥0.5, p.value <0.05) (Figure 1C). Using gene set enrichment analysis (GSEA), on the one hand, we found that the down-regulated genes were significantly enriched in “C5” gene sets associated with ciliary plasm, cilium movement, motile cilium, microtubule-based movement, and cilium organization (Figure 1D). On the other hand, the up-regulated genes were significantly enriched in “hallmark” gene sets associated with epithelial mesenchymal transition (Figure 1D). These results indicated that PM2.5 exposure could suppress cilium-related gene expression and induce epithelial mesenchymal transition (EMT).
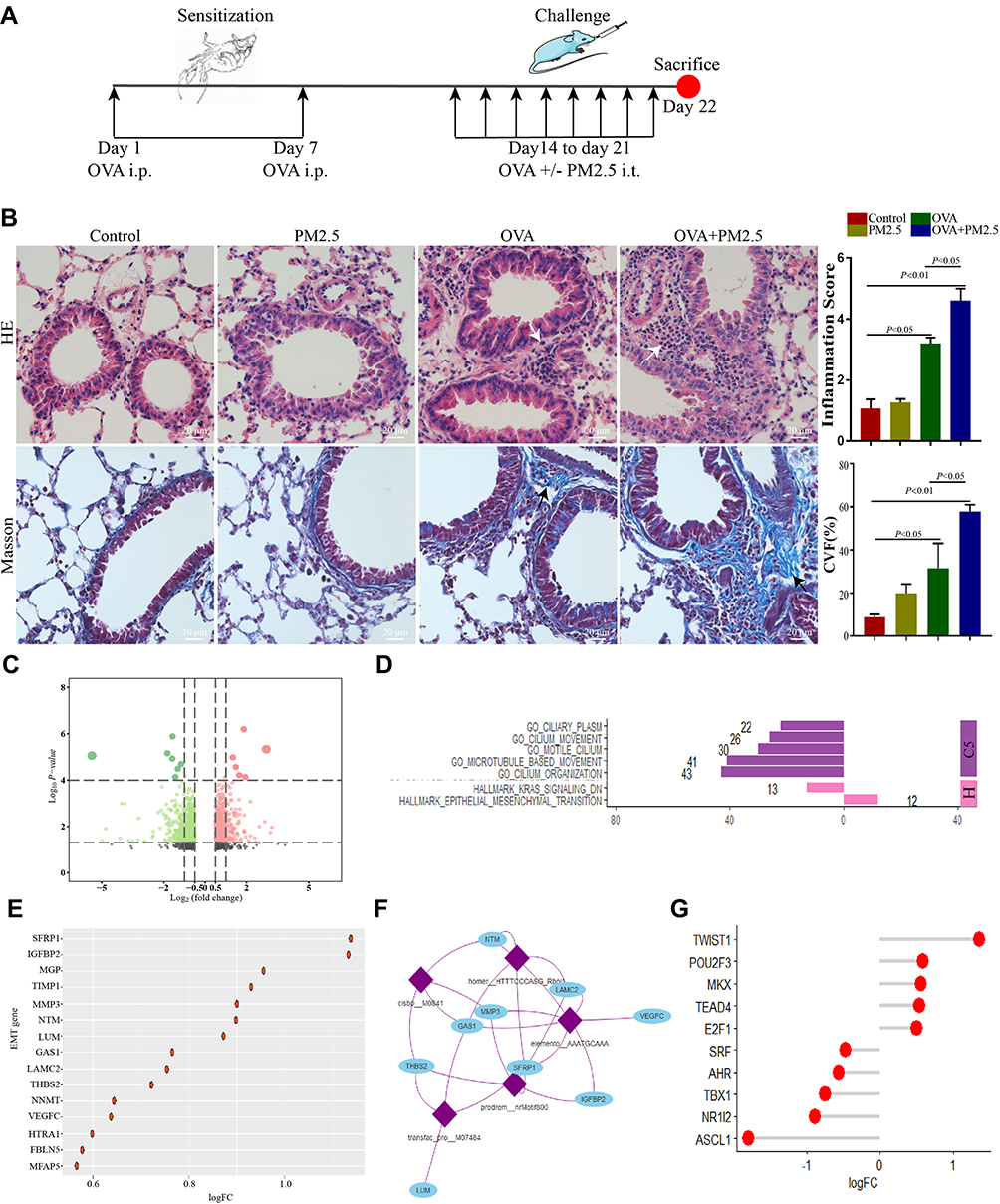 |
Figure 1 PM2.5 exposure aggravated airway inflammation and epithelial mesenchymal transition in asthmatic mice. (A) Animal models used in this study. (B) Lung sections from different groups are stained with hematoxylin and eosin [H&E] and Masson trichrome. White arrows indicated immune cells infiltration. Black arrows indicated collagen production. (C) Volcano plot showed differentially expressed genes between OVA-induced group and OVA-induced exposed to PM2.5 group. (D) GSEA analysis, down-regulated genes were associated with cilium movement and up-regulated genes were associated with EMT. (E) logFC of genes involved in EMT gene set. (F) Enriched transcription factor binding motifs and gene regulatory network of EMT. (G) logFC of transcription factors regulated genes involved in EMT gene set. |
EMT could contribute to airway remodeling and fibrosis, which is associated with asthma severity.20 The logFC of genes in EMT gene set was shown in Figure 1E. It showed that SFRP1 and IGFBP2 were highly expressed in OVA-induced mice exposed to PM2.5 (Figure 1E). We next identified enriched transcription factor binding motifs in EMT gene set, and built the gene regulatory network of EMT using the motifs database for RcisTarget (v1.2.1) according to previous research (Figure 1F).21 We found the expression levels of these transcription factors (TWIST1, POU2F3, MKX, TEAD4, and E2F1) were higher in OVA-challenged mice exposed to PM2.5 (Figure 1G).
PM2.5 Increased Apoptosis in Asthmatic Mice
To determine subtle pathway activity changes in OVA-challenged mice exposed to PM2.5, we performed pathway analysis using GSEABase package with the molecular signature database. By contrasting pathway expression levels between OVA group and OVA plus PM2.5 group, the heat-map showed strong increases in EMT, IL2 STAT5 signaling, apoptosis, P53 pathway, oxidative phosphorylation in OVA-challenged mice exposed to PM2.5 (Figure 2A). We also found strong reductions of Myc target V1, notch signaling and unfolded protein response in OVA-challenged mice exposed to PM2.5 compared with those in OVA-challenged mice (Figure 2A). Besides, OVA-challenged mice exposed to PM2.5 displayed consistently higher score of apoptosis, angiogenesis, EMT, mTORC1 signaling, TGF-beta signaling, oxidative phosphorylation, P53 pathway, and protein secretion (Figure 2B).
 |
Figure 2 PM2.5 increased apoptosis in asthmatic mice. (A and B) Heat-map and dot plot showed apoptosis was increased in OVA plus PM2.5 group compared with it in OVA group. (C) TUNEL assay showed apoptosis in different group. The data represent means±s.d. All data are representative of three experiments. |
To further verify that PM2.5 exposure was related to the apoptosis, we determined airway epithelial apoptosis by TUNEL assay. There were no changes in fluorescence intensities between control and PM2.5 group (Figure 2C). However, the fluorescence intensities of airway epithelial cells in OVA-challenged mice were dramatically increased after PM2.5 exposure (Figure 2C).
FADD Positively Correlated to Apoptosis and Negatively Correlated to Tight Junction in GEO and TCGA Database
Previous studies showed that FADD has emerged as a new actor in innate immunity, inflammation, and cancer development.22 We wondered whether there were distinct molecular features after PM2.5 exposure. To answer this question, we performed GSEA analysis in GSE108134 cohorts. Based on NES score system, the pathways were classified into activated- and suppressed-pathways. In GSE108134 cohorts, FADD was tightly associated with those activated pathways including epithelial mesenchymal transition and suppressed pathways including cilium-related pathways (Figure 3A), which were consistent with our RNA-seq data. In addition, we assessed FADD expression by immunofluorescence (IF) assay in lung tissue in the animal models. It showed that the fluorescence intensities of FADD were weak in the control group and PM2.5 group (Figure 3B). The fluorescence intensities of FADD were significantly stronger in OVA-challenged mice exposed to PM2.5 than those in OVA-challenged mice. Moreover, we checked the transcript level of FADD in human bronchial epithelial cells. The result also showed that PM2.5 exposure induced FADD expression at 3 hrs and peaked at 24 hrs in vitro (Figure 3E).
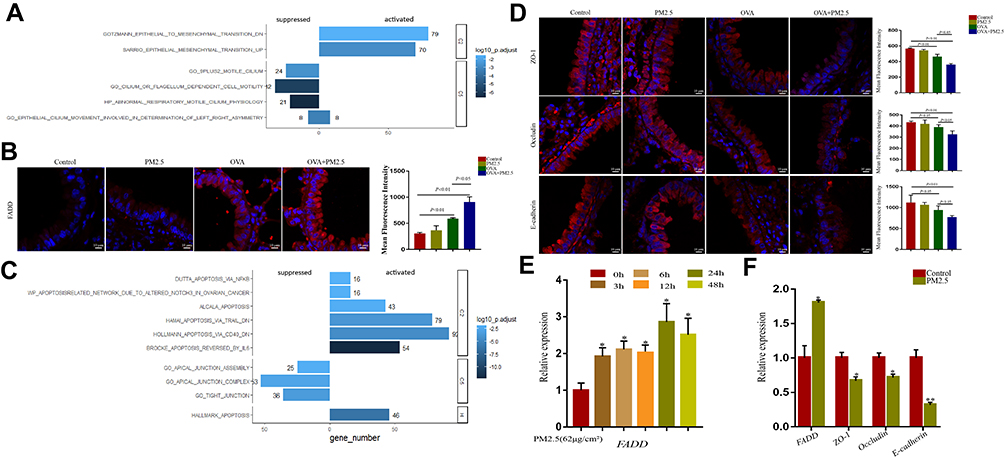 |
Figure 3 FADD positively correlated to apoptosis and negatively correlated to tight junction in GEO and TCGA database. (A) GSEA analysis showed FADD was associated with epithelial mesenchymal transition and cilium related pathways in GSE108134 database. (B) Immunofluorescence imaging showing FADD expression in different groups. (C) GSEA analysis showed FADD positively correlated to apoptosis and negatively correlated to tight junction in TCGA database. (D) Immunofluorescence imaging showing ZO-1, Occludin and E-cadherin expression in different groups. (E) Relative expression of FADD in HBE cells exposed to PM2.5 at different time-points. (F) Relative expression of FADD, ZO-1, Occludin and E-cadherin in HBE cells exposed to PM2.5. The data represent means±s.d. All data are representative of three experiments. *p < 0.05. **p < 0.01. |
In the meantime, we performed FADD single gene GSEA according to FADD-related genes in adjacent normal tissues deposited at The Cancer Genome Atlas (TCGA) adenocarcinoma database. As a result, we found that FADD was tightly associated with activation of apoptosis pathways. Meanwhile, FADD was also associated with those suppressed pathways including apical junction assembly, apical junction complex, and tight junction (Figure 3C). To verify the impact of PM2.5 exposure on tight junction, we examined the expression levels of E-cadherin, ZO-1 and Occludin in vivo and in vitro. Compared with those in other groups, PM2.5 exposure led to a significant decrease in the expression of these markers in OVA-induced mice (Figure 3D). Meanwhile, we found that PM2.5 exposure inhibited the mRNA expression of E-cadherin, ZO-1 and Occludin in vitro with the increased expression of FADD (Figure 3F).
PM2.5 Aggravated HDM-Induced Apoptosis and Tight Junction Disruption in Human Airway Epithelial Cells
To further confirm the apoptosis enhanced by PM2.5 in airway epithelial cells, HBE cells were exposed to PM2.5 and/or HDM in vitro. We found that PM2.5 exposure significantly induced apoptosis in HBE assessed by TUNEL staining. Similarly, the fluorescence intensities in cells exposed to HDM plus PM2.5 were significantly higher than those in cells exposed to PM2.5 or HDM alone (Figure 4A). In addition, to furnish more credible information about the apoptosis induced by PM2.5, we used the Scan R high content screening (HCS) technique to detect related apoptosis indicators. We showed that PM2.5 exposure was associated with increased apoptosis. Furthermore, PM2.5 exposure increased the number of apoptosis cells treated with HDM (Figure 4B). We also detected the expression of FADD in HBE cells by immunofluorescence. It showed that PM2.5 exposure was associated with increased fluorescence intensity of FADD in HBE cells as well. PM2.5 exposure significantly increased the HDM-induced fluorescence intensities of FADD in HBE cells (Figure 4C). Furthermore, we examined the protein expression levels of E-cadherin, ZO-1 and Occludin by Western blot. We found that PM2.5 exposure significantly reduced the expression of E-cadherin, ZO-1 and Occludin in HBE cells compared with those in control group. Meanwhile, the expressions of E-cadherin, ZO-1 and Occludin in HDM-treated HBE exposed to PM2.5 were further decreased (Figure 4D). Our results indicated that PM2.5 aggravated HDM-induced apoptosis and tight junction disruption, which might be associated with the increased expression of FADD.
 |
Figure 4 PM2.5 aggravated HDM-induced apoptosis and tight junction disruption in human airway epithelial cells. (A) TUNEL assay showed apoptosis in different group. (B) Apoptosis in different group detected by Scan R high content screening (HCS) technique. (C) Immunofluorescence imaging showing FADD expression in different groups. (D) The expression of ZO-1, Occludin and E-cadherin in HBE cells were detected by Western blot. The data represent means±s.d. All data are representative of three experiments. |
Down-Regulation of FADD Attenuates PM2.5-Induced Apoptosis and Tight Junction Disruption in Human Airway Epithelial Cells
To deeply investigate the role of FADD in PM2.5-mediated apoptosis and tight junction disruption, we knocked down the expression of FADD in HBE cells. The immunofluorescence showed FADD expression was significantly down-regulated by FADD shRNA (Figure 5B). Compared with that in mock cells, the number of apoptosis cells was significantly decreased in FADD-knockdown cells after PM2.5 plus HDM treatment (Figure 5A). Consistently, the apoptosis signal in PM2.5 puls HDM group was decreased by inhibition of FADD (Figure 5B). To further analyze the effects of FADD on epithelial surface markers after PM2.5 plus HDM exposure, the expression levels of E-cadherin, ZO-1 and Occludin were assessed by immunofluorescence. We found that the expression levels of E-cadherin, ZO-1 and Occludin in PM2.5 plus HDM group were recovered after down-regulation of FADD (Figure 5C).
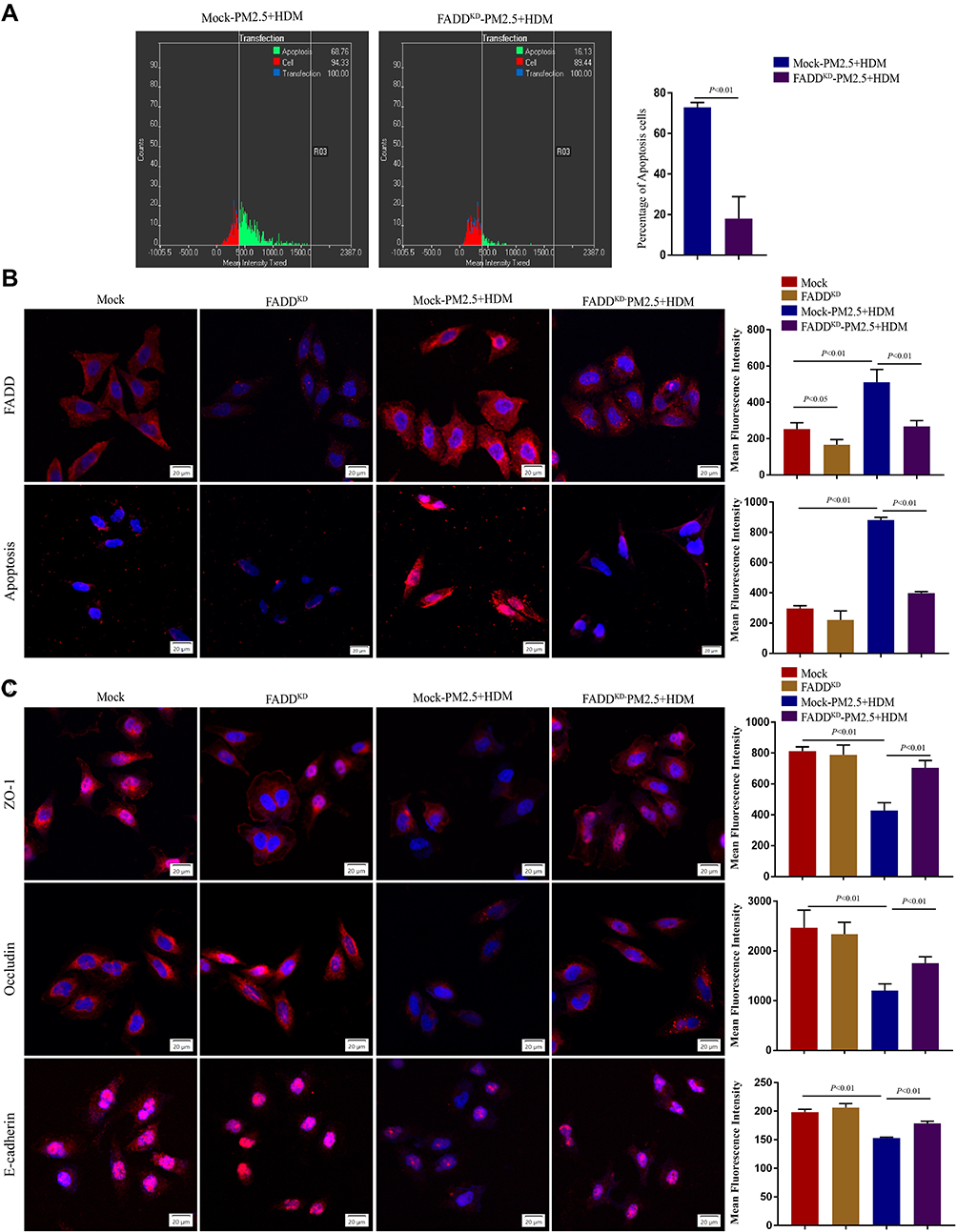 |
Figure 5 Down-regulation of FADD attenuates PM2.5-induced apoptosis and tight junction disruption in human airway epithelial cells. (A) Apoptosis in different group detected by Scan R high content screening (HCS) technique. (B) Immunofluorescence imaging showing FADD expression and TUNEL assay showing apoptosis in different groups. (C) Immunofluorescence imaging showing ZO-1, Occludin and E-cadherin expression in different groups. Mock, HBE cells were infected with GFP-only virus control. FADDKD, HBE cells were infected with recombinant lentivirus containing shFADD. The data represent means±s.d. All data are representative of three experiments. |
To further confirm the role of FADD in epithelial tight junction after PM2.5 plus HDM treatment, we measured the transepithelial electrical resistance (TEER) value in FADD knockdown cells. The result showed that the TEER value was decreased in mock cells after PM2.5 plus HDM treatment. Importantly, compared with mock, down-regulation of FADD restored the TEER value (Figure 6A). Together, these results strongly indicated that down-regulation of FADD attenuated PM2.5-induced apoptosis and tight junction disruption in human airway epithelial cells.
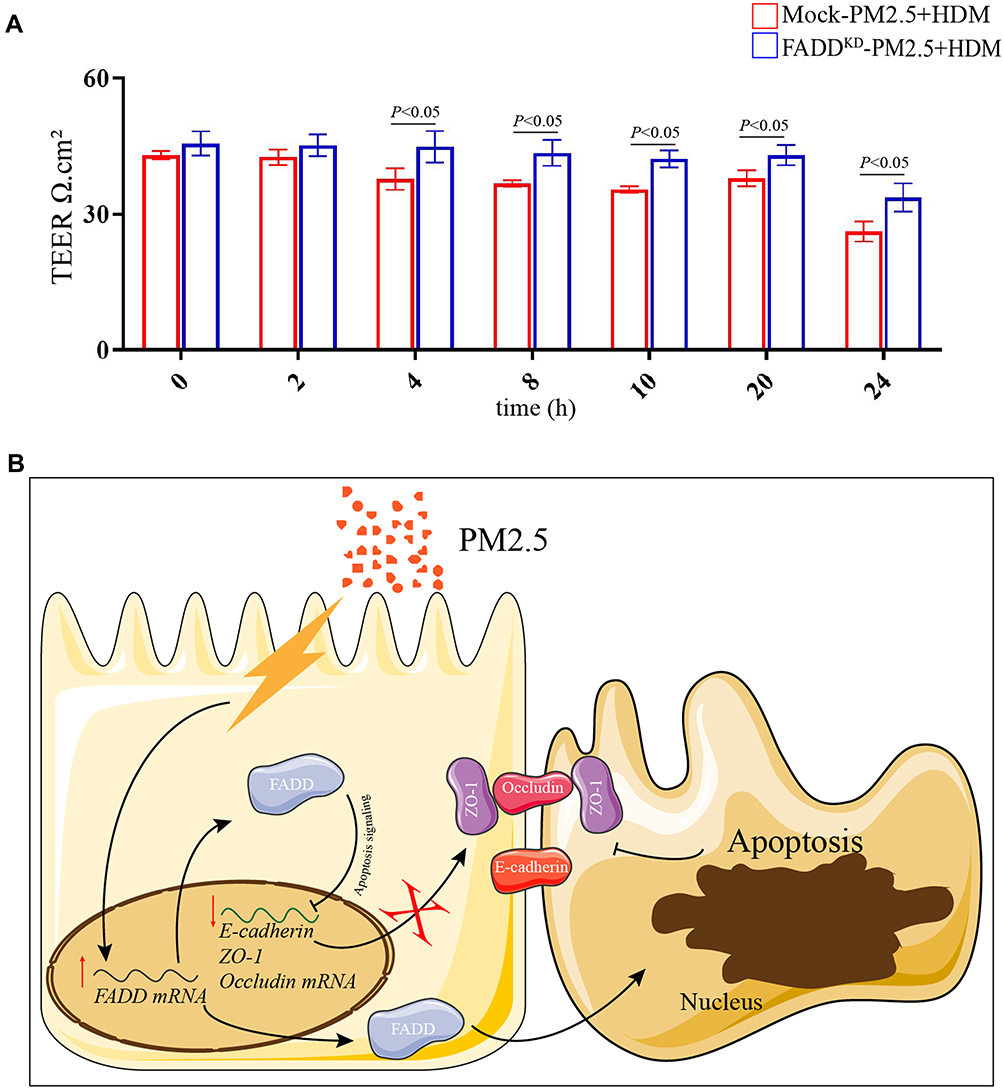 |
Figure 6 TEER value and Schematic model. (A) TEER value in Mock and FADD knock-down cells after PM2.5 plus HDM treatment at different time-points. (B) Schematic model, PM2.5 exposure increased the transcript and protein levels of FADD. On one hand, the increased FADD inhibited the transcript levels of ZO-1, Occludin and E-cadherin through apoptosis signaling, which decreased the protein levels of ZO-1, Occludin and E-cadherin. On the other hand, the increased FADD aggravated apoptosis. Eventually, tight junction was disrupted by PM2.5. |
Discussion
Asthma is a common, chronic respiratory disease affecting varieties of the population in different countries. It is characterized by bronchial hyper-responsiveness and airway inflammation. Increasing evidence has shown that the inhalation of PM2.5 was associated the adverse health effects throughout the respiratory and the circulatory system.23–25 However, there is limited experimental evidence addressing the relationship between exposure to air pollution and development of asthma.26 In this study, we aimed to test the effects of PM2.5 exposure and explore the mechanism on asthma severity. Our results showed that PM2.5 exposure disrupted airway epithelial barrier function by decreasing the expression of tight junction proteins, resulting in the exacerbation of airway inflammation in OVA-induced asthmatic mice through FADD-regulated apoptosis.
To investigate the potential contribution of PM2.5 exposure to the increased development of asthma, an in vivo mouse model of asthma was adopted. Interestingly, we found that administration of OVA together with PM2.5 could trigger the development of severe airway inflammation in vivo. Previous studies have shown that PM2.5 stimulated T-helper type 2 cell differentiation and increased inflammatory responses of allergic diseases.27–29 Consistent with previous studies, our results showed up-regulated inflammatory responses and increased infiltration of inflammatory cells in OVA-challenged mice exposed to PM2.5 compared with those in OVA-induced mice.30,31 Although PM2.5 administration alone could also induce mild airway inflammation, the pathological features are very different.32 Significant epithelial basement membrane thickening, accompanied by goblet cell metaplasia and mucus production, could be observed only in mice subjected to PM2.5 exposure combined with OVA. However, these pathological features were absent in the mice subjected to PM2.5 administration alone.
When tissue is damaged/wounded or invaded by foreign antigens, a series of signaling cascades activate the immune system, resulting in inflammatory responses that lead to EMT.33 In agreement with this, our RNA-Seq and GSEA results suggested that PM2.5 exposure could suppress cilium-related genes and induce EMT in OVA-induced mice. In addition, the results from TCGA and GEO database also revealed that FADD related to EMT and apoptosis. EMT could contribute to airway remodeling and fibrosis, which is associated with asthma severity. EMT is also known to disrupt cell–cell contact as tight junctions (TJs) disband. The loss of apicobasal cell polarity is also characteristic of EMT and permits intermingling of apical and basolateral membrane components.34,35 We also found the expression of these transcription factors (TWIST1, POU2F3, MKX, TEAD4, and E2F1) were higher in OVA-challenged mice exposed to PM2.5. They might be the regulator of EMT in OVA-induced mice exposed to PM2.5, which need to be further investigated.
Excessive apoptosis of epithelial cells increases the destruction of the epithelial barrier. Tight junction disruption (TJD) is usually thought of as a downstream consequence of caspase cleavage during the apoptotic process, which has an important role in several pathologies including viral and bacterial infection,36,37 inflammation,38,39 and tumor progression.40 A study has reported that green tea polyphenols induced apoptosis in intestinal epithelia mediated by caspase-8 through a FADD-dependent pathway.41 Thus, we wondered whether FADD pathway affects the airway epithelial barrier. In this study, the results suggested PM2.5 exacerbated airway epithelial barrier damage in asthmatic mice by increasing epithelial cell apoptosis and inhibiting tight junction protein expression through regulation of FADD expression. The integrity of the airway epithelial barrier relies on the expression of several actin cytoskeleton-bound proteins, including E-cadherin, β- catenin, ZO-1, claudins and occludins.42 As is shown in the results, PM2.5 caused the apoptosis of airway epithelial cells and the down-regulation of the expressions of ZO-1, Occludin and E-cadherin, which were closely related to the airway epithelial barrier, leading to the barrier dysfunction. By down-regulating the expression of FADD, we found that the apoptosis of airway epithelial cells was significantly improved. More importantly, the expression of tight junction protein was significantly restored in airway epithelial barrier as knocking down FADD. These results suggest that PM2.5 may aggravate the apoptosis of asthmatic airway epithelial cells through the apoptotic pathway dependent on FADD. In future studies, it will be of interest to assess whether the loss of FADD is also associated with allergic sensitization.
In conclusion, the present study established that PM2.5 exposure led to the specific degradation of the tight junctions that regulated paracellular permeability, which was mediated by FADD through apoptosis signalling, resulting in the disruption of airway epithelial barrier function in an OVA-induced model (Figure 6B). With the continuous development of economy and industry, air pollution has become a problem that cannot be ignored. This study is to provide some theoretical basis for better prevention and treatment of related respiratory diseases, and to study the partial mechanism of PM2.5 aggravating the airway epithelial barrier damage of experimental asthma.
Data Sharing Statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics Approval and Consent to Participate
All animal experiments were performed and approved in accordance with the guidelines of the Institutional Animal Care and Use Committee in Southwest Jiaotong University.
Acknowledgments
We thank Yi Zhang, Linqiao Tang, and Jinkui Pi from Research Core Facility of West China Hospital for technical assistance.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the National Natural Science Foundation of China (81970026, 82000029), Health Commission of Sichuan Province (19ZD002, 20PJ208), Science and Technology Department of Sichuan Province (2018JY0380), Health Commission of Chengdu (2021011, 2021021, 2021040), Chengdu High-level Key Clinical Specialty Construction Project (ZX20201202020), Sichuan Province Administration of Traditional Chinese Medicine (2020JC0118), Chengdu Science and Technology Bureau (2021-YF09-00102-SN, 2020-YF05-00003-SN), the program for combination of Medical and Engineering of Southwest Jiaotong University (2682021ZTPY007, 2682020ZT8).
Disclosure
The authors declare that they have no conflict of interest.
References
1. Dhami S, Nurmatov U, Agache I, et al. Allergen immunotherapy for allergic asthma: protocol for a systematic review. Clin Transl Allergy. 2016;6(1):1–5.
2. Nawijn MC, Hackett TL, Postma DS, Oosterhout A, Heijink IH. E-cadherin: gatekeeper of airway mucosa and allergic sensitization. Trends Immunol. 2011;32(6):248–255. doi:10.1016/j.it.2011.03.004
3. Le S. Tight junctions on the move: molecular mechanisms for epithelial barrier regulation. Ann N Y Acad Sci. 2012;1258:9–18. doi:10.1111/j.1749-6632.2012.06613.x
4. Kazunari S, Kenji K. Tight junctions in the development of asthma, chronic rhinosinusitis, atopic dermatitis, eosinophilic esophagitis, and inflammatory bowel diseases. J Leukoc Biol. 2020;107(5):749–762. doi:10.1002/JLB.5MR0120-230R
5. Folkerts G, Nijkamp FP. Airway epithelium: more than just a barrier! Trends Pharmacol Sci. 1998;19(8):334–341. doi:10.1016/S0165-6147(98)01232-2
6. Goto Y, Uchida Y, Nomura A, et al. Dislocation of E-cadherin in the airway epithelium during an antigen-induced asthmatic response. Am J Respir Cell Mol Biol. 2000;23(6):712. doi:10.1165/ajrcmb.23.6.4031
7. Tourneur L, Chiocchia G. FADD: a regulator of life and death. Trends Immunol. 2010;31(7):260–269. doi:10.1016/j.it.2010.05.005
8. Kirrane EF, Luben TJ, Benson A, et al. A systematic review of cardiovascular responses associated with ambient black carbon and fine particulate matter. Environ Int. 2019;127:305–316. doi:10.1016/j.envint.2019.02.027
9. Li R, Kou X, Hong G, Xie J, Dong C. Effect of ambient PM(2.5) on lung mitochondrial damage and fusion/fission gene expression in rats. Chem Res Toxicol. 2015;28(3):408–418. doi:10.1021/tx5003723
10. Holgate ST, Wenzel S, Postma DS, Weiss ST, Renz H, Sly PD. Asthma. Nat Rev Dis Primers. 2006;1(1):15025. doi:10.1038/nrdp.2015.25.
11. O’ Connor GT, Neas L, Vaughn B, et al. Acute respiratory health effects of air pollution on children with asthma in US inner cities. J Allergy Clin Immunol. 2008;121(5):1133–1139.e1131. doi:10.1016/j.jaci.2008.02.020
12. Andersen ZJ, Loft S, Ketzel M, et al. Ambient air pollution triggers wheezing symptoms in infants. Thorax. 2008;63(8):710–716. doi:10.1136/thx.2007.085480
13. Lei Z, Xiang H, Ying XC, et al. Transcriptome-wide profiling discover: PM2.5 aggravates airway dysfunction through epithelial barrier damage regulated by Stanniocalcin 2 in an OVA-induced model. Ecotoxicol Environ Saf. 2021;220:12408.
14. He X, Zhang L, Xiong A, Ran Q, Li G. PM2.5 aggravates NQO1-induced mucus hyper-secretion through release of neutrophil extracellular traps in an asthma model. Ecotoxicol Environ Saf. 2021;218(12):112272. doi:10.1016/j.ecoenv.2021.112272
15. Song Q, He X, Xiong Y, Wang J, Li G. The functional landscape of Golgi membrane protein 1 (GOLM1) phosphoproteome reveal GOLM1 regulating P53 that promotes malignancy. Cell Death Discov. 2021;7(1):42. doi:10.1038/s41420-021-00422-2
16. Xie T, Luo GY, Zhang Y, et al. Rho-kinase inhibitor fasudil reduces allergic airway inflammation and mucus hypersecretion by regulating STAT6 and NFκB. Clin Exp Allergy. 2015;45(12):1812–1822.
17. Liang X, He X, Li Y, et al. Lyn regulates epithelial–mesenchymal transition in CS-exposed model through Smad2/3 signaling. Respir Res. 2019;20(2):1–2.
18. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102(43):15545–15550. doi:10.1073/pnas.0506580102
19. Heijink IH, Kuchibhotla VN, Roffel MP, et al. Epithelial cell dysfunction, a major driver of asthma development. Allergy. 2020;75(8);1902–1917.
20. Haddad A, Gaudet M, Plesa M, et al. Neutrophils from severe asthmatic patients induce epithelial to mesenchymal transition in healthy bronchial epithelial cells. Respir Res. 2019;20:1–4.
21. Aibar S, González-Blas C, Moerman T, et al. SCENIC: single-cell regulatory network inference and clustering. Nat Methods. 2017;14(11):1083–1086.
22. Mouasni S, Tourneur L. FADD at the crossroads between cancer and inflammation. Trends Immunol. 2018;14(11):39.
23. Ai S, Qian ZM, Guo Y, et al. Long-term exposure to ambient fine particles associated with asthma: a cross-sectional study among older adults in six low- and middle-income countries. Environ Res. 2019;168:141–145. doi:10.1016/j.envres.2018.09.028
24. Tian Y, Xiang X, Juan J, et al. Fine particulate air pollution and hospital visits for asthma in Beijing, China. Environ Pollut. 2017;230:227. doi:10.1016/j.envpol.2017.06.029
25. Khreis H, Kelly C, Tate J, et al. Exposure to traffic-related air pollution and risk of development of childhood asthma: a systematic review and meta-analysis. Environ Int. 2017;100:1–31.
26. Eisner M, Anthonisen N, Coultas D, et al. An official American Thoracic Society public policy statement: novel risk factors and the global burden of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2010;182(5):693–718. doi:10.1164/rccm.200811-1757ST
27. Papi A, Brightling C, Pedersen SE, Reddel HK. Asthma. Manag Care Interface. 2017;10(9):80–82.
28. Shukla A, Timblin C, Berube K, Gordon T, Mossman BT. Inhaled particulate matter causes expression of nuclear factor (NF)- κ B–related genes and oxidant-dependent NF- κ B activation in vitro. Am J Respir Cell Mol Biol. 2000;23(2):182–187. doi:10.1165/ajrcmb.23.2.4035
29. Ogino K, Zhang R, Takahashi H, et al. Allergic airway inflammation by nasal inoculation of Particulate Matter (PM2.5) in NC/Nga mice. PLoS One. 2014;9(3):3. doi:10.1371/journal.pone.0092710
30. Yu PF, Pang LL, Mao QS, et al. Dose dependency PM2.5 aggravated airway inflammation in asthmatic mice via down-regulating expression of ITGB4. Eur Rev Med Pharmacol Sci. 2019;23(4):1688–1697.
31. He M, Ichinose T, Yea Y. Urban PM2.5 exacerbates allergic inflammation in the murine lung via a TLR2/TLR4/MyD88-signaling pathway. Sci Rep. 2017;7(1):11027. doi:10.1038/s41598-017-11471-y
32. Wang H, Song L, Ju W, et al. The acute airway inflammation induced by PM2.5 exposure and the treatment of essential oils in Balb/c mice. Sci Rep. 2017;7(1):44256. doi:10.1038/srep44256
33. Qian S, Jie F, Billiar TR, Scott MJ. Inflammasome and autophagy regulation: a two-way street. Mol Med. 2017;23(1):188–195.
34. Jiménez-Salazar J, Posadas-Rodríguez P, Lazzarini-Lechuga RC, et al. Membrane-initiated estradiol signaling of epithelial-mesenchymal transition-associated mechanisms through regulation of tight junctions in human breast cancer cells. Horm Cancer. 2014;5(3):161–173. doi:10.1007/s12672-014-0180-3
35. Micalizzi DS, Farabaugh SM, Ford HL. Epithelial-mesenchymal transition in cancer: parallels between normal development and tumor progression. J Mammary Gland Biol Neoplasia. 2010;15(2):117–134. doi:10.1007/s10911-010-9178-9
36. Liu S, Wei Y, Le S, Turner JR, Wang T. Tight junction proteins claudin-1 and occludin control hepatitis C virus entry and are downregulated during infection to prevent superinfection. J Virol. 2009;83(4):2011–2014. doi:10.1128/JVI.01888-08
37. Evans MJ, Hahn TV, Tscherne DM, et al. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature. 2007;446(7137):801–805. doi:10.1038/nature05654
38. Bruewer, Matthias, Utech, Bruewer M, Utech M, Ivanov AI, et al. Interferon-γ induces internalization of epithelial tight junction proteins via a macropinocytosis-like process. FASEB J. 2005;19(8):923–933. doi:10.1096/fj.04-3260com.
39. Prasad S, Mingrino R, Kaukinen K, et al. Inflammatory processes have differential effects on claudins 2, 3 and 4 in colonic epithelial cells. Lab Invest. 2005;85(9):1139–1162.
40. Martin TA, Jiang WG. Loss of tight junction barrier function and its role in cancer metastasis. Biochim Biophys Acta. 2009;1788(4):872–891. doi:10.1016/j.bbamem.2008.11.005
41. Oz HS, Ebersole JL. Green tea polyphenols mediated apoptosis in intestinal epithelial cells by a fadd-dependent pathway. J Cancer Ther. 2010;1(3):105.
42. Wan H, Winton HL, Soeller C, et al. The transmembrane protein occludin of epithelial tight junctions is a functional target for serine peptidases from faecal pellets of Dermatophagoides pteronyssinus. Clin Exp Allergy. 2001;31(2):279–294. doi:10.1046/j.1365-2222.2001.00970.x
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.